

Abstract
Glioblastoma is the most common and lethal primary malignant brain tumor in adults. Patients die from recurrent tumors that have become resistant to therapy. New strategies are needed to design future therapies that target resistant cells. Recent genomic studies have unveiled the complexity of tumor heterogeneity in glioblastoma and provide new insights into the genomic landscape of tumor cells that survive and initiate tumor recurrence. Resistant cells also co-opt developmental pathways and display stem-like properties; hence we propose to name them recurrence-initiating stem-like cancer (RISC) cells. Genetic alterations and genomic reprogramming underlie the innate and adaptive resistance of RISC cells, and both need to be targeted to prevent glioblastoma recurrence.
Glioblastoma therapeutic challenge: preventing and treating tumor recurrence
Patients with glioblastoma multiforme (GBM) have a median overall survival of approximately 15 months (1). Standard therapy for GBM encompasses maximally safe surgical resection followed by radiation and chemotherapy (1–4). While many patients’ tumors initially respond to these treatments, no current regimen can overcome inevitable tumor recurrence, after which patient survival drops to less than 6 months. Personalized therapies against molecular targets that drive the growth of the bulk of primary tumors (5, 6) have so far also been unsuccessful in clinical trials, warranting new approaches.
GBM recurs locally around the surgical cavity without evidence of tumor growth into other organs, despite evidence for extracranial tumor cell dissemination (7, 8). Hence, the failure to control tumor growth at the primary site is the major cause of patient demise. GBM patients have poor prognosis due to tumor cells that survive initial therapies and cause tumor regrowth/recurrence. Tumor heterogeneity is an important reason for the failure of conventional and molecularly targeted therapies (9–12). Consequently, it is essential to identify and characterize which types of cancer cells can evade therapies and become recurrence-initiating cancer cells so that they can be targeted. New knowledge derived from studying the genetic evolution of cancer cell populations in response to therapy, as well as the ability of cancer cells to adopt stem-like characteristics, has provided us with unprecedented new insights to tackle this major challenge.
Complexity of tumor heterogeneity in GBM
Intertumoral heterogeneity.
Recent advances in genomic analyses provide us with a comprehensive view of the tumor-to-tumor complexity of GBM. Subgroups have been defined based on distinct genetic and epigenetic alterations and gene expression profiles (13–16). Differences in cell of origin may further underlie intertumoral heterogeneity (17), and neural stem cells, several CNS progenitor populations, and even mature astrocytes and neurons have been proposed as cells of origin for GBM (18–22). The specific phenotypes of tumors may depend on both the cells of origin and subsequent genetic and epigenetic alterations to these cells (Figure 1A), but our understanding of these matters is still incomplete. The tumorigenic processes within GBM subgroups are being gradually unraveled, particularly in those carrying isocitrate dehydrogenase (IDH) gene mutations (23, 24). Gliomas that carry mutations in the IDH1/2 enzymes produce excessive amounts of hydroxyglutarate (2-HG), which interferes with the function of several epigenetic enzymes, leading to genomic reprogramming (25, 26). Subgroup-specific therapeutic designs will need to be implemented based on the final biological phenotype, which integrates all sources of tumor heterogeneity. For example, drugs that inhibit epigenetic modifiers could reprogram the genomes of gliomas with mutant IDH1/2 enzymes (25), while the use of temozolomide in such tumors may need to be reconsidered, as it induces a hypermutated phenotype (27–29).
Figure 1. The complexity of inter- and intratumoral heterogeneity in glioblastoma.
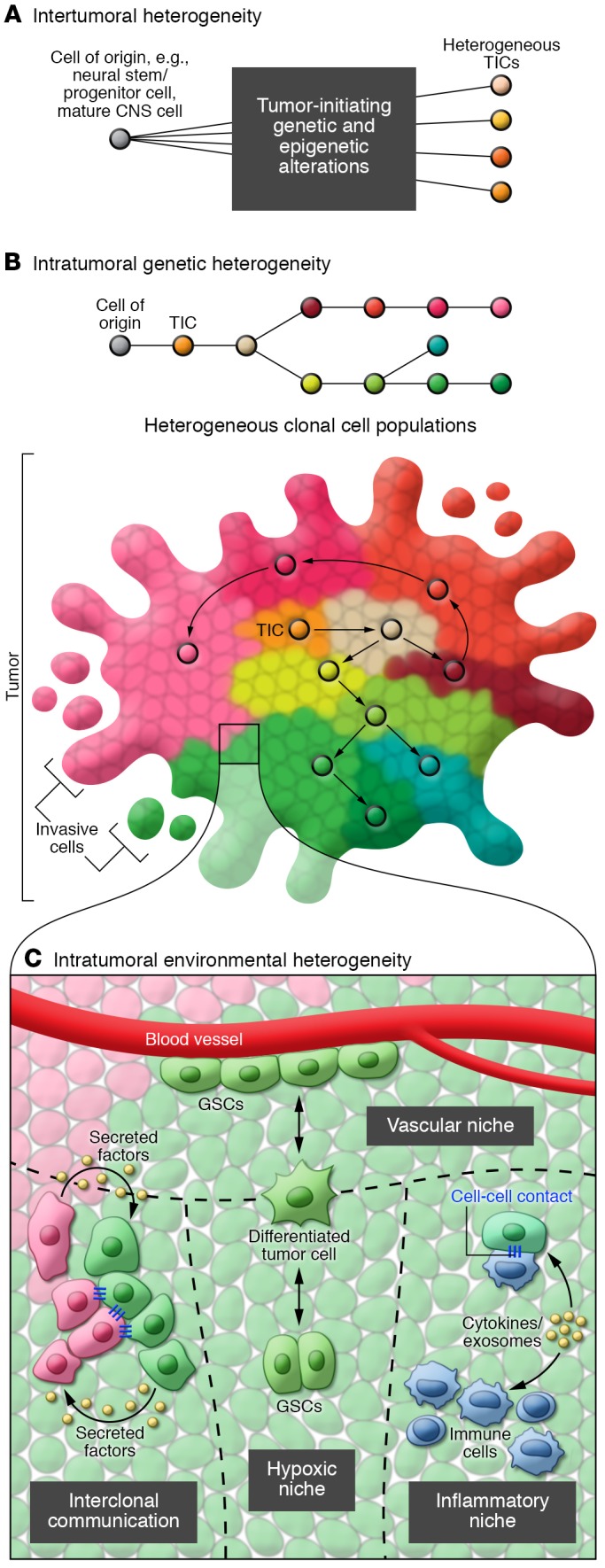
(A) Heterogeneity between individual patient tumors stems from both the cell of origin and the subsequent major epigenetic and genetic alterations. These variations produce different types of tumor-initiating cells (TICs). (B) TICs expand and establish genetically divergent clonal cell populations. During this clonal evolution process, cellular offspring acquire diverse genetic alterations and engender a variety of clones. Cells with similar types of genetic alterations exist in close spatial proximity, but their invasive properties will lead to clonal mixing and normal brain invasion. (C) Further heterogeneity at the cellular level is added by environmental factors. Proximity to blood vessels (vascular and hypoxic niches), paracrine signals between tumor cells, and immune responses (inflammatory niche), will influence individual tumor cell biology, including regulating stemness versus differentiation state of glioma stem cells (GSCs).
