

Abstract
SCOPe (Structural Classification of Proteins – extended, http://scop.berkeley.edu) is a database of relationships between protein structures that extends the Structural Classification of Proteins (SCOP) database. SCOP is an expert-curated ordering of domains from the majority of proteins of known structure in a hierarchy according to structural and evolutionary relationships. SCOPe classifies the majority of protein structures released since SCOP development concluded in 2009, using a combination of manual curation and highly precise automated tools, aiming to have the same accuracy as fully hand-curated SCOP releases. SCOPe also incorporates and updates the ASTRAL compendium, which provides several databases and tools to aid in the analysis of the sequences and structures of proteins classified in SCOPe. SCOPe continues high quality manual classification of new superfamilies, a key feature of SCOP. Artifacts such as expression tags are now separated into their own class, in order to distinguish them from the homology-based annotations in the remainder of the SCOPe hierarchy. SCOPe 2.06 contains 77,439 PDB entries, double the 38,221 structures classified in SCOP.
Graphical abstract

Background
Nearly all proteins have structural similarities with other proteins and, in many of these cases, share a common evolutionary origin. The Structural Classification of Proteins (SCOP) database [1–4] is a manually curated hierarchy of domains from proteins of known structure, organized according to their structural and evolutionary relationships. Work on the SCOP version 1 series concluded in 2009 with the release of SCOP 1.75. To continue its development, we created the SCOPe (SCOP–extended) database, which provides ongoing updates and classification of new protein structures [5]. The initial version of SCOPe imported the SCOP 1.75 classification to build upon. We use a combination of manual curation and a rigorously validated software pipeline [5] to add new structures from the Protein Data Bank (PDB) [6,7], and we have also developed software to identify errors in SCOP, which are then corrected in new releases of SCOPe. SCOPe is backward compatible with SCOP, providing the same parseable files and a history of changes between all stable SCOP and SCOPe releases.
The SCOPe hierarchy, inherited from SCOP, classifies domains from experimentally determined protein structures. The hierarchy comprises the following levels: Species, representing a distinct protein sequence and its naturally occurring or artificially created variants; Protein, grouping together similar sequences of essentially the same functions that either originate from different biological species or represent different isoforms within the same species; Family containing proteins with similar sequences but often distinct functions; and Superfamily bridging together protein families with common functional and structural features inferred to be from a common evolutionary ancestor. Near the root, the basis of classification is purely structural: structurally similar superfamilies are grouped into Folds, which are further arranged into Classes based mainly on their secondary structure content and organization.
SCOPe incorporates and updates the ASTRAL compendium [8–10]. ASTRAL is a collection of software and databases used to aid in the analysis of the protein structures classified in SCOP and SCOPe, particularly through the use of their sequences. ASTRAL provides sequences and coordinate files for all SCOPe domains, as well as sequences for all PDB chains that are classified in SCOPe. Chemically modified amino acids are translated back to the original sequence. Because the vast majority of sequences in the PDB are very similar to others, ASTRAL provides representative subsets of proteins that span the set of classified protein structures or domains in order to alleviate bias towards well-studied proteins. The highest quality representative in each subset is chosen using AEROSPACI scores [10], which provide a numeric estimate of the quality and precision of crystallographically-determined structures.
Since our initial publication describing SCOPe [5], we have released three stable versions of the database, and made a number of improvements, described in detail in this manuscript. We initially prioritized the development and use of highly accurate automatic classification methods. Starting with SCOPe 2.04 (July 2014), we also re-introduced manual curation, focusing on the largest unclassified protein families. Thus, while the first three public releases of SCOPe (versions 2.01 – 2.03) did not add manually curated entries at the Family level or above, the three most recent stable releases (versions 2.04 – 2.06) added 28 new folds, 52 new superfamilies, and 79 new families. SCOPe 2.06 (February 2016) added a new class (Artifacts) outside of the main SCOPe hierarchy (i.e., the first seven classes) in order to record cloning artifacts, such as expression tags, that we could identify in the solved structures based on sequence data and metadata annotations. Including such artifacts in the classified domains can result in spurious similarity between non-homologous sequences, so their removal from the main hierarchy also results in more accurate representative ASTRAL subsets. Finally, we implemented a new, mobile-friendly, version of the SCOPe website, and modified the automated classification protocol slightly in order to accurately classify more PDB entries.
