

Abstract
Epidermal growth factor receptor (EGFR) is a key player in proliferation and metastasis of various cancers. Discovery of novel EGFR inhibitors is still an urgent clinical oncology unmet need. Pachycladins are eunicellin-based diterpenoids isolated from the soft coral Cladiella species. This study evaluated the anticancer activity of pachycladins A-E against diverse breast and cervical cancer cells. Pachycladin A (1) potently inhibited the proliferation of multiple cancer cell lines, without being cytotoxic to non-cancerous cells. The antiproliferative activity of 1 is mediated through cytostatic mechanisms rather than inducing apoptosis, as evidenced by lack of TUNEL response. Additionally, 1 arrested cell cycle in either G1 or G2/M phase, according to the cancer type, which induced caspase-dependent and independent apoptosis only after prolonged treatment. Meanwhile, 1 potently decreased microvessel formation and endothelial cell migration, suggesting its potential antiangiogenic activity. Different kinase profiling platforms revealed the exquisite potency and selectivity of 1 towards EGFR, even compared to other members of the EGFR family. In cancer cells, the antiproliferative activity of 1 was associated with suppression of EGFR activation and its downstream effectors. Interestingly, 1 significantly inhibited the drug-resistant T790M EGFR mutant, which is believed to be an attractive feature of EGFR inhibitors. Docking studies characterized the structural determinants required for efficient wild and mutant EGFR inhibition. Overlay studies of 1 with known EGFR inhibitors provided future guidance to chemically improve its binding affinity. Together, the anticancer activity of 1 is mediated by direct effects on tumor growth and angiogenesis, selectively via deactivating EGFR signaling, providing an excellent scaffold to control EGF-dependent cancers.
Keywords: Pachycladin A, EGFR, proliferation, angiogenesis, breast cancer, cervical cancer
Chemical compounds studied in this article: Pachycladin A (PubChem CID: 46832819), Pachycladin B (PubChem CID: 46833115), Pachycladin C (PubChem CID: 46833116), Pachycladin D (PubChem CID: 46833117), Pachycladin E (PubChem CID: 46833118)
Graphical Abstract
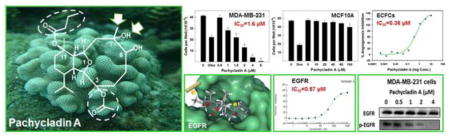
1. Introduction
Epidermal growth factor receptor (EGFR) is a member of the erythroblastosis oncogene B (ErbB) family of receptor tyrosine kinases (RTKs) together with three human epidermal growth factor receptors; HER2, HER3, and HER4 [1]. Upon ligand binding, EGFR undergoes homo- or heterodimerization with other ErbB family members leading to the phosphorylation of specific tyrosine residues within its cytoplasmic tail, which subsequently activates a variety of intracellular downstream signaling pathways that promote cell proliferation, differentiation, angiogenesis, and motility [2, 3]. EGFR is frequently overexpressed and correlated with poor prognosis in many types of human malignancies, including glioblastomas, breast, ovarian, and non-small-cell lung cancers (NSCLC) [3]. EGFR deregulation can be also caused by activating mutations, gene amplification, or oncogenic viruses [1]. These findings rendered EGFR an attractive molecular target for selective cancer therapy. To date, two small molecule reversible tyrosine kinase inhibitors (TKIs) of EGFR have been FDA-approved for NSCLC treatment; gefitinib (Iressa®) and erlotinib (Tarceva®). Although the response rates for gefitinib and erlotinib were significant, they were minor when compared with the overall clinical need and limited only to NSCLC patients [4]. Acquired resistance to gefitinib and erlotinib invariably develops after prolonged clinical use as expected with all targeted anticancer therapies [5]. Therefore, there is a continued need to discover an expanded repertoire of novel EGFR TKI entities.
Recently, there has been a renewed interest in natural product research due to the failure of alternative drug discovery methods to discover novel entities for key therapeutic areas such as immunosuppression and cancer [6]. Nearly 63% of current anticancer drugs are natural products or can be traced back to a natural product origin [7]. In particular, the marine environment has been a productive source of potential anticancer entities. Ecteinascidin-743 was the first marine-derived anticancer drug to be approved in 2007, followed by eribulin mesylate, a synthetic analog of halichondrin B, in 2010, and SGN-30, an antibody drug conjugate incorporating monomethyl auristatin E conjugated to the humanized anti-CD30 monoclonal antibody, which received FDA approval in 2011 for the treatment of Hodgkin’s lymphoma [8].
Soft corals of the class Alcyonaria are among the important marine invertebrate classes that produce a wealth of chemically diverse, unique and bioactive diterpenes, including cembranes [9], norcembranes [10], xeniaenes [11], briaranes [12], and eunicellins [13]. The eunicellin-based diterpenoids displayed a wide range of bioactivities including anti-inflammatory and antitumor activities [14, 15]. Examples of these are the potent microtubule disruptors, sarcodictyins and eleutherobin, which were found to be among the most cytotoxic natural products [16]. The genus Cladiella has been recognized as a rich source of cytotoxic eunicellin diterpenoids [14, 17]. A previous study reported the isolation of five eunicellin diterpenes, pachycladins A-E (1–5), from the Red Sea soft coral Cladiella pachyclados [18]. Pachycladins A (1) and D (4) exhibited significant antimigratory and anti-invasive activities against the human metastatic prostate cancer PC-3 cells [18]. Interestingly, none of these marine-derived natural products showed any effect on the proliferation of PC-3 cells up to 50 μM, suggesting the lack of cytotoxicity towards these cells. Semisynthetic pachycladin analogs showed promising antimigratory and anti-invasive activities against prostate cancer cells but most of them failed to demonstrate better activity than 1 [19].
Despite many reports on eunicellin-based diterpenoids as antitumor agents, pachycladins have not been extensively studied and little is known about their anticancer mechanism. Therefore, the ultimate objective of this study was to evaluate the anticancer activity of pachycladins against human breast and cervical cancer cells, and characterize the possible molecular mechanisms associated with this activity, with focus on 1 as a representative of this class.
2. Materials and methods
2.1. Materials
Pachycladins A-E (1–5) were isolated from the Red Sea soft coral Cladiella pachyclados and identified by spectral analyses [18]. A purity of >95% was established using 1H NMR and TLC analyses. (−)-Oleocanthal was isolated from extra-virgin olive oil (Daily Chef, Italy). Unless otherwise indicated, cell culture reagents were obtained from Life Technologies (Carlsbad, CA). Dulbecco's modified eagle medium (DMEM) and PBS were obtained from Thermo Scientific (Waltham, MA) while endothelial cell growth media EGM-2MV and EGM-2 were purchased from Lonza (Basel, Switzerland). All antibodies were purchased from Cell Signaling Technology (Beverly, MA) and used at a dilution of 1:1000, unless otherwise stated. Antibodies for breast tumor kinase (Brk) and p-Brk were acquired from Abnova (Walnut, CA). Goat anti-rabbit and anti-mouse secondary antibodies were purchased from PerkinElmer Biosciences (Boston, MA). Growth factors were purchased from PeproTech Inc., (Rocky Hill, NJ).
2.2. Cell lines and culture conditions
Human cancer cell lines and non-tumorigenic mammary epithelial MCF10A cells were purchased from the ATCC (Rockville, MD). Breast cancer cell lines (passage 13) were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin G, 0.1 mg/mL streptomycin and 2 mmol/L glutamine (VWR, Suwanee, GA). Human cervical cancer HeLa cells (passage 12) were cultured in DMEM high glucose media supplemented with 10% FBS, 1mM L-glutamine and 1× penicillin-streptomycin solution. MCF10A cells (passage 6) were cultured in DMEM/F12 supplemented with 5% horse serum, 0.5 μg/mL hydrocortisone, 20 ng/mL EGF, 100 U/mL penicillin G, 100 ng/mL cholera toxin, 100 μg/mL streptomycin, and 10 μg/mL insulin (VWR, Suwanee, GA). Human endothelial colony forming cells (ECFCs, Lilly, IN) and adipose-derived stem cells (ADSCs, Lilly, IN) (passage 7) were cultured in EGM-2MV media containing 10% FBS. All cells were maintained at 37°C in a humidified incubator under 5% CO2. Pachycladins were first dissolved in a volume of sterilized DMSO (VWR, Suwanee, GA) to provide final 10 mM stock solutions for all assays. Working solutions at their final concentrations for each assay were prepared in appropriate culture medium immediately prior to use. The vehicle control was prepared by adding the maximum volume of DMSO, used in preparing pachycladins, to the appropriate media type such that the final DMSO concentration was maintained the same in all treatment groups and never exceeded 0.1%. (−)-Oleocanthal and nocodazole (Sigma-Aldrich, St. Louis, MO) were used as positive controls at doses selected based on earlier studies [20, 21].
2.3. Measurement of viable cell number
Viable cell count was determined using the MTT (VWR, Suwanee, GA) colorimetric assay. The optical density was measured at λ 570 nm on a microplate reader (BioTek, VT).The number of cells/well was calculated against a standard curve prepared at the start of each experiment by plating various number of cells (1,000–60,000 cells/well), as determined using a hemocytometer [20].
2.4. Cell viability assays
Breast cancer cells, in exponential growth, were seeded at a density of 1×104 cells/well (6 wells/group) in 96-well plates and allowed to adhere overnight in RPMI-1640 media containing 10% FBS under 5% CO2. Human cervical cancer cells were seeded at a density of 1×104 cells/well onto poly-D-lysine-coated 96-well plates in DMEM high glucose media containing 10% FBS, and incubated for 24 h under 5% CO2. Cancer cells were then divided into different groups and fed various treatments, in appropriate medium containing 40 ng/mL EGF, and re-incubated for 48 or 72 h. Treatment media were then replaced with fresh ones, and 50 μL/well of MTT solution (1 mg/mL) was added and plates were re-incubated for 4 h at 37 ºC. The color reaction was stopped by removing the media and adding 100 μL DMSO in each well and incubating at 37°C for 20 min to ensure complete dissolution of the formed formazan crystals. Absorbance was determined at λ570 nm using a plate reader (BioTek, VT). The % cell survival was calculated as follows: % cell survival = (Cell No.treatment/Cell No.DMSO) x 100.
The cytotoxic effect of 1 was evaluated against the non-tumorigenic mammary epithelial cell line MCF10A. Cells in exponential growth were seeded at a density of 1×104 cells/well into 96-well plates, maintained in DMEM/F12 media containing 5% horse serum, and allowed to attach overnight under 5% CO2. Cells were then divided into different triplicate groups, treated with vehicle control, doxorubicin (Sigma-Aldrich, St. Louis, MO) or 1 at designated concentrations in fresh serum-free defined media, and re-incubated under 5% CO2 for 24 h. Viable cell number was determined using the MTT assay.