Clonal evolution of cancer cell populations drives intratumoral heterogeneity.
Pathological analyses of GBM have long provided evidence of extensive intratumoral heterogeneity (30–32), including variable amounts of necrosis (sometimes with perinecrotic pseudopalisading cells), evidence for intratumoral hemorrhage and thrombosis, glomeruloid microvascular proliferation, and pleomorphic tumor cells (30–32). Immunohistochemistry and molecular biology studies have shown heterogeneous patterns of tumor marker expression, and uneven cellular distribution of genetic alterations (33–35). Nonetheless, a deeper understanding of the complexity of intratumoral heterogeneity has remained elusive because of limitations in technology, which have restricted the ability to trace different tumor cell populations within a human tumor mass. New genomic analyses on separate surgical samples from the same tumors have revealed that multiple clones harboring a variety of genetic alterations coexist within the same tumor. Segregating clones based on the presence of independent or shared mutations has revealed part of the tumor development process (27–29, 35–37). The clonal evolution model posits that tumor formation is initiated in a cell of origin and is followed by the accumulation of single or multiple somatic genetic alterations, leading to advantages in survival or growth (38). Knowledge derived from genetic syndromes (39) and GWAS studies (40, 41) has shown that an inherited genetic component may accelerate this process. The fittest cell populations likely establish precancerous clones, although we have little direct evidence to support this process in human GBM because of limitations in detecting the early steps in brain cell transformation (42, 43). Divergent genetic alterations in early transformed cells can subsequently give rise to a variety of clones under the selective pressure of the evolutionary ecosystem in the tumor (27–29, 35–37). A cell’s spatial location in the tumor is related to its divergent genomic profile, and clones with similar types of genetic alterations are more proximal to each other than those with dissimilar profiles (Figure 1B and refs. 35, 37, 44).
A high capacity for dissemination is one of the defining features of gliomas (45, 46), and this invasion process renders tumors more complex. Invading tumor cells escape at the periphery of the tumor mass and diffusely infiltrate the normal brain parenchyma (47). Deeply infiltrated tumor cells are more likely to escape surgery, and we do not know whether infiltration is a property of a more resilient cell population that initiates and drives tumor recurrence.
There are still many unanswered questions related to what is the exact cell of origin, how fast individual clonal populations develop, and how clonal populations define intratumoral heterogeneity and societal interactions such as competition and cooperation between individual clones and stromal cells. To address these unknowns, further in-depth genetic analyses on hundreds of samples from single patient tumors are warranted. These analyses will define the relative distribution of cell populations in the tumor so that a 3D reconstruction and model of tumor formation can be envisioned and related to imaging (48, 49).
Intratumoral heterogeneity at the single-cell level.
Molecular heterogeneity exists even at the cellular level between cells that carry similar genetic alterations (50, 51). Recently, single-cell transcriptional profiling of 430 cells from five different primary GBM tumors uncovered intratumoral heterogeneity at the cellular level. Individual cells displayed different transcription signatures for genes that regulate cell cycle, hypoxia, immune response, and stemness (50). Expanding single cells into clonal populations further demonstrated unique functionality including proliferation, differentiation abilities, and different sensitivities to temozolomide (51). The complexity of intercellular heterogeneity also relates to variations in epigenetic or transcriptional programs, which define a hierarchy of cells showing variable degrees of differentiation and stemness. The molecular phenotype of each cell is further altered by its unique position relative to other cells and local environment. Hypoxia (52), vascular niche for maintaining stemness (53, 54), and secreted factors produced by other tumoral or stromal cells all influence the molecular phenotypes of tumor cells (55–57). Heterogeneity at the cellular level adds another layer of complexity (Figure 1C). Astoundingly, although these observations hint at each tumor cell being unique, tumor cells can be connected in a network that responds to a therapeutic insult in a coordinated fashion (47). Regional heterogeneity in molecular properties of tumor cells is thus governed by local variation in environmental selection forces.
Glioma stem cells
Glioma stem cells (GSCs) are defined as tumor cells capable of forming heterogeneous glial tumors. They are endowed with specific properties including high tumorigenic ability, unlimited self-renewal potential, and capacity for multipotent differentiation, e.g., generating a diversity of progeny (58, 59). The existence of a hierarchy of cells within gliomas, including some with GSC characteristics, is recognized, although many questions related to their number, dynamics, and physiology remain, in part due to limitations in biomarkers.
Certain GSC populations display higher intrinsic chemo- and radioresistance than non-GSCs, indicating that a fraction of the primary tumor GSC population can survive the initial therapy and initiate recurrent tumor formation (58–62). GSCs can overcome the damage induced by chemotherapy and radiotherapy not only through innate properties (e.g., genetic heterogeneity), but also through adaptive resistance pathways (20, 55, 63). How well studies that are based on known cell surface markers represent GSC populations and behavior is being debated (58, 59). For example, CD133, a commonly used GSC marker, fails to identify all tumor cells capable of self-renewal and tumor-initiating ability (64, 65). The GBM single-cell analysis also revealed that a surprisingly large subpopulation of cells (~40%) had a stemness signature (50). How many of these cells may display self-renewal and tumor initiation is unknown, and they may encompass both GSCs and their hierarchical progeny, such as cancer cells with transit-amplifying or progenitor cell properties. This subpopulation had low expression of cell cycle genes, suggesting slower growth than the remainder of tumor cells. There is emerging evidence that GSCs vary in different GBM subtypes (66–68), between treatment-naive and recurrent GBM (51, 55, 63, 69), and even within alternate niches in the same tumor (i.e., perivascular and hypoxic) (54, 66, 70). Which subpopulations of GSCs can initiate tumor recurrence needs further clarification.
We need to also continually reevaluate and refine the concept of GSCs, taking into account several unanswered questions: can we identify stem cell markers that define specific GSC subpopulations; is there plasticity between non-GSCs and GSCs, such as cell identity drift; and what are the molecular mechanisms underlying the maintenance of GSC stem properties? These limitations are obstacles in defining which tumor cell populations are the most important contributors to tumor recurrence. New technical capabilities in single-cell analyses (50, 51) will soon quantify and characterize cells in GBM that are capable of initiating tumor recurrence.