An overview of changes to SCOP and SCOPe design since the introduction of stable identifiers (SCOP 1.55, July 2001) is shown in Fig. 1. Statistics on all SCOP and SCOPe releases, summaries and a full history of changes and other information are available from the SCOPe website (https://scop.berkeley.edu/) together with parseable files containing all current and historic SCOPe, SCOP, and ASTRAL data. With the current releases of SCOPe, we aim to best meet the inferred needs of SCOP users [11], focusing on a classification consistent with that developed over the past 22 years, while maintaining outstanding classification accuracy, and being as comprehensive as possible.
Fig. 1. Changes to SCOP(e) design and size.
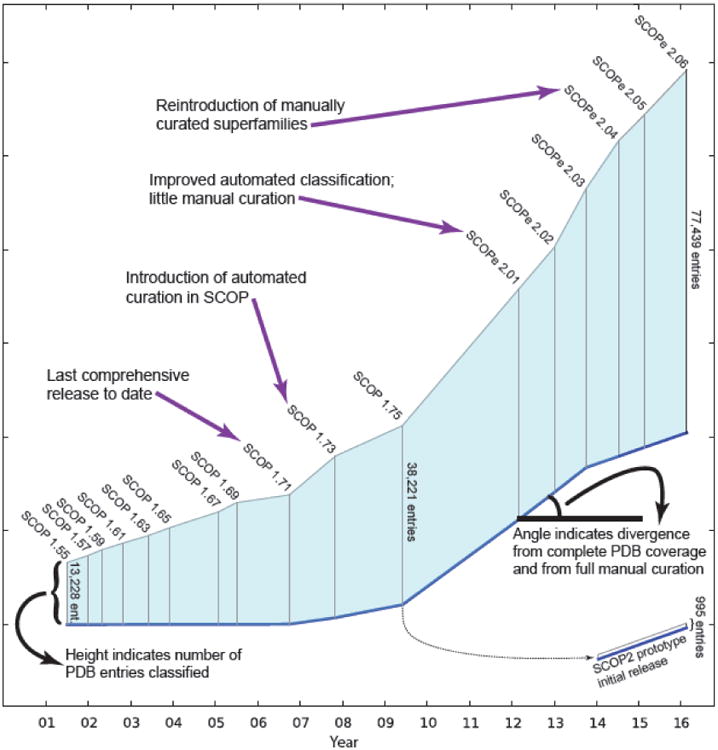
All stable SCOP and SCOPe releases since the introduction of stable identifiers (SCOP 1.55, July 2001) are shown. The height of the vertical line for each release represents the number of PDB entries classified. The angle of the blue baseline between releases reflects the degree of divergence from comprehensive and fully manually curated releases. SCOP2 [31] is a major redesign of SCOP that enables curators to annotate a richer set of evolutionary relationships between proteins, providing a more precise and accurate characterization of protein relationships. SCOP2 is currently available as a prototype that classifies 995 proteins. A dashed line indicates that the SCOP2 prototype is partially based on SCOP 1.75.
Manual Curation
Manual curation of superfamilies is a key feature of SCOPe, in which proteins with similar three-dimensional (3D) structure and no recognizable sequence similarity are divided into homologs and possible analogs at the superfamily level on the basis of the expert biological insight of human curators. Like SCOP, SCOPe is unique among current structural classification databases in that the hierarchy above the Species level is completely defined by expert curators, with automation used only to identify newly structurally characterized members of existing groups. Based on a study of 571 recent articles that cited SCOP [11], we found that our largest category of users are biologists who use SCOP or SCOPe as a “gold standard” for benchmarking computational algorithms, or to create training sets to aid in setting algorithmic parameters. For these users, manual curation consistent with SCOP standards is necessary in order to include newly structurally characterized protein families in the classification without compromising the utility of SCOPe-derived benchmark datasets. To date, computational methods alone have not been able to classify structures with sufficient accuracy: even specialized methods designed to classify new structures into SCOP, such as SCOPmap [12], proCC [13] and SUPERFAMILY [14], report that they fail to classify between 5% and 12% of domains in the correct SCOP superfamily [12,13,15]. Manually curated structures are also used as a basis for further classification by our automated tools, and the resulting increased classification of PDB structures, together with rapid synchronization with PDB releases, benefit all our users [11].