2.5. G2M cell cycle assay
HeLa cells were seeded at a density of 1×103 cells/well in poly-D-lysine coated 384-well plates and incubated in appropriate media type for 24 h under 5% CO2. Different concentrations of pachycladins were then added in triplicates and incubated for 48 h. Cells were fixed with Prefer (Anatech, Battle creek, MI) for 45 min, washed with PBS, treated with 0.1% Triton X-100 (TX-100, Sigma-Aldrich, St. Louis, MO) for 15 min, re-washed and treated with RNAse (50 μg/mL, Sigma-Aldrich, St. Louis, MO) in PBS. After 1 h, cells were washed with PBS, primary antibodies directed against cyclin B (BD 554177, BD Biosciences, San Jose, CA) and phosphorylated histone H3 (pHH3) in PBS containing 1% BSA were added and allowed to incubate overnight at 4°C. Samples were washed with PBS and Alexa-488 labeled secondary antibodies (Invitrogen, Carlsbad, CA) in PBS were added. After 1 h, samples were washed with PBS and nuclei were stained with propidium iodide (PI, Sigma-Aldrich, St. Louis, MO) at 10 μg/mL for 20 min. Plates were read on an Acumen® laser cytometer at λ 488 nm/530 nm for cyclin B+pHH3, and λ 488 nm/>650 nM for PI. Histograms of cell number verses total PI fluorescence, peak PI fluorescence, and total Alexa 488 fluorescence provide information on cellular DNA content, DNA condensation, and levels of cyclin B+pHH3, respectively. Discrimination gates were set on the basis of population shifts between maximally and minimally mitotic control samples treated with 200 nM nocodazole or 0.5% (v/v) DMSO, respectively [21].
2.6. Analysis of cell cycle progression using flow cytometry
To study treatment effects of 1 on cell cycle, MDA-MB-231 cells were seeded at a density of 1×106 cells/100 mm culture plates in RPMI-1640 media supplemented with 10% FBS and allowed to adhere overnight. Cells were then washed twice with PBS and starved in serum-free medium containing 0.5% FBS for 48 h to synchronize the cells in G1 phase. Cells were then fed various doses of 1 in serum-free defined media containing 40 ng/ml EGF as the mitogen for 24 h. Cells in different treatment groups were then isolated with trypsin and resuspended in ice cold PBS, fixed with cold (−20°C) 70% ethanol, and stored at 4°C for 2 h. Cells were then rehydrated with ice cold PBS and incubated with DNA staining buffer (sodium citrate 1 mg/ml, Triton-X 100 3 μl/ml, propidium iodide 100 μg/ml, ribonuclease A 20 μg/ml) for 30 min at 4°C in dark. Finally, DNA content was analyzed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). For each sample, 10,000 events were recorded, and histograms were generated using CellQuest software (BD Biosciences, San Jose, CA).
2.7. TUNEL assay
TUNEL analysis was performed on HeLa cells, after treatment with different doses of 1 in triplicates for 48 and 72 h, using an in situ cell death detection kit (Roche #11684795910, Basel, Switzerland) according to the manufacturer’s protocol. Apoptotic cells were fixed, permeabilized, and incubated with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and fluorescein-dUTP. During incubation, TdT catalyzes the attachment of fluorescein-dUTP to the free 3’OH ends in the DNA. Finally, the incorporated fluorescein was visualized using a flow cytometer.
2.8. Caspase-3 activity assay
The activity of caspase-3 was determined using a caspase-3 assay kit (Roche #12012952001, Basel, Switzerland) according to the manufacturer’s protocol. The assay uses a fluorometric immunosorbent enzyme assay (FIENA) principle, which allows specific and quantitative detection of caspase-3 activity in cellular lysates. HeLa cells were plated at a density of 2×106 cells/100 mm culture plates and allowed to attach overnight. Cells were then incubated in the respective control or 1-treated medium for 48 and 72 h. Cells in each treatment group were then washed, pelleted, and incubated in lysis buffer to prepare cell lysates. Meanwhile, a microplate was coated with anti-caspase-3 solution and blocked for nonspecific binding with a blocking buffer which was then removed by extensive washing with incubation buffer. To assay the protease activity, 100 μL of each lysate, in triplicates, was transferred into the anti-caspase-3- coated wells and incubated for 1 h at 37°C. The immobilized antibody-caspase-3 complexes were then washed and a freshly prepared caspase substrate solution (Ac-DEVD-AFC) was added and incubated for 3 h at 37°C to allow its proteolytic cleavage into free fluorescent 7-amino-4- trifluoromethylcoumarin (AFC) which was measured fluorometrically at λ400/505 nm.
2.9. Detection of apoptosis inducing factor (AIF)
HeLa cells (7×106/100 mm culture plate) were treated with different doses of 1 for 72 h. Cells were then suspended in lysis buffer and homogenized using a Teflon homogenizer. To perform subcellular fractionation, homogenates were centrifuged at 1,000 g for 10 min at 4°C. The pellet was used to prepare the nuclear fraction. The supernatant was re-centrifuged at 10,000 g for 30 min; the resulting pellet represented mitochondrial fraction. Pellets for nuclear and mitochondrial fractions were lysed, sonicated and aliquots in triplicates were used to measure the AIF level by an ELISA kit (Abnova, Taiwan) according to the manufacturer’s instructions. The absorbance was read at λ450 nm using a microplate reader (BioTek, VT).
2.10. Angiogenesis assay
ECFCs and ADSCs were prepared as previously described [21]. ADSCs were seeded at a density of 5×103 cells/well in a 384 well Cell Bind plate, incubated at rt for 5 min, then under 5% CO2 overnight. ECFCs (5×102 cells/well) were then overlaid onto the ADSC feeder layer and re-incubated under 5% CO2 for 2 h. The recombinant vascular endothelial growth factor (rhVEGF, 10 ng/mL) or a proangiogenic factors mixture (VEGF 10 ng/mL, EGF 20 ng/mL, and fibroblast growth factor FGF 10 ng/mL), along with different concentrations of 1 or 3 were added in triplicates and incubated for 96 h. Co-cultures were then fixed with 3.7% formaldehyde (Sigma- Aldrich, St. Louis, MO) in PBS for 20 min, treated with 0.1% TX-100 in PBS for 20 min, and incubated with 63 ng/mL mouse anti-human CD31 primary antibody in PBS containing 1% BSA at 4°C overnight. Samples were then incubated with 3μg/mL Alexa 488 goat secondary antibody in PBS for 1 h and stained for cellular DNA with Hoechst (2 μg/mL). ECFC-ADSC co-cultures were then analyzed with an Array Scan VTI (Thermo Scientific, Waltham, MA) using a 5× objective and collecting four non-adjacent frames/well; nuclei and CD31 were visualized using the XF93-Hoechst and XF93-Alexa-488 dichroic mirror emission filter pairs, respectively. Endothelial tube features were determined using the Cellomics Tube Formation V3 application by analyzing the CD31 channel. Relative cell death was measured by loss of valid nuclear objects as determined using the Target Activation V3 application. The maximum and minimum responses corresponded to rhVEGF+0.5% (v/v) DMSO and rhVEGF+500 nM sutent (Biovision, Milpitas, CA), an antiangiogenic drug inhibiting vascular endothelial growth factor receptor 2 (VEGFR2), respectively [22].
2.11. Dead/live cells assay
The ratio of dead/live ECFCs was determined using the MultiTox-Fluor Multiplex cytotoxicity assay (Promega, WI). Briefly, ECFCs were seeded in a 96-well plate at a density of 1×103 cells/well. After 4 h, ECFCs were treated with various concentrations of 1 in triplicates for 72 h prior to the addition of assay substrates directly to the media for 2 h to allow the reaction to complete. The fluorescence was then measured on SpectraMax plate reader at λ400 nm/505 nm and 485 nm/520 nm, to detect the products of live and dead cell-associated proteases, respectively.
2.12. Oris™ cell migration assay
ECFCs were seeded at a density of 3×104 cells/well in their appropriate media on Oris™ cell migration-collagen I coated plates, containing cell seeding stoppers (Platypus Tech, Madison, WI). Cells were allowed to attach and spread for 20 h prior to stoppers removal. Media were replaced with optimized growth medium containing 10% FBS, and either DMSO or different concentrations of 1 or dasatinib (Sigma-Aldrich, St. Louis, MO) as a positive control in triplicates for 24 h. Cells were then washed and incubated in 100 μL of Prefer fixative. After 30 min, cells were permeabilized with PBS containing 0.1% Triton X-100 for 15 min. Cells were then washed with PBS, and 50 μL of 15 μM PI in RNase-free PBS was added. Images were captured on the Acumen™ Explorer using a 488 nm laser and >655 nm filter. A square around the cell-free zone defined the migration region whereas a rectangle encompassing the confluent monolayer from the same well defined the cytotoxicity region. Total fluorescent cellular area (μm2) in the migration and cytotoxicity regions were used to calculate percent migration inhibition (Equation 1) and percent cell death (Equation 2), respectively, where AreaDMSO, Areacompound 1, and Areamax dasatinib are the cellular areas within the analysis region in the presence of 0.4% DMSO, 1, and a maximum inhibitory dose of dasatinib (40 μM), respectively.
| Equation 1 |
| Equation 2 |
2.13. Kinase profiling
Compound 1 was profiled at 2 μM in triplicates against a structurally diverse panel of 24 known kinases, at the physiological ATP concentration of each kinase, in through-plate mode using the CerepLANCE kinase assays. The service was made by Eli Lilly and protocols are available at http://www.cerep.fr/cerep/users/pages/productsservices/kinasePlatform.asp.
2.14. Biochemical kinase assays
The Z’-LYTE™ Kinase Assay-Tyr4 Peptide kit (Thermo Scientific, Waltham, MA) was used to evaluate the ability of 1 to inhibit the catalytic activity of wild-type EGFR (Product# PV3872) and its four oncogenic variants, L858R (Product# PV4128), L861Q (Product# PV3873), T790M (Product# PV4803), and T790M L858R (Product# PV4879). Briefly, 20 μL/well reactions (6 wells/group) were set up in 96-well plates containing kinase buffer, 200 μM ATP, 4 μM Z-LYTE™ Tyr4 Peptide substrate, 2500 ng mL−1 EGFR kinase, and compound 1 as an inhibitor at indicated doses. After incubation for 1 h at rt, 10 μL development solution containing site-specific protease was added to each well and incubation was resumed for 1 h. The reaction was then stopped, and the fluorescent signal ratio of 445 nm (coumarin)/520 nm (fluorescein) was measured on a plate reader (BioTek FLx800™, VT), which reflects the peptide substrate cleavage status and/or the kinase inhibitory activity in the reaction. Inhibition rate (%) was determined using the following equation: % Inhibition = 100 − [(Activity of enzyme with tested compound − Min)/(Max − Min)] × 100 (Max: the enzyme activity measured in the presence of enzyme, substrates, and cofactors; Min: the enzyme activity in the presence of substrates, cofactors and in the absence of enzyme).
The LANTHASCREEN™ EU Kinase Binding Assay (Thermo Scientific, Waltham, MA) was used to assess the ability of 1 to inhibit the catalytic activity of EGFR kinase with deletion mutation d746-750 (Product# PV6178). Briefly, 16 μL/well reactions were set up in a low-volume 384-well plate, including 3.84 μL kinase buffer, 8 μL 2x EGFR kinase/antibody mixture, 4 μL 4x Alexa Fluor™ conjugate or Tracer, and 160 nL 100x compound 1 in 100% DMSO as an inhibitor. After incubation for 1 h at rt, the fluorescent signal ratio of 665 nm (AF647 emission)/615 nm (europium emission) was measured on a plate reader (BioTek FLx800™, VT), which reflects the degree of tracer/antibody binding to EGFR and/or the kinase inhibitory activity in the reaction.