Because of their tumor-sustaining capacity and resistance to conventional therapies, GSCs represent an important target in the quest to find more effective therapies for GBM. New genomic analyses have uncovered how therapeutic intervention alters the dynamics of glioma cell populations (27–29, 35–37, 44), and an increase in the size of the GSC population after radiation or chemotherapy has been suggested (71, 72). Studies have identified important signaling pathways that are required for the biological maintenance of GSCs (58, 73), and they represent potential new targets to prevent tumor recurrence.
The process of tumor recurrence in GBM
Clinical treatment.
Although the clinical course of each GBM patient is unique, and influenced by tumor location, age, and complications, we present an outline of the standard of treatment. When possible, GBM patients receive a maximally safe surgical resection, and the best outcome is called gross total resection, where the entire tumor has been physically removed and only minimal residual disease remains that is invisible on postoperative contrast-enhanced MRI (ref. 74 and Figure 2, top). Thereafter, a standard protocol of radiotherapy (5 d/wk at 1–2 Gy/d) focused on the tumor mass and adjacent margin is delivered in combination with the alkylating chemotherapeutic temozolomide (75 mg/m2) over the course of 6 weeks (1–4). After this initial treatment phase, the subset of patients with gross total resection often exhibit stabilization of their disease, with no radiological evidence of further tumor growth (1, 3). Nearly all patients receive further adjuvant temozolomide during this radiographic progression-free phase, even though remnant tumor cells are likely undergoing active biological progression (75, 76). This intermediate phase is typically short-lived (a few months), and most patients develop radiological evidence of local recurrence around the surgical cavity (Figure 2, top and refs. 3, 44). Once the tumor grows back, patients may receive further therapies, including additional tumor resection, bevacizumab (an anti-VEGF antibody), different chemotherapy, or additional radiation therapy focused exclusively on the tumor site. Yet there is little evidence that such salvage treatments increase survival. Tumor cells resistant to multiple therapies persist in the brain parenchyma around the tumor cavity and underlie tumor repopulation, making them a critical target to overcome tumor recurrence.
Figure 2. Treatment and tumor recurrence in glioblastoma.

Top: MRI scans of a patient with a primary glioblastoma before treatment, after initial gross total resection followed by chemo- and radiotherapy, and after tumor recurrence. Middle: A cartoon rendering of the associated changes in clonal populations in the tumor at each stage. Surgery successfully removes the tumor and eliminates many subclones. Postoperative chemo- and radiotherapies can further reduce tumor burden around the surgical cavity. However, a small fraction of tumor cells survive and initiate the formation of the recurrent tumor. The length of each line is proportional to the number of mutations acquired between each clone and branching indicates acquisition of divergent mutations. We have proposed calling these surviving cells recurrence-initiating stem-like cancer (RISC) cells. Bottom: Phylogenetic tree showing the process of clonal evolution in the primary tumor, the survival of RISC cells that have acquired adaptive resistance to therapy after initial treatments, and their evolution into a recurrent tumor. The length of each line is proportional to the number of mutations acquired between each clone, and branching indicates acquisition of divergent mutations.
How do heterogeneous tumors respond to therapeutic intervention?
Genomic landscape analyses of pre- and post-treatment GBM pairs from the same patients have demonstrated that recurrent tumors display variable degrees of genetic relatedness to the original tumor (clonal evolution), but also have acquired new mutations (subclonal evolution) (27–29, 35–37, 44). Recurrence is a complex process, with a diversity of evolutionary trajectories broadly classified into linear recurrences that share extensive genetic similarity with the primary tumor, and branched evolution leading to the formation of divergent cell populations (subclones) (27–29, 35–37, 44). Recurrence shows a high degree of variability and can originate from one subpopulation that branched off early during tumorigenesis or much later. In most patients, 50–200 clonal or subclonal mutations are found at relapse (27, 29, 44), and this number can increase to more than 1,000 in cases with mismatch repair gene alterations (hypermutated tumors). This information provides for the first time a detailed genetic portrait of the impact of therapies on GBM.
These data further suggest that the efficacy of surgery in prolonging patient survival (74, 77, 78) occurs through both a reduction in the physical burden of tumor and an alteration in the dynamics of tumor cell populations. Surgical debulking reduces intratumoral heterogeneity by removing many subclones from the tumor. This conclusion is based on two observations: first, there is regional diversity in primary tumor clones, suggesting limited cell mixing (27–29, 36, 37, 44), and second, comparison of primary/recurrent tumors of patients having only received surgery shows that the recurrences diverge early from the primary tumor and lack the end mutations found in the primary tumor (27, 36). Postoperative chemo- and radiotherapies can likely also reduce clonal diversity when only biopsy or partial surgical resection is possible, but the main documented clinical benefit of these treatments after tumor debulking is probably the reduction of the infiltrative tumor cells left by surgery. Despite these treatments, small fractions of clones originating from beyond the surgical margin survive and lead to recurrence (Figure 2, middle).
What type of clone can initiate tumor recurrence?
Of the many primary tumor clones, it is important to determine which can engender recurrence-initiating cells. Deep sequencing of multiple sectors of primary/recurrent GBM pairs has shown that recurrent clones can variably originate from a clone positioned early, in the middle, or at the end of the clonal evolution process of the primary tumor (Figure 2, bottom, and refs. 27, 44). While the branching position for the start of each recurrent clone was variable in each patient, it was surprising that recurrent clones did not all originate from clones at the end point of the clonal selection process as was observed in other cancers (79, 80). The dominant clone at recurrence usually was not a lineal progeny from the main clone in the primary tumor. It is unclear whether this is because the dominant clones in the primary tumor were removed or killed by therapy, whether not all tumor cells can act as recurrence-initiating cells, or whether recurrent cells are more invasive and deeply infiltrated in the surrounding normal brain, protecting them from surgical removal. Thus, the number of genomic alterations is not simply correlated with therapeutic resistance in GBM, and recurrences either share most primary tumor mutations with accumulation of additional genetic alterations or diverge early genetically and evolve rather separately, with little in common with the primary tumor. Consequently, targets identified based on the analysis of the primary tumor may not be informative in treating the recurrence. Moreover, analyses combining the branching pattern with estimates of evolutionary rate suggest that subclones associated with recurrence were already present years before diagnosis, which implies that many of the unique genetic alterations found in the cells initiating recurrence were not caused by the treatment (29).