Several other resources also classify a large fraction of protein structures using partial manual curation. CATH [16] and ECOD [17] are similar to SCOPe, but rely more heavily on automated classification tools to assign protein domains and place them in the hierarchy. Classifications in CATH have been compared to SCOP [18–20], with the conclusion that while the majority of assignments are consistent, there are significant inconsistencies caused by deliberate design differences. The ECOD authors also note that their domain partition strategy is different from SCOPe, resulting in alternative domain assignments for some structures common to both databases. We compared ECOD (version develop146, 12 July 2016) to SCOPe (version 2.06-stable), finding that 153,553 protein chains classified in both databases have consistent domain partitioning (which we define as the same number of domains, with no N- or C-terminal domain boundary changed by more than 10 residues), perhaps because early versions of ECOD were partially derived from SCOP [17]. However, 25,606 protein chains have inconsistent partitioning between SCOPe and ECOD, with ECOD defining an average of 2.6 domains for these chains vs. 1.3 domains per chain in SCOPe. One example of a manually curated SCOPe superfamily that is not consistent with ECOD (bulge domains from archaeal A-type ATP Synthase) is discussed below. We expect that that a more thorough comparison of annotations from independently curated databases may be valuable for identifying highly confident annotations (e.g., [20]), and for distinguishing philosophical design differences from errors.
In addition to adding new superfamilies, manual curation can also involve other changes to the SCOPe hierarchy. If two distinct superfamilies are later discovered to be related, for example on the basis of a newly discovered structure of an evolutionary intermediate, our curator would merge the two superfamilies into one.
Manual curation is also used to make changes to domain boundaries, including splitting a single domain into multiple domains. This is because SCOPe defines a domain as an evolutionarily conserved unit (as opposed other common definitions of a domain, e.g., based on structural compactness), so a superfamily composed of large domains may be split into multiple superfamilies of smaller domains if these domains are discovered in other evolutionary contexts. Examples of merging superfamilies and splitting domains are discussed below.
We prioritized manual curation of new structures by focusing on Pfam [21] families with the most structures not classified in SCOP or SCOPe. To identify such families, we used HMMER 3 [22] to identify Pfam (version 28.0) families in all protein chains in the PDB. We considered only matches that scored at or above the trusted cutoff for each Pfam family, for which the alignment comprised at least 75% of the Pfam model. We found 2,433 Pfam families had been structurally characterized but not yet classified in SCOPe. This large backlog is a consequence of the fact that SCOP has not comprehensively classified every protein in the PDB since SCOP 1.71, which classifies all proteins released by the PDB prior to 18 January 2005. Although some new families were manually classified in SCOP versions 1.73 and 1.75, none were classified between the release of SCOP 1.75 in June 2009 and the release of SCOPe 2.04 in June 2014. Recent advances in high resolution cryo-electron microscopy have contributed to this backlog; for example, the structure of the yeast spliceosome [23] represented the first structural characterization of 15 different Pfam families. We prioritized manual classification of the largest unclassified Pfam families for two reasons: first, because having at least one manually classified structure from a Pfam family allows our automated tools to work on many other members of that family, and second, because a large number of solved structures is a crude proxy for scientific impact, and therefore we expect larger families to be of potentially greater interest to SCOPe users.