Compound 1 was screened at 10 μM against two members of the EGFR family, HER2 (Product# PV3366) and HER4 (Product# PV3626) kinases, using SelectScreen® Kinase Profiling Selectivity Testing Services (Life Sciences, Carlsbad, CA). Percent inhibition for each kinase was determined using a time-resolved fluorescence resonance energy transfer assay. Protocols are available at https://www.thermofisher.com/us/en/home/products-and-services/services/custom-services/screening-and-profiling-services/selectscreen-profiling-service/selectscreen-kinase-profiling-service.html.
2.15. Western blot analysis
Human breast cancer MDA-MB-231, MDA-MB-468, and BT-474 cells were treated according to the method previously described [20]. The whole-cell extracts were prepared in RIPA buffer (Qiagen Sciences Inc., CA). Protein concentration was determined using the bicinchoninic acid assay (Bio-Rad, Hercules, CA) according to the manufacturer's protocol. Equivalent amounts of protein were electrophoresed on SDS-polyacrylamide gels (Bio-Rad Hercules, CA). Gels were then electroblotted onto polyvinylidene fluoride membranes (Bio-Rad Hercules, CA). Membranes were blocked with 2% BSA in 10 mM Tris-HCl (VWR, Suwanee, GA), containing 50 mM NaCl (Sigma-Aldrich, St. Louis, MO) and 0.1% Tween 20 (Sigma- Aldrich, St. Louis, MO), pH 7.4 (TBST) and then, probed with the indicated specific primary antibodies overnight at 4 ºC. Membranes were then washed extensively with TBST and incubated with respective horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies in 2% BSA in TBST for 1 h at rt followed by rinsing with TBST for 5 times. Blots were then visualized using an enhanced chemiluminescence according to the manufacturer’s instructions (Pierce, Rockford, IL). Images of protein bands from all treatment groups within a given experiment and scanning densitometric analysis were acquired using Kodak Gel Logic 1500 Imaging System (Carestream Health Inc., CT). All experiments were repeated three times and a representative Western blot image from each experiment is shown in Fig. 10.
Fig. 10.


Western blot analysis showing pachycladin A treatment effects on MDA-MB-231 breast cancer signaling after 48 h treatment in human MDA-MB-231 cancer cells. (A) Treatment with 1 caused a dose-dependent inhibition of EGFR phosphorylation without any effect on its total levels, compared to vehicle-treated control group. (B-E) Treatment with 1 caused a dose-dependent phosphorylation inhibition of EGFR downstream signaling proteins, including Akt, MAPK, FAK, and Brk, without any effect on their total levels, compared to vehicle-treated control group. (F) No significant inhibition was observed on c-Met signaling. Scanning densitometric analysis was performed on all blots done in triplicate and the integrated optical density of each band was normalized with corresponding β-tubulin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± S.D., *P <0.05 as compared with vehicle-treated controls.
2.16. Molecular modeling
In silico experiments were carried out using Schrödinger molecular modeling software package (Schrödinger, New York, NY). Two X-ray crystal structures of the EGFR kinase domain (PDB codes: 2ITW and 4WKQ) were retrieved from the Protein Data Bank (www.rcsb.org). The Protein Preparation Wizard was implemented to prepare the kinase domain of EGFR protein. The protein was reprocessed by assigning bond orders, adding hydrogens, and optimizing H-bonding networks using PROPKA (Jensen Research Group, Denmark). Finally, energy minimization with RMSD value of 0.2 °A was applied using optimized potentials for liquid simulation (OPLS) force field (OPLS_2005, Schrödinger, NY). The chemical structure of 1 was sketched on the Maestro 9.3 panel interface (Maestro, version 9.3, 2012, Schrödinger, NY). The Ligprep 2.3 module (Ligprep, version 2.3, 2012, Schrödinger, NY) was implemented to generate the three-dimensional structure and to search for different conformers. The OPLS force field was applied to geometrically optimize the ligand structure and to compute partial atomic charges. Finally, 32 poses per ligand were generated with different steric features for subsequent docking studies. The prepared X-ray crystal structures of EGFR kinase domain were used to generate receptor energy grids applying the default value of the protein atomic scale (1 °A) within a cubic box centered on the co-crystallized ligand of each crystal structure. 1 was then docked using the Glide 5.8 module (Glide, 2012, Schrödinger, NY) in extra-precision (XP) mode.
2.17. Statistical analysis
Results are presented as means ± S.D. of three independent experiments. Differences among various treatment groups were determined by ANOVA followed by Dunnett’s test using PASW statistics® version 18 (Quarry Bay, Hong Kong). A difference of P<0.05 was considered statistically significant compared to the vehicle-treated control group. IC50/EC50 values were determined applying non-linear regression using GraphPad Prism version 6 (La Jolla, USA).
3. Results
3.1. Chemical diversity of tested pachycladins and their effect on breast cancer cell viability
Five known eunicellin-based diterpenoids (1–5, Fig. 1) have been identified and screened for their antiproliferative activity against the human triple negative breast cancer (TNBC) MDA-MB- 231 cells in MTT assay. The structures represent diverse eunicellin diterpenes possessing various oxygen functionalities, exemplified by free hydroxy groups and ester moieties at C-3, C- 6, C-7, and C-11, with or without exomethylene substitutions at C-7 and C-11. In this assay, at least four different concentrations per compound were tested and used to assess an IC50 (Table 1, Fig. 2 and 3A). The natural olive secoiridoid, oleocanthal, was used as a positive control with known activity [20]. Results showed that pachycladin A (1) inhibited the MDA-MB-231 cell growth in a dose-responsive manner, with an IC50 value of 1.6 μM. However, pachycladins 2–5 were shown to have no remarkable effect on the growth of MDA-MB-231 cells up to 10 μM, compared to the vehicle-treated group (Table 1). Chemically, 1 possesses eunicellin skeleton with oxygen functionalities at C-3, C-6, C-7, and C-11 [18]. Two of these oxygenated carbons, C-6 and C-7, bear free hydroxy groups while the remaining carbons, C-3 and C-11, have acetate and butyrate ester moieties, respectively. Replacing the C-7 hydroxy in 1 with an exomethylene functionality in 2–5 caused a drastic reduction in the activity, with IC50 >10 μM for all compounds (Table 1). This suggested the crucial role of the C-7 hydroxy in 1 for maintaining the growth inhibitory activity. Similarly, pachycladin D (4), with an additional exomethylene group at C-11 substituting the butyrate moiety in 1, demonstrated a significant drop in the activity (Table 1). In addition, replacing the C-3 acetate in 1 with a hydroxy group in 4 or having an additional C-13 hydroxy substituent in 3 did not show activity improvement (Table 1). This clearly implied the significance of the acetate and butyrate moieties at C-3 and C-11, respectively, for optimal antiproliferative activity.
Fig. 1.

Chemical structure of pachycladins.
Table 1.
Antiproliferative activities of 1–5 against the human breast cancer MDA-MB-231 cells.
| Compound | Antiproliferative activity MDA-MB-231
|
|---|---|
| IC50 (μM) ± S.D. (n = 3/dose) | |
| Oleocanthala | 10.2 ± 1.6 |
| 1 | 1.6 ± 0.5 |
| 2 | 15.2 ± 0.8 |
| 3 | 31.4 ± 1.4 |
| 4 | >40 |
| 5 | >40 |
The olive-derived oleocanthal was used as a positive standard control and its IC50 value was calculated for activity comparison.
Fig. 2.

Effect of various doses of 2 and 3 on the viability of MDA-MB-231 breast cancer cells, compared to DMSO as vehicle control. Viable cell count was determined using MTT assay. Error bars indicate the S.D. of N=3/dose. Oleocanthal was used as positive control at 10μM [20].
Fig. 3.

Evaluation of the antiproliferative effects of 1 in cancer cells. Effect of pachycladin A treatment on the growth of (A) MDA-MB-231, (B) MDA-MB-468, (C) BT-474, (D) MCF-7, (E) SKBR3 and (F) T-47D breast cancer cells after 48 h. Effect of pachycladin A treatment on the growth of HeLa cervical cancer cells after (G) 48 and (H) 72 h. Viable cell count was determined using MTT assay. Error bars indicate the S.D. of N=3/dose. Oleocanthal and nocodazole were used as positive controls at doses selected based on earlier studies [20, 21].
3.2. Effect of pachycladin A on the growth of human breast and cervical cancer cells
The remarkable antiproliferative potency of 1 against the MDA-MB-231 cells prompted the investigation of its effect across a panel of human breast and cervical cancer cells. Treatment with 1 significantly inhibited the growth of the human breast cancer cell lines MDA-MB-231, MDA-MB-468, BT-474, MCF-7, SKBR3, and T-47D in a dose-dependent manner, with IC50 values ranging from 1.6 to 10.6 μM (Fig. 3A–F and Table 2). Similarly, treatment with 1 caused a dose-dependent suppression of human cervical cancer HeLa cells proliferation after 48 and 72 h culture periods, compared to their vehicle-treated groups, with IC50 values of 1.7 and 1.3 μM, respectively (Figs. 3G and 3H and Table 2). The activity of 1 was several-fold that of oleocanthal in all investigated cancer cell lines, indicating its potential as a novel antiproliferative entity (Fig. 3).
Table 2.
IC50 values for 1 in different human breast and cervical cancer cell lines.
| Cell Line | Antiproliferative activity of pachycladin A
|
|---|---|
| IC50 (μM)a ± S.D. (n = 3/dose) | |
| MDA-MB-231 | 1.6 ± 0.5 |
| MDA-MB-468 | 2.1 ± 1.2 |
| MCF-7 | 3.5 ± 0.9 |
| BT-474 | 7.1 ± 1.6 |
| SKBR3 | 9.5 ± 1.4 |
| T-47D | 10.6 ± 0.7 |
| HeLa (48h) | 1.7 ± 0.4 |
| HeLa (72h) | 1.3 ± 0.8 |
IC50 values were calculated using non-linear regression analysis using GraphPad Prism software.
3.3. Cytotoxic activity against the non-tumorigenic MCF10A mammary epithelial cells
To evaluate the relative selectivity of 1 towards malignant cells, the non-tumorigenic MCF10A human mammary epithelial cells were treated with various concentrations of 1 for 24 h. Results demonstrated that 1 was non-toxic to MCF10A cells at concentrations several-fold higher than its corresponding IC50 values in malignant cell proliferation assays (Fig. 4). Doxorubicin treatment (10 μM) resulted in 60% reduction of MCF10A cell viability. In contrast, 1 showed no remarkable effect on the viability of MCF10A cells up to 80 μM, while it exhibited IC50 values of 1.6 and 2.1 μM against the breast cancer cells MDA-MB-231 and MDA-MB-468, respectively (Fig. 4 and Table 2). These results suggested the ability of 1 to exhibit preferential growth inhibitory effects toward cancer cells over non-tumorigenic cells.
Fig. 4.

Cytotoxic activity of pachycladin A treatment against the non-tumorigenic human mammary epithelial MCF10A cells, compared to DMSO as a vehicle control. Error bars indicate the S.D. of N=3/dose. Doxorubicin was used as a positive control at 10 μM.