The above findings demonstrate that the tumor cells leading to recurrence differ from GSCs that initiated and maintained the primary tumor: they took a divergent evolutionary path and need to be studied separately. We hypothesize that recurrence-initiating cancer cells emerged from the residual tumor cell population that survived therapy and have stem-like properties, because they can initiate a recurrent GBM with a diversity of tumor cells. Therefore, we propose to call them recurrence-initiating stem-like cancer (RISC) cells. Early studies support this model. Cells with GSC properties can be isolated from recurrent GBM, and can generate heterogeneous GBM when transplanted in mice (66). Such cells are more aggressive than primary tumor–derived GSCs (51, 63, 69), consistent with the shorter survival of patients with recurrent GBM (81), and display different markers (loss of CD133 and gain of CD15, BMI1, and SOX2) (63). Human and mouse GSCs acquire therapeutic resistance following repeated chemo- and radiotherapy (55, 63), and temozolomide treatment in a transgenic mouse model of glioma showed that recurrent tumors originated from quiescent glioma cells with stem cell features (20). Therefore, we further hypothesize that RISC cells are a subset of GSCs that developed increased innate resistance to treatment through further genetic mutations, and acquired further adaptive resistance through epigenetic alterations during the course of therapy. Whether RISC cells are direct descendants from the GSCs in the primary tumor or have emerged from more differentiated progeny remains to be established (82). Comparisons of GSCs from the primary tumor with RISC cells from the recurrent tumor of the same patients will further help in defining the ontology of RISC cells. Understanding the genomic alterations and molecular architecture of RISC cells is critical for the development of successful therapies that could be deployed immediately after surgery, thereby preventing their adaptive resistance and expansion into a new tumor mass.
Molecular pathways implicated in therapeutic resistance of RISC cells
A growing number of molecular pathways have been associated with therapeutic resistance in GSCs and should be particularly relevant to treatment of RISC cells (58, 73).
Extracellular signaling pathways.
These signaling pathways are activated through autocrine or paracrine secretion of growth factors/cytokines, as well as homotypic tumor cell contacts and heterotypic tumor-stroma interactions, involving tumor cells, endothelial cells, and immune cells (Figure 1C). The most studied include the Wnt/β-catenin (83–85), Notch (86), receptor tyrosine kinase (RTK)/PI3K (87–92), NF-κB (93), SHH/GLI (94, 95), and JAK/STAT signaling pathways (96, 97), and others were recently reviewed (58, 73). These pathways maintain stemness in several types of normal and cancer stem cells (58, 73).
Transcription factors implicated in GSC maintenance.
The above signals are integrated through the activation of a limited number of transcription factors that control a variety of functions underlying GSC maintenance, including survival, self-renewal, proliferation, metabolism, and stemness state. They include OLIG2 (98, 99), c-Myc (100, 101), BMI1 (102, 103), SOX2 (104), NANOG (105), OCT4 (106), and ID1 (107). Some transcription factors were already known to maintain several types of normal stem cells (108–110), and c-Myc and OCT4 can help induce the formation of GSCs from astrocytes (111). Expression of all these transcription factors can be increased in GSCs (101–104), and is controlled by extracellular signaling pathways, superenhancers (112, 113), epigenetic regulation (102), and microRNAs (114, 115). They also activate DNA damage repair pathways that contribute to the therapeutic resistance of GSCs (58).
DNA damage repair and other resistance mechanisms.
The activation status of intrinsic or adaptive DNA damage pathways is an important determinant in chemo- and radioresistance of cancer cells. DNA damage checkpoint proteins can render human GSCs more resistant to radiation-induced apoptosis through increased efficiency in repair of damaged DNA. GSCs display increased expression of ataxia telangiectasia mutated (ATM), the cell cycle checkpoint protein RAD17, and the checkpoint kinases CHK1 and CHK2 (61). O-6-Methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme whose expression level is regulated epigenetically at the gene promoter, correlates with resistance to chemotherapies (116), and renders GSCs resistant to temozolomide (62). Secretion of exosomes, activation of autophagy (117), cell metabolism (66, 118), ROS production (119, 120), drug efflux (121), and microRNA expression (114, 115) are also altered in GSCs and can further enhance therapeutic resistance.
Which molecule should be the next target for clinical trials?
Mining resistance pathways is an important step to identify the next set of clinical targets, and several clinical trials have already been conducted to identify GSC resistance pathways. However, the antitumor effects observed in these trials were limited and did not prevent tumor recurrence (122–126). Further understanding of the role of these signaling pathways in the different tumor cell populations is needed to optimize their targeting. For example, adaptive radioresistance in mouse GSCs is associated with autocrine IGF-1 receptor activation and downregulation of Akt/ERK signaling, leading to a slow-growth/high–self-renewal phenotype (55). Additional targets for clinical testing need to be identified as well.
Targeting transcription factors for GSC maintenance.
Most clinical trials have targeted the ligand or receptors that initiate extracellular signaling. In contrast, the targeting of important transcription factors for GSC maintenance has not been achieved, largely because of inherent difficulties in designing small molecules to target them. Because multiple extracellular signaling pathways regulate the transcription factors that maintain GSCs, if one major pathway is inhibited, alternative pathways can substitute for their activation and lead to resistance (127–129). Soon, advances in drug design or different approaches (e.g., RNAi, stapled peptides, ref. 130; or artificial transcription factors, ref. 131) will allow targeting of the transcription factors that control developmental signaling pathways that GSCs hijack for self-maintenance.
Slow-growth state.
The effectiveness of radiation and chemotherapy is in part dependent on cell proliferation rate, which underlies the increased sensitivity of cancer cells over normal cells (132, 133). Ergo, GBM cells in a state of slow growth could play an important role in recurrence as suggested by human and mouse studies (20, 55, 134, 135). About 40% of the tumor cells in human GBM have a high-stemness and low-proliferation gene expression profile (50), yet the molecular mechanisms that maintain the GSC population and control their state of slow growth are still unknown. In normal stem cells, this is regulated by niche factors that ensure balanced self-renewal and differentiation through asymmetrical cell division, but this process is disrupted in gliomas (136–138). Studying and finding an effective therapy for slow-growing clones is challenging, because they are dispersed within the bulk of the tumor. Yet inroads into the slow-growing mechanisms of GSCs are being made (20, 55). Improved knowledge of the signaling mechanisms maintaining the slow-growth status will unveil new RISC cell targets.
Adaptive resistance to therapeutic intervention.
Prospective analysis of the molecular features of RISC cells in human tumors is challenging. The tissue surrounding the resection cavity has the appearance of normal brain parenchyma, and although autopsy studies have shown that it contains infiltrating cells (135, 139), their numbers are small and further surgical removal cannot be justified. Thus, we lack a sample of the tumor cells from the location of the recurrent tumor, and just have cells from the resected tumor bulk. This is important, as the biological features of deeply infiltrated cells may be different from those from the resected primary tumor (134, 135). To overcome this limitation, mouse models have been used to garner data on the molecular changes associated with radio- (55) and chemotherapies (20). Mouse GSCs can overcome the damage of repeated irradiation through gradual activation of IGF1R-dependent resistance pathways (55), and repeated chemo- and radiotherapies render human GSCs more aggressive and enrich their stem cell features (63, 140). GBM recurrence is also associated with a transition from glial to mesenchymal phenotype and is related to poor outcome (141, 142). Autophagy is another player in adaptive radioresistance mechanisms in GSCs (117). Such adaptive processes are driven by molecular alterations induced by epigenetic or genetic cues. These data demonstrate that adaptation mechanisms represent an important strategy for tumor cell survival and repopulation in response to therapeutic stress.
Tumor-stroma interactions and microenvironment.
Disrupting the tumor-stroma interactions that support GSC survival is another potential approach for antagonizing GSCs. Self-renewing GSCs are known to interact closely with endothelial cells (53, 143) and pericytes (144) in the perivascular niches. Microenvironmental changes such as hypoxia can also render tumors more resistant to conventional therapies. Hypoxic cells display increased radiation resistance because oxygen radicals play a major role in the damage generated by irradiation and because hypoxia-inducible transcription factor (HIF) alters the DNA damage response (145–147). Cytotoxic chemotherapies delivered via the bloodstream diffuse into the tumors from functional blood vessels, and the hypoxic areas are the furthest removed from these vessels. The hypoxic niche further promotes the self-renewal capacity of GSCs through HIF-mediated activation of the inducible nitric oxide synthase (iNOS) (148). Both HIF-1 and HIF-2 are important for GSC maintenance and tumor angiogenesis (52). GSCs display plasticity in the metabolic pathways they use to adapt to nutrient limitations in their microenvironment (66, 149, 150). Although the complexity of metabolic alteration in GSCs is not fully understood, metabolic inhibitors could be developed to target them in the future. Some studies suggest that GSCs preferentially use oxidative phosphorylation while the rest of the tumor is glycolytic, suggesting targeting with mitochondrial inhibitors (149, 150).
Immune suppression of tumor cells.
Cancer cells can also evade the immune response by activating immune checkpoint receptors (PD-1 and CTLA-4) on effector T cells, thus blocking the antitumor immune response (151, 152). Programmed death-1 (PD-1) is a T cell receptor that, upon activation by its ligand (PD-L1), can negatively regulate the T cell–mediated immune response. Tumor cells, including GBM, overexpress PD-L1 (153). Blockade of the PD-1 pathway in mice also promotes killing of GSCs by NK cells (140). Anti–PD-1 and –CTLA-4 antibodies have shown impressive efficacy with high response rates and tumor remissions in several cancer types in the clinic (154–156), and clinical trials are currently ongoing in GBM (NCT02667587, NCT02017717, NCT02311920; ClinicalTrials.gov). Patients with hypermutated tumors may benefit from immune checkpoint inhibitors because of increased neoantigen load and have slightly longer survival (27, 29, 157). GSCs also modulate the immune system by recruiting microglia/macrophages, modulating their function toward tumor growth and generating an immunosuppressive phenotype through the secretion of IL-10 and TGF-β (57, 158). These new findings support targeting immune checkpoints to activate an immune response against RISC cells.
Future therapeutic strategy
With our improved understanding of resistance mechanisms in the recurrent tumor (Figure 2), we can now envision new therapeutic strategies. At the risk of oversimplifying, we propose that current clinical therapy has three major shortcomings (Figure 3A), and we outline new strategies to overcome them (Figure 3, B and C). We also propose that future molecularly targeted therapies should be designed for at least four types of cancer cells based on their different properties and response to therapies: primary GSCs, RISC cells, and both the proliferating and the postmitotic fractions of non-GSCs. To facilitate this process, we have attempted to summarize their known and hypothetical properties (Table 1) and suggested ways to target them (Figure 3, B and C, and Table 2).
Figure 3. Opportunities for future molecularly targeted therapy for glioblastoma.

(A) The primary tumor is heterogeneous, and composed of several abundant cell subpopulations: cells with variable proliferation properties (pink and green), glioma stem cells (GSCs) (tan), and recurrence-initiating stem-like cancer (RISC) cells (red). Initial treatments successfully reduce the bulk tumor volume and its heterogeneity. However, subpopulations of RISC cells survive therapeutic intervention though intrinsic and adaptive resistance mechanisms (indicated by a blue ring). RISC cells initiate tumor recurrence through a second round of clonal evolution that repopulates the tumor. The current clinical strategies for GBM could be strengthened by adding molecular therapies to target: (i) initially resistant clones, (ii) adaptive resistance mechanisms, and (iii) the tumor when its population diversity and cell numbers are at their lowest. (B) Schematic showing the 4 proposed different cancer cell populations in the tumor (GSCs [tan], RISC cells [red], non-GSCs [light blue], and proliferating non-GSCs [dark blue]) and putative ways to target them (see also Table 2). RISC cells likely represent a subset of the GSC population with both innate and acquired resistance. The precise proportion of each cell population remains to be established. (C) Proposed optimal timing for each therapeutic option during the initial (primary treatment) and intermediate (stabilization/remission period before recurrence) phases of therapy.
Table 1. Working model to define different cell populations in the primary GBM (IDH1 wild type).

Table 2. Proposed therapies for different GBM cell populations.

Shortcomings of current clinical therapy.
Improved tumor targeting during the intermediate phase that follows the primary treatment period and ends at evidence for tumor recurrence could be developed. In this phase of a few months, imaging often provides evidence for remission/stabilization, but resistant cancer cells are left behind untreated, given ample time to recover from the initial treatment, and provided with the opportunity to grow and progress into a new tumor. Currently, aggressive treatment is not delivered in this phase, except for maintenance temozolomide (75, 76). Within this therapeutic window, the tumor is most vulnerable, as the heterogeneity and number of tumor cells are most reduced and they might not yet have acquired full resistance.
A second opportunity lies in targeting the intrinsically resistant subpopulation in the initial tumor. The basic effect of radiation and alkylating chemotherapy is DNA damage, and this effect is mainly (but not exclusively) dependent on the speed of cell division (132, 133). Slow-growing tumor cells are important targets to prevent chemo- and radioresistance (20, 55, 132, 133, 159–161). However, in the majority of molecularly targeted therapies, the targets of interest were selected based on highly expressed molecules found on a large fraction of tumor cells, such as the tyrosine kinase receptors, and their downstream signaling effectors (162–164). While these therapies are efficient in controlling the initial tumor, they may do little to prevent recurrence because they target mainly proliferating cells.