3.4. Screening of pachycladins in Lilly’s phenotypic drug discovery program (PD2)
The cell cycle and antiangiogenic activities of 1, 3, and 5 were screened in the PD2 initiative offered by Eli Lilly [21]. The first five phenotypic screening modules available via PD2 are insulin and apolipoprotein E (apoE) secretion assays, Wnt pathway, angiogenesis, and G2M cell cycle assays [21]. Primary screening revealed that tested compounds did not show significant activities in the Wnt, insulin and apoE secretion assays (Table 3). Interestingly, 1 exhibited a promising activity profile in cell cycle and angiogenesis assays, whereas 3 was only active in the angiogenesis module (Table 3). A 10 μM treatment concentration of 1 demonstrated 95% and 130% efficacies in cell cycle and angiogenesis assays, respectively. Meanwhile, 3 showed a 75% efficacy in the angiogenesis assay, whilst lacking promising activity in cell cycle assay (Table 3). Based on these results, dose-response follow-up studies have been conducted for single-point screen hits 1 and 3 to confirm the activity obtained in these phenotypic screens.
Table 3.
Percentage efficacies of 1, 3, and 5 in different modules of the PD2 screening panel.
| Compound | % Efficacy at 10 μM dose
|
||||
|---|---|---|---|---|---|
| Insulin secretion | ApoE secretion | Wnt pathway | G2M cell cycle | Angiogenesis | |
| 1 | 6 % | −3 % | 2 % | 95 % | 130 % |
| 3 | 8 % | −1 % | 3 % | 11 % | 75 % |
| 5 | 6 % | 9 % | 3 % | 1 % | −6 % |
3.5. Effect of pachycladin A on cervical cancer cell cycle progression
The effect of 1 on cancer cell cycle progression was evaluated using high-content cellular imaging [21]. This DNA content assay identifies compounds that arrest cell cycle in G2 or M phase. HeLa cells were treated with various doses of 1 for 48 h prior to fixation and staining. Multiplexed cell cycle parameters were then measured (Fig. 5). Treatment with 1 resulted in a dose-dependent increase in the proportion of HeLa cells in G2/M (4N), and a subsequent dose responsive reduction in the proportion of cells in G1 (2N), with an EC50 value of 0.93 μM (Fig. 5A, 5B and Table 4). Additionally, treatment with 1 resulted in a significant induction of DNA condensation with an EC50 of 0.7 μM, consistent with the potent modulation of DNA content in HeLa cells (Fig. 5C and Table 4). Further studies were conducted to determine the effects of 1 on the relative intracellular levels of cyclin B and pHH3 as biomarkers for G2 and M phases, respectively [21]. Interestingly, 1 resulted in prominent activation of cyclin B and pHH3 levels, with an EC50 of 0.83 μM (Fig. 5D and Table 4). These data clearly indicated the ability of 1 to arrest cell cycle in the G2/M phase, leading to decreased 2N and increased 4N, cyclinB-pHH3, and DNA condensation.
Fig. 5.

Evaluation of 1 in the multiplexed G2/M cell cycle assay using HeLa cells. Effect of treatment with 1 for 48 h on the (A) increase in tetraploid (4N) cell population, (B) reduction in diploid (2N) cell population, (C) induction of DNA condensation, and (D) activation of both cyclin B and pHH3 levels.
Table 4.
IC50/EC50 values of pachycladin A in different oncology assays.
| Assay/Cell Line | Pachycladin A
|
|---|---|
| IC50/EC50 (μM) a | |
| G2/M cell cycle (DNA content, 4N) / HeLa | 0.93 |
| G2/M cell cycle (DNA content, 2N) / HeLa | 0.93 |
| G2/M cell cycle (DNA condensation) / HeLa | 0.7 |
| G2/M cell cycle (cyclin B + pHH3) / HeLa | 0.83 |
| TUNEL after 48 h / HeLa | > 20 |
| TUNEL after 72 h / HeLa | 5.1 |
| Caspase-3 activity after 48 h / HeLa | > 20 |
| Caspase-3 activity after 72 h / HeLa | 3.1 |
| AIF release after 72 h / HeLa | 2.3 |
| Angiogenesis (CD31) / ECFCs + ADSCs co-culture3 | 0.36 |
| Cytotoxicity (nuclear area) / ECFCs + ADSCs co-culture | > 10 |
| Dead/Live cells cytotoxicity / ECFCs | > 40 |
| Oris™ cell migration / ECFCs | 7.5 |
| Oris™ cell cytotoxicity / ECFCs | > 40 |
The reported values were computed by internal fitting algorithms [21].
3.6. Effect of pachycladin A on breast cancer cell cycle progression
The effects of 1 treatment on breast cancer cell cycle progression were also studied using flow cytometry (Fig. 6). MDA-MB-231 cells exposed to various concentrations of 1 demonstrated a dose-dependent increase in the proportion of cells in G1 phase of cell cycle. This increase was nearly 90% with 2 μM pachycladin A-treated cells compared to 50% in the vehicle-treated control group (Fig. 6). Interestingly, no sub-G1 population of cells was seen in any of the treatment groups (0-2 μM), implying that pachycladin A did not initiate apoptosis in MDA-MB- 231 breast cancer cells at the used treatment doses.
Fig. 6.

Flow cytometry and cell cycle progression in control and pachycladin A-treated MDA-MB- 231 cells. Left panel shows histograms generated using CellQuest software (PI staining). Right panel shows percentage of cells in each phase of cell cycle. Vertical bars show the average of three independent experiments.
3.7. Pro-apoptotic effects of pachycladin A in cancer cells
The ability of 1 to induce cancer cell apoptosis was assessed using the TUNEL assay to determine whether the antiproliferative effects of 1 represented cell death or growth arrest. HeLa cells were treated with different doses of 1 for 48 or 72 h and cell death was assessed by measuring the degree of TUNEL staining using flow cytometry. After 72 h, cells underwent significant apoptosis as demonstrated by their increased TUNEL staining, with an EC50 of 5.1 μM (Fig. 7B and Table 4). In contrast, HeLa cells contained virtually no apoptotic fraction after 48 h, as evidenced by the lack of TUNEL response up to 20 μM (Fig. 7A and Table 4). These results indicated that HeLa cells underwent cytostasis and were less susceptible to apoptosis induction after treatment with 1 for 48 h. Meanwhile, relatively high doses of 1 caused a significant induction of apoptosis only after 72 h culture period.
Fig. 7.

Evaluation of the pro-apoptotic effects of 1 in HeLa cells. Effect of 1 on TUNEL staining after (A) 48 h and (B) 72 h using TUNEL assay. Effect of 1 on cleaved caspase-3 levels after (C) 48 h and (D) 72 h using an immunosorbent fluorometric assay. (E) Effect of 1 on AIF release from the mitochondria after 72 h using an ELISA assay.
3.8. Molecular mechanism of the pro-apoptotic effects of pachycladin A in cancer cells
The activity of caspase-3 was determined in HeLa cells using an immunosorbent fluorometric enzyme assay, after treatment with various doses of 1 for 48 and 72 h. Treatment with 1 markedly increased the levels of cleaved caspase-3 (activated) in HeLa cells after 72 h, with an EC50 of 3.1 μM (Fig. 7D and Table 4). However, the cleavage of caspase-3 was not detected after 48 h with doses up to 20 μM (Fig. 7C and Table 4). In addition, an ELISA assay was performed to ascertain whether 1 stimulates AIF release from the mitochondria after prolonged treatment. HeLa cells were treated for 72 h with different doses of 1 and cells were then homogenized and subcellular fractions were prepared. Results revealed a concentration-dependent increase in AIF mitochondrial release, as represented by an increase in the AIF content in the nuclear fraction along with a concomitant decrement in the mitochondria, with an IC50 of 2.3 μM, implying that 1 may induce mitochondrial membrane depolarization leading to AIF release (Fig. 7E and Table 4).
3.9. Antiangiogenic activity of pachycladins in endothelial colony-forming cells (ECFCs)
Based on the promising results in PD2 angiogenesis module, 1 and 3 were further evaluated for modulation of microvessel density assessed by CD31 immunostaining. In this study, a co-culture cord formation system of human ADSCs and ECFCs was treated with VEGF alone or in combination with other pro-angiogenic factors including FGF and EGF. Co-cultures were then treated with different doses of 1 or 3 for 96 h prior to fixation and staining. Finally, multiplexed parameters of endothelial tube and cell nuclei areas were determined. Quantitative dose-response curves of CD31 tube formation revealed that treatment with 1, and to a less extent 3, potently inhibited angiogenesis with IC50 values of 0.36 and 6.3 μM, respectively (Fig. 8A and 8B). Interestingly, 1 fully maintained its antiangiogenic efficacy in cultures treated with a combination of pro-angiogenic factors compared to VEGF-treated cells (Fig. 8A and 8C). Meanwhile, 1 did not affect ECFCs nuclei area up to 10 μM (Table 4). It was evident that replacing the C-7 hydroxy and C-11 butyrate groups of 1 with exomethylene and acetate functionalities, respectively, in 3 caused a drastic reduction in the antiangiogenic activity by almost 18 folds (Fig. 8A and 8B). This clearly highlighted the crucial role of the C-7 hydroxy and C-11 butyrate groups for optimal activity. Taking into consideration the fact that 5 did not show any activity in the angiogenesis assay even at 10 μM (Table 3), and by comparing its structure with 3 which showed moderate activity in the same assay, the importance of the C-3 acetate for 1’s antiangiogenic activity can be also deduced.
Fig. 8.

Effect of pachycladins (A) 1 and (B) 3 on CD31 tube formation inhibition after 96 h in ECFC-ADSC co-cultures treated with a combination of pro-angiogenic factors. (C) Effect of 1 on CD31 tube formation inhibition after 96 h in ECFC-ADSC co-cultures treated with VEGF alone. (D) Cytotoxic effects of 1 on ECFC cultures at different concentrations using LDH cytotoxicity assay. Triton X-100 was used as a positive control to induce the total possible LDH release. (E) Effect of 1 on ECFC migration inhibition after 24 h using the Oris™ cell exclusion zone assay.
The ratio of dead/live ECFCs was also determined using the MultiTox-Fluor assay, after treatment with 1 for 72 h. This assay allows the simultaneous and ratiometric analysis of intra and extracellular proteolytic activities. Cell-associated protease activity is proportional to the number of live cells, whereas the activity of protease in the media correlates with the number of cells with damaged membranes. The ratio of dead/live cells did not increase in the 1-treated cells up to 40 μM (Table 4). Lactate dehydrogenase (LDH) activity measurements produced similar results; the percentage of LDH release in the media did not increase after treating ECFCs with 1 (Fig. 8D).