The third point that merits considerable further attention relates to acquired resistance mechanisms. Prior attempts to develop new molecular therapies aimed at targeting cancer cell resistance mechanisms focused on intrinsic resistance (162–165). However, the precursors to RISC cells can also become radioresistant through several acquired resistance mechanisms during the intermediate phase of disease (20, 55, 63). Identifying novel targets of adaptive resistance is currently a challenge, as there is a paucity of experimental models specifically addressing adaptive resistance in GBM (20, 55, 63). New models need to be developed to discover novel targets and validate appropriate targeting agents. While the brain has remained hermetic to most chemotherapies, novel approaches that open the blood-brain barrier in a sustained manner are becoming available, thus making the CNS accessible to a plethora of drugs already in use for other cancers (166).
Future molecularly targeted therapies: concepts and timing.
Future molecularly targeted therapies should be designed for all cancer subpopulations within the tumor, RISC cells being the most important to prevent tumor recurrence. Appropriate therapies are needed for each cell type (Figure 3B), and they need to be delivered at the right time (Figure 3C). A number of candidate inhibitors for these cellular targets are already being used in clinical trials for other cancers and could be rapidly tested in GBM.
Radiation and chemotherapy are appropriate therapies for proliferative non-GSCs that are sensitive to DNA-damaging agents. Proapoptotic agents could be evaluated for the slower-growing, terminally differentiated non-GSCs (167). To further reduce the dividing non-GSC population, radio- and chemotherapies should continue to be delivered as initial treatments following surgery. Adjuvant chemotherapy is one option to maintain the suppression of cycling non-GSCs in the intermediate/remission phase, as are molecular therapies targeting a mosaic of growth-signaling pathways.
Existing Wnt, SHH, and Notch pathway inhibitors are good candidate therapies for all GSCs, and in the future, transcription factors sustaining stemness could also be targeted. Molecular therapies targeting epigenetic reprogramming (DNA methylation and histone modification) (13, 168), hypoxia-activated pathways, angiogenesis, and metabolic rewiring/reprogramming all hold promise for GSCs (66, 149, 150). They should be applied starting at the end of the initial therapeutic phase and continuously during the intermediate phase, when the population of RISC cells is emerging.
A further type of targeted therapy is needed to eliminate RISC cell populations. This should start early to eliminate intrinsically resistant RISC precursor cells. Targeting of adaptive resistance mechanisms and blocking of immune suppression can be accomplished in the intermediate phase (27, 29, 157). Creatively engineered virotherapies can also be considered to target all GSCs, including RISC cells (169–171).
Acknowledgments
This work was supported in part by grants from the NIH (CA163722, CA180805, CA176001, NS096236, CA138292, and CA212755 to EGVM), the Southeastern Brain Tumor Foundation (to EGVM and SO), and the Japan Society for the Promotion of Science (26830082 and 13J06657 to SO). We thank Daniel Brat, Ichiro Nakano, Jeffrey J. Olson, and Joseph F. Costello for providing suggestions on the manuscript. MRI images were provided by Eiichi Ishikawa (Tsukuba University, Tsukuba, Japan).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(2):415–426. https://doi.org/10.1172/JCI89587.
References
- 1.Stupp R, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Weller M, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15(9):e395–e403. doi: 10.1016/S1470-2045(14)70011-7. [DOI] [PubMed] [Google Scholar]
- 3.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60(3):166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryken TC, Kalkanis SN, Buatti JM, Olson JJ, AANS/CNS Joint Guidelines Committee The role of cytoreductive surgery in the management of progressive glioblastoma : a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118(3):479–488. doi: 10.1007/s11060-013-1336-7. [DOI] [PubMed] [Google Scholar]
- 5.Lau D, Magill ST, Aghi MK. Molecularly targeted therapies for recurrent glioblastoma: current and future targets. Neurosurg Focus. 2014;37(6):E15. doi: 10.3171/2014.9.FOCUS14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastien JI, McNeill KA, Fine HA. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer. 2015;121(4):502–516. doi: 10.1002/cncr.28968. [DOI] [PubMed] [Google Scholar]
- 7.Smith DR, Hardman JM, Earle KM. Contiguous glioblastoma multiforme and fibrosarcoma with extracranial metastasis. Cancer. 1969;24(2):270–276. doi: 10.1002/1097-0142(196908)24:2<270::AID-CNCR2820240210>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan JP, et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014;4(11):1299–1309. doi: 10.1158/2159-8290.CD-14-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baca SC, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordentoft I, et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014;7(5):1649–1663. doi: 10.1016/j.celrep.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 11.Diaz LA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murtaza M, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760. doi: 10.1038/ncomms9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceccarelli M, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.TCGA Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alcantara Llaguno SR, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell. 2015;28(4):429–440. doi: 10.1016/j.ccell.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alcantara Llaguno S, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacques TS, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010;29(1):222–235. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedmann-Morvinski D, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338(6110):1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bachoo RM, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–277. doi: 10.1016/S1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 23.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 24.Li F, et al. NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell. 2015;60(4):661–675. doi: 10.1016/j.molcel.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 25.Flavahan WA, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson BE, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. doi: 10.1038/ng.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bailey P, Cushing H. A Classification of the Tumors of the Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis. Philadelphia, Pennsylvania, USA: JB Lippincott Co.; 1926. [Google Scholar]
- 31.Bigner SH, Mark J. Chromosome and chromosomal progression of human gliomas in vivo, in vitro and in athymic nude mice. Prog Exp Tumor Res. 1984;27:67–82. doi: 10.1159/000408223. [DOI] [PubMed] [Google Scholar]
- 32.Wikstrand CJ, Grahmann FC, McComb RD, Bigner DD. Antigenic heterogeneity of human anaplastic gliomas and glioma-derived cell lines defined by monoclonal antibodies. J Neuropathol Exp Neurol. 1985;44(3):229–241. doi: 10.1097/00005072-198505000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Snuderl M, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Szerlip NJ, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012;109(8):3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazor T, et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28(3):307–317. doi: 10.1016/j.ccell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki H, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 38.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 39.Paraf F, Jothy S, Van Meir EG. Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol. 1997;15(7):2744–2758. doi: 10.1200/JCO.1997.15.7.2744. [DOI] [PubMed] [Google Scholar]
- 40.Rajaraman P, et al. Genome-wide association study of glioma and meta-analysis. Hum Genet. 2012;131(12):1877–1888. doi: 10.1007/s00439-012-1212-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shete S, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41(8):899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fulci G, et al. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germ-line mutation: evidence for a precursor lesion. Cancer Res. 2002;62(10):2897–2905. [PubMed] [Google Scholar]
- 43.Ishii N, et al. Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene. 1999;18(43):5870–5878. doi: 10.1038/sj.onc.1203241. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell. 2015;28(3):318–328. doi: 10.1016/j.ccell.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 45.Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53(8):799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- 46.Sampetrean O, et al. Invasion precedes tumor mass formation in a malignant brain tumor model of genetically modified neural stem cells. Neoplasia. 2011;13(9):784–791. doi: 10.1593/neo.11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osswald M, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528(7580):93–98. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- 48.Cooper LA, et al. Digital Pathology: Data-Intensive Frontier in Medical Imaging: Health-information sharing, specifically of digital pathology, is the subject of this paper which discusses how sharing the rich images in pathology can stretch the capabilities of all otherwise well-practiced disciplines. Proc IEEE Inst Electr Electron Eng. 2012;100(4):991–1003. doi: 10.1109/JPROC.2011.2182074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leder K, et al. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell. 2014;156(3):603–616. doi: 10.1016/j.cell.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyer M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015;112(3):851–856. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15(6):501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calabrese C, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 54.Pietras A, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14(3):357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osuka S, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31(4):627–640. doi: 10.1002/stem.1328. [DOI] [PubMed] [Google Scholar]
- 56.Ye XZ, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189(1):444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 57.Zhou W, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170–182. doi: 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hadjipanayis CG, Van Meir EG. Tumor initiating cells in malignant gliomas: biology and implications for therapy. J Mol Med. 2009;87(4):363–374. doi: 10.1007/s00109-009-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 61.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 62.Liu G, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qazi MA, et al. A novel stem cell culture model of recurrent glioblastoma. J Neurooncol. 2016;126(1):57–67. doi: 10.1007/s11060-015-1951-6. [DOI] [PubMed] [Google Scholar]
- 64.Chen R, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 65.Beier D, et al. CD133(+) and CD133(–) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67(9):4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 66.Mao P, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110(21):8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhat KP, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lottaz C, et al. Transcriptional profiles of CD133+ and CD133– glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70(5):2030–2040. doi: 10.1158/0008-5472.CAN-09-1707. [DOI] [PubMed] [Google Scholar]
- 69.Huang Q, et al. Glioma stem cells are more aggressive in recurrent tumors with malignant progression than in the primary tumor, and both can be maintained long-term in vitro. BMC Cancer. 2008;8:304. doi: 10.1186/1471-2407-8-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piccirillo SG, et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene. 2009;28(15):1807–1811. doi: 10.1038/onc.2009.27. [DOI] [PubMed] [Google Scholar]
- 71.Auffinger B, et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21(7):1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tamura K, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J Neurosurg. 2010;113(2):310–318. doi: 10.3171/2010.2.JNS091607. [DOI] [PubMed] [Google Scholar]
- 73.Schonberg DL, Lubelski D, Miller TE, Rich JN. Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol Aspects Med. 2014;39:82–101. doi: 10.1016/j.mam.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marko NF, Weil RJ, Schroeder JL, Lang FF, Suki D, Sawaya RE. Extent of resection of glioblastoma revisited: personalized survival modeling facilitates more accurate survival prediction and supports a maximum-safe-resection approach to surgery. J Clin Oncol. 2014;32(8):774–782. doi: 10.1200/JCO.2013.51.8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brandes AA, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26(13):2192–2197. doi: 10.1200/JCO.2007.14.8163. [DOI] [PubMed] [Google Scholar]
- 76.Hart MG, Garside R, Rogers G, Stein K, Grant R. Temozolomide for high grade glioma. Cochrane Database Syst Rev. 2013;4:CD007415. doi: 10.1002/14651858.CD007415.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanai N, Polley MY, McDermott MW, Parsa AT, Berger MS. An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg. 2011;115(1):3–8. doi: 10.3171/2011.2.JNS10998. [DOI] [PubMed] [Google Scholar]
- 78.Grabowski MM, et al. Residual tumor volume versus extent of resection: predictors of survival after surgery for glioblastoma. J Neurosurg. 2014;121(5):1115–1123. doi: 10.3171/2014.7.JNS132449. [DOI] [PubMed] [Google Scholar]
- 79.Gundem G, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murugaesu N, et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015;5(8):821–831. doi: 10.1158/2159-8290.CD-15-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma — are we there yet? Neuro Oncol. 2013;15(1):4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar SM, et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012;31(47):4898–4911. doi: 10.1038/onc.2011.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim Y, et al. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest. 2012;92(3):466–473. doi: 10.1038/labinvest.2011.161. [DOI] [PubMed] [Google Scholar]
- 84.Zheng H, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17(5):497–509. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang N, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20(4):427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang J, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28(1):17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharma SV, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sibilia M, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell. 2000;102(2):211–220. doi: 10.1016/S0092-8674(00)00026-X. [DOI] [PubMed] [Google Scholar]
- 89.Kauffmann-Zeh A, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385(6616):544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 90.Soeda A, et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283(16):10958–10966. doi: 10.1074/jbc.M704205200. [DOI] [PubMed] [Google Scholar]
- 91.Joo KM, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72(15):3828–3838. doi: 10.1158/0008-5472.CAN-11-3760. [DOI] [PubMed] [Google Scholar]
- 92.Li Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A. 2011;108(24):9951–9956. doi: 10.1073/pnas.1016912108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim SH, et al. Serine/threonine kinase MLK4 determines mesenchymal identity in glioma stem cells in an NF-κB-dependent manner. Cancer Cell. 2016;29(2):201–213. doi: 10.1016/j.ccell.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bar EE, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25(10):2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Puram SV, et al. STAT3-iNOS signaling mediates EGFRvIII-induced glial proliferation and transformation. J Neurosci. 2012;32(23):7806–7818. doi: 10.1523/JNEUROSCI.3243-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cao Y, et al. Erythropoietin receptor signaling through STAT3 is required for glioma stem cell maintenance. Genes Cancer. 2010;1(1):50–61. doi: 10.1177/1947601909356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ligon KL, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53(4):503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bao S, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68(15):6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455(7216):1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang J, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3(11):e3769. doi: 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29(28):8884–8896. doi: 10.1523/JNEUROSCI.0968-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci. 2010;30(30):10096–10111. doi: 10.1523/JNEUROSCI.1634-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ikushima H, et al. Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J Biol Chem. 2011;286(48):41434–41441. doi: 10.1074/jbc.M111.300863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt SN, Borges I, Ruiz i Altaba A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29(15):2659–2674. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5(5):504–514. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 107.Anido J, et al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18(6):655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 108.Nam HS, Benezra R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell. 2009;5(5):515–526. doi: 10.1016/j.stem.