3.10. Antimigratory activity of pachycladin A in endothelial colony-forming cells (ECFCs)
Since endothelial cell migration is an essential component of angiogenesis, 1 was further assessed for its ability to inhibit the migration of ECFCs across a cell exclusion zone in the Oris™ assay [23]. ECFCs were seeded and allowed to spread in presence of stoppers contacting the plate’s bottom. Subsequent stoppers removal reveals central cell-free zones into which migration can occur. Different doses of 1 were immediately added after stopper removal and incubated for 24 h. 1 was able to significantly inhibit the cell-free zone closure in a dose-dependent manner, with an IC50 of 7.5 μM (Fig. 8E and Table 4). The Oris™ assay defines a second region seeded with ECFCs within each well and determines changes in cellular area, as a relative measure of compound-mediated toxicity. 1 did not demonstrate any signs of cytotoxicity to ECFCs up to 40 μM, which is many folds higher than its cell migration IC50 (Table 4).
3.11. Kinase profiling of pachycladin A
The activity of 1 was evaluated across a structurally diverse cross-section of 24 tyrosine and serine-threonine kinases in Lilly’s kinase profiling assays. Some selected kinases are known to be key players in cell cycle regulation such as cyclin-dependent kinase 1 (CDK1), aurora kinases, and PLK1 [24–26], whereas other kinases are closely linked to angiogenesis such as VEGFR1, VEGFR2, and FGFR1 [27, 28]. Other kinases induce both tumor cell proliferation and angiogenesis such as EGFR and EPHB4 [29–31]. Results were expressed as % inhibition at a single 2 μM dose of 1 (Table 5). Significant inhibition was observed only for EGFR and protein kinase C β2 (PKCβ2), with greatest relative inhibition for EGFR. In contrast, 1 barely inhibited the activity of six other kinases, including VEGFR1, FLT3, p70S6K1, JAK2, aurora kinase A, and TIE2. In addition, 1 was >50-fold selective for EGFR versus the remaining kinases evaluated in the panel.
Table 5.
Initial kinase selectivity profiling of pachycladin A.
| Kinase | Mean inhibition at 2 μM (%) |
|---|---|
| EPHB4 | −13 |
| FGFR1 | −9 |
| EGFR (ERbB1) | 58 |
| FLT1 (VEGFR1) | 5 |
| KDR (VEGFR2) | −9 |
| PDGFRa | −10 |
| CHK2 | −9 |
| FLT3 | 8 |
| Rock2 | −7 |
| Aura | 6 |
| CDC2/CDK1 | −24 |
| PLK1 | −22 |
| ABL1 | −45 |
| ALK4 | −3 |
| CDK5 | −3 |
| GSK3β | 0 |
| JAK2 | 6 |
| JNK1 | −1 |
| KIT | −14 |
| P38a | −1 |
| P70S6K1 | 6 |
| PKCβ2 | 14 |
| TIE2 | 8 |
| ERK2 | 0 |
In an attempt to have a further insight on its selectivity profile, 1 was evaluated against the ErbB family members, HER2 and HER4, which are known to be structurally related and oncogenically relevant to EGFR, through the SelectScreen® Profiling Service (Life Sciences, CA). In this assay, EGFR kinase was also tested for activity comparison and the results were expressed as % inhibition at a 10 μM dose of 1 (Table 6). Compound 1 showed nearly complete blockade of the EGFR catalytic activity at 10 μM dose with a mean inhibition of 91% (Table 6). In contrast to its high potency against EGFR, 1 barely inhibited the catalytic activity of HER2 and HER4 kinases with mean inhibition of 15% and 16%, respectively (Table 6). To further confirm its exquisite selectivity towards EGFR, the HER2-positive BT-474 human breast cancer cells were chosen to evaluate the effect of 1 on HER2 signaling using Western blotting analysis. Cells were treated with various doses of 1 and then, the expression and phosphorylation levels of HER2 proteins were determined in cell lysates (Fig. 9B). Consistent with kinase profiling results, 1 neither affected the total nor the activated HER2 levels compared to the vehicle-treated BT-474 cells (Fig. 9B).
Table 6.
Effect of Pachycladin A on the catalysis of ErbB kinases and EGFR mutant variants.
| Kinase | Mean inhibition at 10 μM (%) |
|---|---|
| EGFR (ERbB1) | 91 |
| HER2 (ERbB2) | 15 |
| HER4 (ErbB4) | 16 |
| EGFR L858R | 98 |
| EGFR L861Q | 93 |
| EGFR T790M | 93 |
| EGFR T790M L858F | 7 |
| EGFR d746-750 |
Fig. 9.

(A) Left panel; Effect of various doses of 1 on the phosphorylation (activation) of wild-type recombinant human EGFR kinase, using the Z’-LYTE™ assay kit. Error bars indicate the S.D. of n = 3/dose; staurosporine was used as a standard positive control at 0.1 μM. Right panel; The complete 10-point dose-response curve of 1 in inhibiting the phosphorylation (activation) of wild-type recombinant human EGFR kinase using the Z’-LYTE™ assay kit and its IC50 shown in red. (B) Left panel; Western blot analysis showing pachycladin A did not have any significant effects on HER2 signaling after 48 h treatment in human BT-474 breast cancer cells. Right panel; Western blot analysis showing pachycladin A treatment caused a dose-dependent inhibition of EGFR phosphorylation without any effect on its total levels, compared to vehicle-treated control group in MDA-MB-468 cells. Scanning densitometric analysis was performed on all blots conducted in triplicate and the integrated optical density of each band was normalized with corresponding β-tubulin, as shown in bar graphs below their respective Western blot images. Vertical bars in the graph indicate the normalized integrated optical density of bands visualized in each lane ± S.D.
3.12. Effect of pachycladin A on the catalytic activity of wild-type EGFR and its oncogenic variants in vitro
Compound 1 was subjected for further validation of its promising EGFR inhibitory activity shown through the single point kinase profiling. The ability of 1 to inhibit EGFR phosphorylation was directly tested on the recombinant kinase domain of EGFR (amino acids 668–1210) that was in vitro phosphorylated to reach the maximum level of intrinsic kinase activity. In this assay, the Z′-LYTE™ Tyr4 peptide was selected as a substrate; thus, the changes in its phosphorylation can directly reflect the EGFR kinase activity. Meanwhile, staurosporine was used as a standard positive control. Compound 1 demonstrated potent inhibition of the recombinant wild-type EGFR kinase in this cell-free assay, suppressing EGFR phosphorylation in a dose-dependent manner, with an IC50 value of 0.57 & mu;M (Fig. 9A).
The promising activity of 1 against wild-type EGFR prompted the investigation of its ability to inhibit EGFR phosphorylation across five EGFR human oncogenic mutant variants, including two activation loop mutants L858R and L861Q, as well as T790M mutant in the hinge region, the double-mutant T790M L858F, and the in-frame deletion of d746-750, which is in the region connecting the P-loop and C helix. Interestingly, 1 maintained its activity against L858R, L861Q, and the gefitinib-resistant T790M mutants, compared with wild-type EGFR at the same 10 & mu;M screening dose (Table 6). In contrast, a marked shift in the potency of 1 was observed against the double-mutant variant T790M L858F and the deletion mutation d746-750 compared with wild-type receptor (Table 6).
3.13. Effect of pachycladin A on EGFR signaling
vIn this study, MDA-MB-231 and MDA-MB-468 human breast cancer cells were chosen to evaluate the effect of 1 on EGFR signaling using Western blot analysis. Cells were treated with various doses of 1 and then, the expression and phosphorylation levels of EGFR and its downstream signaling proteins, including protein kinase B (Akt), ERK/mitogen-activated protein kinase (MAPK), focal adhesion kinase (FAK), and Brk, were determined in cell lysates (Fig. 10). Consistent with its biochemical activity, 1 demonstrated a striking dose-dependent inhibition of EFGR phosphorylation in MDA-MB-231 and MDA-MB-468 cells, compared to the vehicle-treated control group (Figs. 9B and 10A). Meanwhile, 1 did not affect the total EGFR levels at the same treatment doses (Figs. 9B and 10A). Furthermore, treatment with 1 caused a dosedependent inhibition of Akt and ERK/MAPK phosphorylation, compared to the vehicle-treated control group in MDA-MB-231 cells (Fig. 10B and 10C). Meanwhile, 1 did not affect the total levels of Akt and MAPK at the same treatment doses (Fig. 10B and 10C). Consistent with EGFR phosphorylation inhibition, 1 showed a dose-dependent suppression of FAK and Brk phosphorylation, as compared to the vehicle-treated control group, without any effect on their total levels (Fig. 10D and 10E). To assess its off-target effects, 1 has been also tested for its ability to inhibit the mesenchymal-epithelial transition factor (c-Met) RTK signaling in the MDA-MB-231 TNBC model. 1 resulted in a slight or no effect on total and activated c-Met levels compared to the vehicle-treated cells (Fig. 10F). Collectively, these data suggested that 1 is a robust inhibitor of the EGFR signaling axis and its important downstream pathways in TNBC cells.
3.14. Molecular modeling
3.14.1. Binding mode analysis of pachycladin A
In order to elucidate the binding mode of 1 at the ATP binding pocket of EGFR kinase, a detailed docking study was performed. To minimize false positive results due to crystal structure conformational variations, two highly resolved EGFR crystal structures (PDB codes: 2ITW and 4WKQ) were used to investigate the possible binding modes of 1 within the catalytic domain of unphosphorylated EGFR by means of the Schrödinger software (Fig. 11). The visualization of the docked pose of 1 confirmed its perfect shape fitting at the ATP binding pocket of EGFR kinase (Fig. 11A and 11B, right panel). The oxabicycloundecane ring system of 1 was oriented in the back of the ATP-binding pocket, occupying the same region as the ATP purine ring, and its C-7 hydroxy participated in a critical hydrogen bond (HB) with Met793 or Pro794 of the EGFR crystal structures 4WKQ or 2ITW, respectively (Fig. 11A and 11B, left and middle panels). Both Met793 and Pro794 lie in the hinge of the kinase, which connects the N and C lobes [32]. Alternatively, pachycladins 2–5, which lack the C-7 hydroxy due to exomethylene substitution, failed to satisfy such critical HB within the hinge and subsequently showed poor activity in the MTT assay (Table 1). Furthermore, the C-11 butyrate of 1 was uniquely engaged in a strong HB via its carbonyl with the backbone amide of Met793 in crystal structure 2ITW (Fig. 11A, left and middle panels). Additionally, the same butyrate extended into a hydrophobic pocket defined by the side chains of Val726, Ala743, Lys745, Gln791, and Leu792 in the back of the ATP-binding cleft (Fig. 11A, middle panel). Despite the fact that this butyrate group lacked HB interactions in crystal structure 4WKQ, it was able to access the deep end of the ATP-binding pocket, exerting favorable van der Waal interactions with the gatekeepers Ala743, Ile744, Lys745, Met766, Leu788, and Thr790, thus hindering these residues from participating in catalysis (Fig. 11B, left and middle panels). This might explain the significant impact of the C-11 butyrate on increasing the activity level of 1 in cell proliferation and angiogenesis assays. The hydrophobic interactions exerted by this butyrate appeared to impart a substantial portion of binding affinity since pachycladins 3–5, with either C-11 acetate or exomethylene functionalities, had significantly lower or even diminished activities in cell-based assays. The lower binding affinity of 3 and 5 relative to 1 could be partially because their C-11 acetate is not sufficiently long enough for optimal fitting into the hydrophobic pocket. Finally, the C-3 acetate group in 1 extends toward the solvent near Asp800 at the edge of the active site and it is involved in a HB with Cys797 of the extended hinge region in both 2ITW and 4WKQ (Figs. 11A and 11B, left and middle panels). Interestingly, the C-7 methyl group of 1 is buried in the binding pocket and exerting hydrophobic interactions with the side chains of Phe795, Leu718, and Leu844 at an EGFR hydrophobic subpocket (Figs. 11A and 11B, middle panel). This observation may provide another possible explanation for the poor activity of 2–5, since these compounds lack the C-7 methyl, and thus failed to exert such important interactions within the EGFR hydrophobic subpocket. However, the kinase inhibitory activity of 1 is still in the high nanomolar level, perhaps because the C-7 methyl is not sufficiently large enough to fill the hydrophobic subpocket or lack the aromaticity to engage in π-electron interactions with the aromatic residues comprising the pocket (Figs. 11C and 11D, middle panel). Although the unique oxabicycloundecane ring did not show direct binding with the receptor, it ultimately played a crucial role in properly aligning the binding pharmacophores of 1 at the EGFR kinase.
Fig. 11.

(A and B) In silico binding mode of 1 at the ATP binding site of EGFR kinase crystal structures (A) 2ITW and (B) 4WKQ. Left panel; Important interactions of 1 at the EGFR kinase domain, the protein is shown in three-dimensional cartoon presentation. Middle panel; The two-dimensional ligand interaction diagrams (LID) of 1 at the EGFR kinase domain are shown, as generated using the Schrödinger software. Right panel; The transparent protein surface, in aquamarine color, and the solid 1 surface, in spring green color, are shown to emphasize the 1’s shape fitting within the target pocket. (C and D) Left panel; In silico binding pose of 1 at the ATP binding site of EGFR crystal structures (C) 2ITW and (D) 4WKQ, the protein solid surface is shown in aquamarine color with arrows indicating the solvent surface and the deep hydrophobic pocket where the C-11 butyrate group of 1 resides, suggesting the need for bulkier group at this position to improve the 1’s EGFR binding affinity. Middle panel; The hydrophobic subpocket of EGFR crystal structures (C) 2ITW and (D) 4WKQ, where the C-7 methyl group of 1 resides, is indicated by arrows suggesting the need for molecular extension at this position to efficiently fill the EGFR hydrophobic subpocket and improve 1’s EGFR binding affinity. Right panel; Structure overlay for 1 shown in tube with (C) AFN941 and (D) gefitinib conformations shown in thin tube, as obtained from EGFR crystal structures 2ITW and 4WKQ, respectively, via docking simulations. The important HB interactions exerted by 1 and the original ligands towards critical amino acids within the EGFR kinase pocket are also shown. In particular, 1 engaged in a crucial HB with Cys797, conferring T790M EGFR mutant inhibitory activity, unlike the hexahydroindole ring of AFN941 and the propylmorpholino group of gefitinib, which missed this interaction. The protein solid surface for the deep hydrophobic pocket where the C- 11 butyrate of 1 perfectly overlaid with the aniline ring of gefitinib at the back of the ATP-binding cleft is also shown in aquamarine color and the solvent surface is indicated in arrows.
Abbreviations: ATCC, American type culture collection; CD30, cluster of differentiation 30 a type I transmembrane glycoprotein belonging to the TNF receptor superfamily; CD31, platelet endothelial cell adhesion molecule 1; EGF, epidermal growth factor; EPHB4, ephrin type-B receptor 4; ERK, extracellular signal-regulated kinase; FGFR, fibroblast growth factor receptor; FLT3, fms-related tyrosine kinase 3; JAK2, janus kinase 2; L858R, leucine 858 mutated to arginine; L861Q, leucine 861 mutated to glutamine; M, mitosis; MTT, 3-[4,5-dimethylthiazol-2- yl]-2,5-diphenyltetrazolium bromide; PDB, protein data bank; PLK1, polo-like kinase 1; P70S6K1, phosphorylation of 70-kDa ribosomal protein S6 kinase 1; RIPA, radioimmunoprecipitation assay; RMSD, root mean square displacement; RPMI, Roswell park memorial institute; TBST, tris-buffered saline with Tween 20; TIE2, tyrosine kinase with immunoglobulin-like and EGF-like domains 2; T790M, threonine 790 mutated to methionine; TNBC, triple negative breast cancer; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; Wnt, wingless-type MMTV integration site family member.
3.14.2. Binding mode comparison of pachycladin A with other EGFR inhibitors
To validate the docking results, AFN941 and gefitinib, the original co-crystallized ligands for 2ITW and 4WKQ respectively, were docked into their ATP binding pockets using the same parameters which have been applied for docking 1 (Figs. 11C and 11D, right panel). The bound conformation of each ligand was generated with RMSD value of 0.2 Å, validating the docking protocols. In crystal structure 2ITW, 1 partially overlaid AFN941 demonstrating the same critical interaction with Met793 in the hinge (Fig. 11C, right panel). The oxabicycloundecane ring of 1 is partially aligned across one axis of the nearly planar structure of AFN941, with its C-11 butyrate mimicking the binding role of the lactam ring of AFN941 in the back of the pocket, where both the amide carbonyl of AFN941 and the butyrate carbonyl of 1 engage in a HB with the backbone amide of Met793 (Fig. 11C, right panel). Despite the fact that the indoline ring of AFN941 adopted an extended conformation and did not overlap with 1, it was solvent exposed and therefore, should not have a significant impact on EGFR inhibition (Fig. 11C, right panel). The hexahydroindole ring of AFN941 did not provide the same structural extension as that offered by the ring system of 1 with its connected C-3 acetate and C-7 hydroxy groups and thus, AFN941 missed two important interactions offered by 1 in the extended hinge region of the EGFR kinase (Fig. 11C, right panel).
The space occupied by 1 in crystal structure 4WKQ was very similar to that exploited by gefitinib, where the oxabicycloundecane ring partially superimposed with the quinazoline core, and the butyrate perfectly overlaid with the aniline ring of gefitinib in the hydrophobic pocket at the back of the ATP-binding cleft (Fig. 11D, right panel). The C-3 acetate in 1 extended into the solvent and partially overlapped with the propylmorpholino group of gefitinib (Fig. 11D, right panel). This was consistent with the gefitinib structure activity relationship (SAR) data, as this substituent was added to improve its pharmacokinetic properties [33]. In contrast, the C-3 acetate of 1 positively contributed to the EGFR binding affinity via a HB with Cys797 in the extended hinge region (Fig. 11D, right panel).
4. Discussion
Marine natural products are deemed a rich resource of unique chemical entities exhibiting anticancer properties [8]. In this study, the initial screening of five known pachycladin diterpenoids against the human breast cancer MDA-MB-231 cells revealed a spectrum of variable activity, with 1 being the most effective inhibitor of cancer cells’ growth. The results suggested the significance of 1’s acetate, hydroxy, and butyrate groups at C-3, C-7, and C-11, respectively, for optimal activity. Pachycladin A was selected for further examination in a panel of human breast and cervical cancer cell lines. The results provided compelling evidence that 1 has the potential to exert prominent antiproliferative effects across a broad spectrum of aggressive cancer phenotypes with different molecular characteristics. Generally, the TNBC MDA-MB-231 and MDA-MB-468 in addition to the cervical cancer HeLa cells were the most sensitive to 1 treatment. TNBC lacked the expression of estrogen, progesterone, and HER-2 receptors [34]. It represents 17% of all breast cancers, with aggressive clinical behavior and poor prognosis. Chemotherapy stands as the main available treatment option for TNBC patients [34]. Current chemotherapeutic treatments are not completely selective for carcinogenic cells and often induce significant cytotoxic effects on normal tissues, affecting the quality of life of cancer patients. Thus, there is an urgent need to discover new treatments with relatively higher selectivity index for this aggressive subtype, which lack targeted therapy. It is interesting to note that the activity of 1 towards cancer cells was coupled with absence of cytotoxicity to the non-tumorigenic mammary epithelial cells at similar or even higher treatment doses (Fig. 4). In the highly metastatic MDA-MB-231 breast cancer cells, pachycladin A treatment was also associated with the induction of G1 cell cycle arrest. The promising in vitro profile and excellent selectivity index of 1 towards cancerous cells suggested its potential as a new and selective antiproliferative entity. It is worth noting that this is the first report for the antiproliferative activity of 1 against any type of cancer.
To further characterize the antiproliferative effects of 1, the cell cycle arrest position was also defined in HeLa cervical cancer cells using the G2/M marker cyclin B, the mitotic marker pHH3, and TUNEL staining. PHH3 is a marker for chromosomal condensation promoting the recruitment of condensin, while cyclin B activates CDK1 and allows progression through G2 into mitosis [35]. TUNEL staining identifies apoptotic cells with DNA strand breaks in situ using TdT to add fluorescein-dUTP to cleaved ends of DNA and detect the directly incorporated nucleotides with a fluorescence microscope or flow cytometer [36]. After treatment with 1, HeLa cells were unable to progress through mitosis and have been arrested in G2, expressing high levels of cyclin B and pHH3. The ability of 1 to modulate DNA content and DNA condensation with high and equal potency suggested that the antiproliferative activity of 1 in cervical cancer cells is mediated through cytostatic mechanisms rather than inducing apoptosis, which was further evidenced by lack of TUNEL response at the in vitro tested doses. Thus, pachycladin A treatment is associated with cytostatic activity and either G1 or G2/M cell cycle arrest, according to the cancer type, the findings that contribute to the growth inhibitory activity of this compound in breast and cervical cancer cells. It is well established that blockade of cell cycle progression can initiate programmed cell death in different cancer types. Results of the present study showed that the cell cycle arrest found upon 1 treatment likely led to cancer cell apoptosis only after prolonged treatment periods (72 h). To unravel the molecular mechanism underlying the apoptotic effects of 1, the activity of caspase-3 and AIF mitochondrial release were assessed. Caspase-3 functions as an executioner caspase, cleaving various substrates that causes the morphological and biochemical changes seen in apoptotic cells [23]. On the other hand, AIF is a caspase-independent pro-apoptotic protein released from the mitochondria after the cell has committed to die [36]. AIF then translocates to the nucleus, where it induces DNA fragmentation, chromatin condensation, and nuclear shrinkage [37]. The remarkable increase in the levels of cleaved caspase-3 and AIF mitochondrial release in HeLa cells after treatment with 1 for 72 h, without any significant change in their levels after 48 h, corroborated the TUNEL assay results and suggested that cancer cell sensitivity partially links to the ability of 1 to induce cell death after prolonged treatment periods via both the caspase-dependent and -independent (AIF-mediated) apoptotic pathways. Importantly, these findings distinguish 1 from the potent cytotoxic microtubule disruptor natural products, sarcodictyins and eleutherobin, and clearly imply that 1 is a potential antiproliferative entity which doesn’t work via tubulin polymerization-depolymerization stabilization mechanism.