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109(1):61–73. doi: 10.1016/S0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 110.Chambers I, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113(5):643–655. doi: 10.1016/S0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 111.Li F, Liu X, Sampson JH, Bigner DD, Li CY. Rapid reprogramming of primary human astrocytes into potent tumor-initiating cells with defined genetic factors. Cancer Res. 2016;76(17):5143–5150. doi: 10.1158/0008-5472.CAN-16-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin CY, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530(7588):57–62. doi: 10.1038/nature16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chipumuro E, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Godlewski J, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68(22):9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 115.Xia H, et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J Biol Chem. 2012;287(13):9962–9971. doi: 10.1074/jbc.M111.332627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Esteller M, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 117.Lomonaco SL, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125(3):717–722. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 118.Saga I, et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014;16(8):1048–1056. doi: 10.1093/neuonc/nou096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim SH, Kwon CH, Nakano I. Detoxification of oxidative stress in glioma stem cells: mechanism, clinical relevance, and therapeutic development. J Neurosci Res. 2014;92(11):1419–1424. doi: 10.1002/jnr.23431. [DOI] [PubMed] [Google Scholar]
- 120.Yang W, Shen Y, Wei J, Liu F. MicroRNA-153/Nrf-2/GPx1 pathway regulates radiosensitivity and stemness of glioma stem cells via reactive oxygen species. Oncotarget. 2015;6(26):22006–22027. doi: 10.18632/oncotarget.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bleau AM, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu R, et al. Molecular and clinical effects of notch inhibition in glioma patients: a phase 0/I trial. Clin Cancer Res. 2016;22(19):4786–4796. doi: 10.1158/1078-0432.CCR-16-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sloan AE, et al. Targeting glioma-initiating cells in GBM: ABTC-0904, a randomized phase 0/II study targeting the Sonic Hedgehog-signaling pathway. J Clin Oncol. 2014;32(suppl):2026 [Google Scholar]
- 124.Sampson JH, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sandmann T, et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide: retrospective analysis of the AVAglio trial. J Clin Oncol. 2015;33(25):2735–2744. doi: 10.1200/JCO.2015.61.5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Westphal M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer. 2015;51(4):522–532. doi: 10.1016/j.ejca.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 127.Sunayama J, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28(11):1930–1939. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- 128.Stommel JM, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 129.Jun HJ, Bronson RT, Charest A. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells. 2014;32(2):338–348. doi: 10.1002/stem.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moellering RE, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stolzenburg S, et al. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012;40(14):6725–6740. doi: 10.1093/nar/gks360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Skvortsova I, Debbage P, Kumar V, Skvortsov S. Radiation resistance: Cancer stem cells (CSCs) and their enigmatic pro-survival signaling. Semin Cancer Biol. 2015;35:39–44. doi: 10.1016/j.semcancer.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 133.Vogin G, Foray N. The law of Bergonié and Tribondeau: a nice formula for a first approximation. Int J Radiat Biol. 2013;89(1):2–8. doi: 10.3109/09553002.2012.717732. [DOI] [PubMed] [Google Scholar]
- 134.Munthe S, Petterson SA, Dahlrot RH, Poulsen FR, Hansen S, Kristensen BW. Glioma cells in the tumor periphery have a stem cell phenotype. PLoS One. 2016;11(5):e0155106. doi: 10.1371/journal.pone.0155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sabit H, et al. Characterizing invading glioma cells based on IDH1-R132H and Ki-67 immunofluorescence. Brain Tumor Pathol. 2014;31(4):242–246. doi: 10.1007/s10014-013-0172-y. [DOI] [PubMed] [Google Scholar]
- 136.Mukherjee S, et al. Drosophila brat and human ortholog TRIM3 maintain stem cell equilibrium and suppress brain tumorigenesis by attenuating notch nuclear transport. Cancer Res. 2016;76(8):2443–2452. doi: 10.1158/0008-5472.CAN-15-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen G, et al. Human Brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Cancer Res. 2014;74(16):4536–4548. doi: 10.1158/0008-5472.CAN-13-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sugiarto S, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20(3):328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34(3):333–351. [Google Scholar]
- 140.Huang BY, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One. 2015;10(8):e0134715. doi: 10.1371/journal.pone.0134715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Halliday J, et al. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A. 2014;111(14):5248–5253. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cheng WY, Kandel JJ, Yamashiro DJ, Canoll P, Anastassiou D. A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS One. 2012;7(4):e34705. doi: 10.1371/journal.pone.0034705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bao S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 144.Cheng L, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–1416. [PubMed] [Google Scholar]
- 146.Burroughs SK, Kaluz S, Wang D, Wang K, Van Meir EG, Wang B. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med Chem. 2013;5(5):553–572. doi: 10.4155/fmc.13.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yin S, et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1α interaction with cofactors p300/CBP. Clin Cancer Res. 2012;18(24):6623–6633. doi: 10.1158/1078-0432.CCR-12-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Soeda A, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene. 2009;28(45):3949–3959. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- 149.Kathagen A, et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013;126(5):763–780. doi: 10.1007/s00401-013-1173-y. [DOI] [PubMed] [Google Scholar]
- 150.Vlashi E, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108(38):16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 152.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 153.Berghoff AS, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064–1075. doi: 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bouffet E, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 158.Wu A, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lechman ER, et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. 2016;29(2):214–228. doi: 10.1016/j.ccell.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fidoamore A, et al. Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int. 2016;2016:6809105. doi: 10.1155/2016/6809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Touil Y, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res. 2014;20(4):837–846. doi: 10.1158/1078-0432.CCR-13-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Brown PD, et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008;26(34):5603–5609. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hainsworth JD, et al. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer. 2010;116(15):3663–3669. doi: 10.1002/cncr.25275. [DOI] [PubMed] [Google Scholar]
- 164.Kreisl TN, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92(1):99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 165.Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol. 2013;10(1):14–26. doi: 10.1038/nrclinonc.2012.204. [DOI] [PubMed] [Google Scholar]
- 166.Leuthardt EC, et al. Hyperthermic laser ablation of recurrent glioblastoma leads to temporary disruption of the peritumoral blood brain barrier. PLoS One. 2016;11(2):e0148613. doi: 10.1371/journal.pone.0148613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ziegler DS, Kung AL, Kieran MW. Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol. 2008;26(3):493–500. doi: 10.1200/JCO.2007.13.9717. [DOI] [PubMed] [Google Scholar]
- 168.Suvà ML, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157(3):580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chiocca EA, Blair D, Mufson RA. Oncolytic viruses targeting tumor stem cells. Cancer Res. 2014;74(13):3396–3398. doi: 10.1158/0008-5472.CAN-14-0290. [DOI] [PubMed] [Google Scholar]
- 170.Post DE, et al. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res. 2004;10(24):8603–8612. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 171.Post DE, et al. Targeted cancer gene therapy using a hypoxia inducible factor dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007;67(14):6872–6881. doi: 10.1158/0008-5472.CAN-06-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]