In addition to its promising antiproliferative effects, the results of this study revealed the potent antiangiogenic potential of 1 via inhibiting endothelial tube formation and migration in a co-culture of human ADSCs and ECFCs. The implemented co-culture model has been chosen for three reasons; firstly, ECFCs tend to form tube-like structures that express CD31 when cocultured on an ADSC feeder layer ; secondly, the model allows the simultaneous measurement of total tube area, as the most sensitive measure of endothelial tube formation, and cell nuclei area which provides an important measure of relative cytotoxicity to readily differentiate active hits from false-positives in the angiogenesis assay [21]; thirdly, it may better mimic the interplay of signaling pathways that occur during neovascularization and provides the opportunity to identify compounds that inhibit angiogenesis by novel mechanisms distinct from current VEGF pathway-directed agents [21]. In addition to the dominant proangiogenic factor VEGF, growth factors such as FGF, EGF, and hepatocyte growth factor (HGF) have been included in this assay because of their potential to modulate angiogenesis and confer resistance to the already existing VEGF inhibitors [38]. The potential of 1 to maintain its antiangiogenic activity in cultures treated with a pro-angiogenic factors cocktail and/or VEGF, without exhibiting cytotoxicity to ECFCs at concentrations many folds higher than its angiogenesis IC50, clearly implied its true and unique antiangiogenic profile. MultiTox-Fluor and LDH cytotoxicity assays confirmed the lack of any undesirable cytotoxic mechanisms towards ECFCs and emphasized the potential of 1 as a true antiangiogenic hit which can control aggressive cancer phenotypes. Meanwhile, the antiangiogenic SAR observed for 1, 3, and 5 was parallel to their antiproliferative activities in MTT assay, suggesting a common molecular mechanism for these anticancer effects.
The second part of the study attempted to unravel the molecular signaling pathway underlying the remarkable antiproliferative and antiangiogenic activities of 1. Two different kinase profiling platforms revealed the exquisite inhibitory activity of 1 towards EGFR RTK at pharmacologically relevant concentrations (Tables 5 and 6). Consistently, a follow-up dose-response quantitative study demonstrated the nanomolar efficacy of 1 in inhibiting the wild-type EGFR catalytic activity (Fig. 9A). An interesting feature of 1 is its high degree of selectivity towards EGFR when profiled against the highly homologous HER2 and HER4 kinases (6-fold selectivity difference versus EGFR, Table 6), which was further corroborated by Western blotting analysis, where 1 did not show any significant effect on HER2 signaling in the HER2- driven BT-474 breast cancer cells (Fig. 9B). Importantly, 1 did not show any appreciable inhibition towards VEGFRs 1 and 2, supporting the hypothesis that the antiangiogenic activity of 1 is taking place via novel mechanism of action. On the molecular level, the growth inhibitory effect of 1 in TNBC MDA-MB-231 and MDA-MB-468 cells was associated with suppression of EGFR phosphorylation and subsequent inhibition of downstream EGFR effectors in MDA-MB- 231 cultures (Figs. 9B and 10). These include: Akt and MAPK signaling pathways which are involved in mediating cell growth and proliferation, respectively [39, 40]; FAK, along with other ErbB signaling molecules, which participate in angiogenesis, cell motility, adhesion, and organogenesis [40]. Results of this study also showed the ability of 1 to inhibit Brk tyrosine kinase phosphorylation in a dose-dependent manner. Despite the fact that Brk is an important downstream molecule for some RTKs that are commonly dysregulated in breast cancer such as c-Met [41], it is not yet considered as a “classic” substrate of EGFR kinase activity. However, inhibition of Brk phosphorylation can be considered as a very desirable feature for an EGFR small-molecule inhibitor such as 1 for three reasons; firstly, due to a novel regulatory mechanism of EGFR signaling shown by Li et al, where Brk sustained ligand-activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR, which may contribute to the limited efficacy of EGFR targeted therapy in breast cancer clinical trials [42]. Secondly, Brk is activated by EGF stimulation of cells, and experimental elevation of Brk sensitized immortalized non-cancerous human mammary epithelial cells to EGF-induced mitogenic effects and protected cells from anoikis [43, 44]. Brk can also interact with EGFR downstream substrates, such as Akt and paxillin in breast cancer cells, promoting mammary cancer cell growth and motility [45, 46]. Therefore, inhibiting Brk phosphorylation by pachycladin A can be considered as an advantageous off-target effect as a small-molecule EGFR kinase inhibitor specifically in breast cancer therapy due to the direct and diverse interactions between EGFR and Brk. Given the critical role of c-Met RTK in controlling breast cancer cell proliferation, the possibility that the antiproliferative effects of 1 might be associated with the suppression of c-Met signaling was eliminated, implying a degree of specificity towards EGFR (Fig. 10F). Taken together, the antiproliferative and antiangiogenic activities of 1 in cancer cells were associated with the inhibition of EGFR phosphorylation and its downstream signaling pathways mediating proliferation, angiogenesis, and motility. Though EGFR overexpression is observed in all breast cancer subtypes, solid clinical evidence showed that it is more frequently overexpressed (50% of cases) in the highly aggressive TNBC leading to reduced survival and poor clinical outcomes [47]. In addition, it has been recently found that EGFR expression in breast cancer inversely correlated with hormonal receptor expression, particularly estrogen receptor [48]. This suggests that TNBC which lacks estrogen and progesterone receptors will demonstrate higher levels of EGFR expression and dependency compared to non-TNBCs. The MTT results revealed that the TNBC cells, with higher levels of EGFR expression and dependency, were the most sensitive to treatment with 1 supporting the hypothesis that EGFR inhibition can be one of the possible mechanisms for its anticancer effects. Further, a recent study has shown that targeting EGFR enhances the chemosensitivity of TNBC cells by rewiring their apoptotic signaling networks [49]. Thus, small-molecule EGFR inhibitors, such as 1, might have a potential therapeutic role, as chemosensitizers or anticancer agents, in TNBC.
Consistent with biochemical data and Western blot results, 1 showed complete fitting when docked at the EGFR kinase domain (Fig. 11). Interestingly, the C-3 acetate, C-7 hydroxy, and C- 11 butyrate of 1 were virtually proved to be the main anchoring groups at the kinase domain, justifying EGFR kinase inhibition and well-matching the SAR results of 1 in cell-based assays (Fig. 11). Structural overlay docking studies suggested that the presence of the 1’s butyrate group in a hydrophobic pocket at the back of the ATP-binding cleft is not sufficient for optimal EGFR inhibition (Figs. 11C and 11D, left panel), and that increasing the bulkiness at this site by replacing the butyrate with a substituted aromatic ring, mimicking the structure of gefitinib, would probably enhance the EGFR binding affinity and subsequent cellular potency (Fig. 11D, right panel). Another possible direction for future optimization of 1 as a novel EGFR inhibitor can be implemented through the replacement of the C-7 methyl group either with bulkier aliphatic or aromatic functionalities to efficiently fill the EGFR hydrophobic subpocket, where the C-7 methyl resides, and engage in favorable interactions with the residues present in this pocket (Figs. 11C and 11D, middle panel). Together, docking and overlay studies were able to explain the established SAR of 1 in angiogenesis and proliferation assays, being a result of EGFR kinase inhibition, and characterized the key pharmacophores likely to impart higher binding affinity towards EGFR that would be of great importance in guiding the future optimization of 1 as a potential EGFR inhibitor.
Emergence of resistant mutations in the EGFR kinase domain represents a serious challenge to the NSCLC targeted chemotherapy. It is now established that the efficacy of gefitinib and erlotinib is of limited duration due to the development of acquired drug resistance conferred by a secondary mutation of the gatekeeper residue T790M in the kinase domain [50, 51]. This mutation maintains the catalytic activity of the enzyme but substantially suppresses the inhibitory effects of gefitinib and erlotinib through changing the topology of the ATP-binding pocket [50, 51]. In contrast, L858R, L861Q, and d746-750 EGFR mutations resulted in enhanced kinase catalysis and improved sensitivity to gefitinib at the target protein level [52]. However, T790M mutation can also confer resistance to any of these gefitinib-sensitive EGFR mutants when double mutation takes place [50]. Addressing this unmet medical need requires the discovery of novel entities capable of targeting T790M as a common EGFR TKIs-resistant mutation. In addition to the potent activity against the wild-type EGFR, 1 significantly inhibited its T790M point mutation, along with L858R and L861Q, in a panel of EGFR mutant kinases screened in this report, which could prove to be a very desirable feature in new small-molecule EGFR inhibitors (Table 6). The predominant strategy to overcome T790M-mediated resistance has been the use of irreversible inhibitors, which usually incorporate a Michael acceptor group that forms a key covalent bond with Cys797 within the EGFR kinase domain, conferring both potency and kinase selectivity [53, 54]. Due to their reactive functional groups, the clinical efficacy of irreversible EGFR inhibitors was limited by the associated off-target skin rash and gastrointestinal toxicity, suggesting the dire need for drugs that don’t act via covalent adducts formation with Cys797 for neither potency nor selectivity [55, 56]. Interestingly, the docking and overlay studies presented herein suggested the ability of the 1’s terminal C-3 acetate to non-covalently bind with Cys797, unlike the propylmorpholino group of gefitinib which missed this interaction, justifying 1’s EGFR inhibitory potency against the gefitinib-resistant T790M mutant in kinase assays (Fig. 11D, right panel). It is worth noting that 1 was able to exert the same interaction with Cy797 in the EGFR crystal structure 2ITW while the crystal structure AFN941 lacked it, adding further justification for the results of kinase assays (Fig. 11C, right panel). Meanwhile, the mutations L858R and L861Q lie in the EGFR kinase domain about 12 Å away from 1. This may explain the negligible effect of these mutations on 1’s EGFR inhibitory activity in kinase assays. Although non-covalent inhibition of T790M mutant sounds like attractive strategy for overcoming the acquired drug resistance whilst lacking the confounding off-target toxicity due to irreversible EGFR inhibition, brigatinib has been the only known non-covalent EGFR T790M inhibitor [57]. Unfortunately, brigatinib is a dual anaplastic lymphoma kinase (ALK)/EGFR inhibitor and the primary focus of its clinical applications appears to be in ALK-driven lung cancers [57]. These data renders 1 as one of the novel reversible EGFR T790M mutant inhibitors known to date, with possible therapeutic potential to prevent relapse and recurrences in EGFR-dependent malignancy patients after the successful completion of gefitinib or erlotinib regimen.
Overall, this study comprehensively characterized the anticancer properties of 1 in different cancer models with novel pharmacophoric features. Results indicated that the anticancer activity of 1 is mediated by direct effects on tumor growth and antiangiogenic mechanisms, selectively via deactivating EGFR signaling.
Acknowledgments
This work was supported in-part by the National Cancer Institute of the National Institutes of Health under Award Number R15CA167475. The Lilly’s Open Innovation Program is greatly acknowledged for different biological assays.
Footnotes
Conflict of interest statement The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Galvani E, Alfieri R, Giovannetti E, Cavazzoni A, La Monica S, Galetti M, Fumarola C, Bonelli M, Mor M, Tiseo M, Peters GJ, Petronini PG, Ardizzoni A. Epidermal growth factor receptor tyrosine kinase inhibitors: Current status and future perspectives in the development of novel irreversible inhibitors for the treatment of mutant non-small cell lung cancer. Current Pharmaceutical Design. 2013;19:818–832. [PubMed] [Google Scholar]
- 2.Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5:390–401. doi: 10.1016/j.apsb.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. Journal of Clinical Oncology. 2008;26:1742–1751. doi: 10.1200/JCO.2007.12.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roskoski RJ. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacological Research. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Ou SH. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): A better mousetrap? A review of the clinical evidence. Critical Reviews in Oncology/Hematology. 2012;83:407–421. doi: 10.1016/j.critrevonc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Lahlou M. The success of natural products in drug discovery. Pharmacol Pharm. 2013;4:17–31. [Google Scholar]
- 7.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cragg GM, Grothaus PG, Newman DJ. New horizons for old drugs and drug leads. J Nat Prod. 2014;77:703–723. doi: 10.1021/np5000796. [DOI] [PubMed] [Google Scholar]
- 9.Sheu JH, Wang GH, Duh CY, Soong K. Pachyclavulariolides M-R, six novel diterpenoids from a Taiwanese soft coral Pachyclavularia violacea. Journal of Natural Products. 2003;66:662–666. doi: 10.1021/np020603j. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed AF, Shiue RT, Wang GH, Dai CF, Kuo YH, Sheu JH. Five novel norcembranoids from Sinularia leptoclados and S. parva. Tetrahedron. 2003;59:7337–7344. [Google Scholar]
- 11.Ahmed AF, Su JH, Shiue RT, Pan XJ, Dai CF, Kuo YH, Sheu JH. New β-caryophyllene-derived terpenoids from the soft coral Sinularia nanolobata. J Nat Prod. 2004;67:592–597. doi: 10.1021/np030286w. [DOI] [PubMed] [Google Scholar]
- 12.Sung PJ, Hu WP, Wu SL, Su JH, Fang LS, Wang JJ, Sheu JH. Briaexcavatolides X-Z, three new briarane-related derivatives from the gorgonian coral Briareum excavatum. Tetrahedron. 2004;60:8975–8979. [Google Scholar]
- 13.Wang GH, Sheu JH, Duh CY, Chiang MY. Pachyclavulariaenones D-G, new diterpenoids from the soft coral Pachyclavularia violacea. Journal of Natural Products. 2002;65:1475–1478. doi: 10.1021/np020095d. [DOI] [PubMed] [Google Scholar]
- 14.Chen TH, Lu MC, Chang YC, Su YD, Chen YH, Lin NC, Fang LS, Wu YC, Sung PJ. Discovery of new eunicellin-based diterpenoids from a Formosan soft coral Cladiella sp. Mar Drugs. 2013;11:4585–4593. doi: 10.3390/md11114585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tai CJ, Su JH, Huang MS, Wen ZH, Dai CF, Sheu JH. Bioactive eunicellin-based diterpenoids from the soft coral Cladiella krempfi. Mar Drugs. 2011;9:2036–2045. doi: 10.3390/md9102036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindel T, Jensen PR, Fenical W, Long BH, Casazza AM, Carboni J, Fairchild CR. Eleutherobin, a new cytotoxin that mimics paclitaxel (Taxol) by stabilizing microtubules. J Am Chem Soc. 1997;119:8744–8745. [Google Scholar]
- 17.Huang TZ, Chen BW, Huang CY, Hwang TL, Uvarani C, Dai CF, Sung PJ, Su JH, Sheu JH. Eunicellin-based diterpenoids, Hirsutalins S-V, from the Formosan soft coral Cladiella hirsuta. Marine Drugs. 2015;13:2757–2769. doi: 10.3390/md13052757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassan HM, Khanfar MA, Elnagar AY, Mohammed R, Shaala LA, Youssef DT, Hifnawy MS, El Sayed KA. Pachycladins A-E, prostate cancer invasion and migration inhibitory eunicellin-based diterpenoids from the red sea soft coral Cladiella pachyclados. J Nat Prod. 2010;73:848–853. doi: 10.1021/np900787p. [DOI] [PubMed] [Google Scholar]
- 19.Hassan HM, Elnagar AY, Khanfar MA, Sallam AA, Mohammed R, Shaala LA, Youssef DT, Hifnawy MS, El Sayed KA. Design of semisynthetic analogues and 3D-QSAR study of eunicellin-based diterpenoids as prostate cancer migration and invasion inhibitors. Eur J Med Chem. 2011;46:1122–1130. doi: 10.1016/j.ejmech.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 20.Akl MR, Ayoub NM, Mohyeldin MM, Busnena BB, Foudah AI, Liu Y-Y, El Sayed KA. Olive phenolics as c-Met inhibitors: (–)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models. PloS One. 2014;9:e97622. doi: 10.1371/journal.pone.0097622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JA, Chu S, Willard FS, Cox KL, Husain S, Palkowitz AD, Moxham CM. Open innovation for phenotypic drug discovery: The PD2 assay panel. J Biomol Screen. 2011;16:588–602. doi: 10.1177/1087057111405379. [DOI] [PubMed] [Google Scholar]
- 22.Atkins M, Jones CA, Kirkpatrick P. Sunitinib maleate. Nat Rev Drug Discov. 2006;5:279–280. doi: 10.1038/nrd2012. [DOI] [PubMed] [Google Scholar]
- 23.Gough W, Hulkower KI, Lynch R, Mcglynn P, Uhlik M, Yan L, Lee JA. A quantitative, facile, and high-throughput image-based cell migration method is a robust alternative to the scratch assay. J Biomol Screen. 2011;16:155–163. doi: 10.1177/1087057110393340. [DOI] [PubMed] [Google Scholar]
- 24.van de Weerdt BC, Medema RH. Polo-like kinases: a team in control of the division. Cell Cycle. 2006;5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 25.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 26.Fu J, Bian M, Jiang Q, Zhang C. Roles of aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 27.Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fons P, Gueguen-Dorbes G, Herault JP, Geronimi F, Tuyaret J, Frederique D, Schaeffer P, Volle-Challier C, Herbert JM, Bono F. Tumor vasculature is regulated by FGF/FGFR signaling-mediated angiogenesis and bone marrow-derived cell recruitment: This mechanism is inhibited by SSR128129E, the first allosteric antagonist of FGFRs. J Cell Physiol. 2015;230:43–51. doi: 10.1002/jcp.24656. [DOI] [PubMed] [Google Scholar]
- 29.Fraguas S, Barberan S, Cebria F. EGFR signaling regulates cell proliferation, differentiation and morphogenesis during planarian regeneration and homeostasis. Dev Biol. 2011;354:87–101. doi: 10.1016/j.ydbio.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 30.van Cruijsen H, Giaccone G, Hoekman K. Epidermal growth factor receptor and angiogenesis: Opportunities for combined anticancer strategies. Int J Cancer. 2005;117:883–888. doi: 10.1002/ijc.21479. [DOI] [PubMed] [Google Scholar]
- 31.Martiny-Baron G, Korff T, Schaffner F, Esser N, Eggstein S, Marme D, Augustin HG. Inhibition of tumor growth and angiogenesis by soluble EphB4. Neoplasia. 2004;6:248–257. doi: 10.1593/neo.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Henthorn L, Richards L. Studies leading to the identification of ZD1839 (IRESSA): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–1914. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan HG, Malmgren JA. Impact of triple negative phenotype on breast cancer prognosis. Breast J. 2008;14:456–463. doi: 10.1111/j.1524-4741.2008.00622.x. [DOI] [PubMed] [Google Scholar]
- 35.Low J, Chakravartty A, Blosser W, Dowless M, Chalfant C, Bragger P, Stancato L. Phenotypic fingerprinting of small molecule cell cycle kinase inhibitors for drug discovery. Curr Chem Genomics. 2009;3:13–21. doi: 10.2174/1875397300903010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baritaud M, Boujrad H, Lorenzo HK, Krantic S, Susin SA. Histone H2AX: The missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle. 2010;9:3166–3173. doi: 10.4161/cc.9.16.12887. [DOI] [PubMed] [Google Scholar]
- 38.Folkman J. Angiogenesis: An organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 39.Kitano H, Chung JY, Ylaya K, Conway C, Takikita M, Fukuoka J, Doki Y, Hanaoka J, Hewitt SM. Profiling of phospho-AKT, phospho-mTOR, phospho-MAPK and EGFR in non-small cell lung cancer. Journal of Histochemistry and Cytochemistry. 2014;62:335–346. doi: 10.1369/0022155414523022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opinion on Therapeutic Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Locatelli A, Lofgren KA, Daniel AR, Castro NE, Lange CA. Mechanisms of HGF/Met Signaling to Brk and Sam68 in Breast Cancer Progression. Hormones & Cancer. 2012;3:14–25. doi: 10.1007/s12672-011-0097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, Hung MC, Fan Z. Brk/PTK6 Sustains Activated EGFR Signaling through Inhibiting EGFR Degradation and Transactivating EGFR. Oncogene. 2012;31:4372–4383. doi: 10.1038/onc.2011.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamalati T, Jolin HE, Mitchell PJ, Barker KT, Jackson LE, Dean CJ, Page MJ, Gusterson BA, Crompton MR. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. Journal of Biological Chemistry. 1996;271:30956–30963. doi: 10.1074/jbc.271.48.30956. [DOI] [PubMed] [Google Scholar]
- 44.Irie HY, Shrestha Y, Selfors LM, Frye F, Iida N, Wang Z, Zou L, Yao J, Lu Y, Epstein CB, Natesan S, Richardson AL, Polyak K, et al. PTK6 regulates IGF-1- induced anchorage-independent survival. PloS One. 2010;5:e11729. doi: 10.1371/journal.pone.0011729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang P, Ostrander JH, Faivre EJ, Olsen A, Fitzsimmons D, Lange CA. Regulated association of protein kinase B/Akt with breast tumor kinase. Journal of Biological Chemistry. 2005;280:1982–1991. doi: 10.1074/jbc.M412038200. [DOI] [PubMed] [Google Scholar]
- 46.Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Molecular and Cellular Biology. 2004;24:10558–10572. doi: 10.1128/MCB.24.24.10558-10572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Changavi AA, Shashikala A, Ramji AS. Epidermal Growth Factor Receptor Expression in Triple Negative and Nontriple Negative Breast Carcinomas. Journal of Laboratory Physicians. 2015;7:79–83. doi: 10.4103/0974-2727.163129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 52.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 53.Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, Bradbury RH, Brown SJ, Butterworth S, Campbell A, Chorley C, Colclough N, Cross DA, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. Journal of Medicinal Chemistry. 2014;57:8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 54.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discovery. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janne P, Schellens J, Engelman J, Eckhardt S, Millham R, Denis L, Britten C, Wong S, Boss D, Camidge D. Preliminary activity and safety results from a phase I clinical trial of PF-00299804, an irreversible pan-HER inhibitor, in patients with NSCLC. Journal of Clinical Oncology. 2008;26:8027. [Google Scholar]
- 56.Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, Eaton K, Zacharchuk C, Freyman A, Powell C, Ananthakrishnan R, Quinn S, Soria JC. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2010;28:3076–3083. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 57.Heald R, Bowman KK, Bryan MC, Burdick D, Chan B, Chan E, Chen Y, Clausen S, Dominguez-Fernandez B, Eigenbrot C, Elliott R, Hanan EJ, Jackson P, et al. Noncovalent mutant selective epidermal growth factor receptor inhibitors: A lead optimization case study. Journal of Medicinal Chemistry. 2015;58:8877–8895. doi: 10.1021/acs.jmedchem.5b01412. [DOI] [PubMed] [Google Scholar]