In producing SCOPe versions 2.04 – 2.06, we curated structures from the 126 largest Pfam families not classified in SCOP, using the same principles previously employed by the SCOP curator to identify domains and classify them in the hierarchy [24]. As we expected, the relationship between Pfam families and SCOPe families (or superfamilies) is not 1:1. Among the classified structures from 103 non-ribosomal protein families, 28 (27%) had at least one domain classified into a new SCOPe fold, 24 (23%) into a new superfamily in an existing fold, 29 (28%) into a new family within an existing superfamily, and 22 (21%) as new proteins within an existing family. These results are similar to the novelty of newly classified structures in SCOP ten to twenty years ago, for structures that did not have significant sequence similarity with previously classified structures [25,26]. Since ∼50% of newly classified Pfam families correspond to a new SCOPe fold or superfamily, we project that over 1,000 new folds and superfamilies are harbored in the more than 2,000 Pfam families that are structurally characterized but still unclassified in SCOPe.
Example of a new superfamily
FoF1-ATPases function as ATP synthases in mitochondoria, chloroplasts, and bacteria, by coupling proton gradients to ATP hydrolysis or synthesis through a rotary catalytic mechanism. The alpha and beta subunits of the water-soluble F1 part of ATP synthase have been classified in SCOP since the earliest version with stable identifiers; each contains three domains. A structure of the V1 subunit of vacuolar-type ATPase, which regulates the acidic environments of cells and compartments in a variety of organisms, was recently solved [27]. Top and side views of V1 and F1 complexes are shown in Fig. 2A. The V1 ATPase A and B subunits are clearly homologous to the alpha and beta subunits of F1, except for the insertion of a “bulge” domain between the first two conserved (with F1) domains in the catalytic A subunits. The bulge domain is structurally similar to other structures in the “Barrel/sandwich hybrid” fold that contain 8 β-strands (the unique SCOPe concise classification string identifier, or sccs, for this fold is b.84; the b indicates this fold is in the all-β class). However, there is no evidence of homology with members of the four other superfamilies in that fold. Therefore, the V1 bulge domain was classified as a new superfamily in SCOPe 2.06. We also classified structures of A1 subunits of archaeal A-type ATP synthase, which have domains homologous to the “bulge” domain, but lack domains homologous to the N-terminal domain of F1 ATPase subunits [28]. We note that ECOD does not classify the “bulge” domains consistently: in A1 structures, these domains are merged into the ATP-binding central domain, while in V1 structures they are split from the central domain.
Fig. 2. Examples of manual curation in SCOPe.
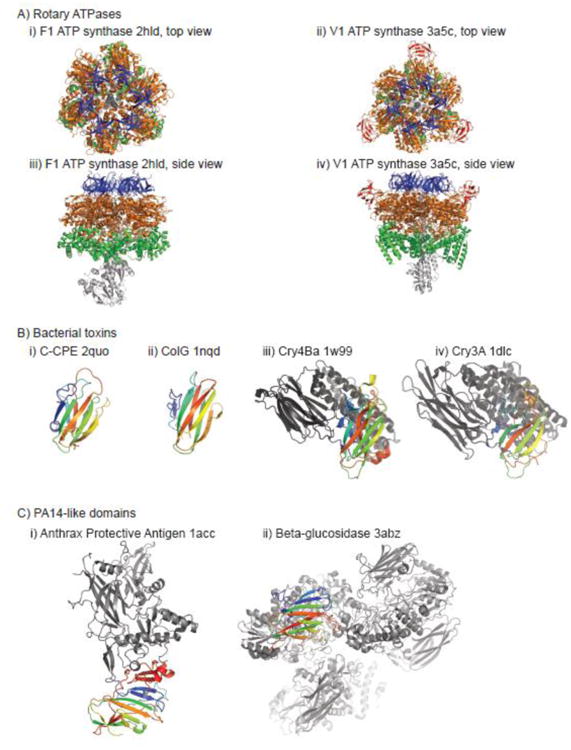
A) Top and side views of F1 and V1 ATP synthase subunits; the “top” view is oriented towards the membrane. Conserved N-terminal, middle, and C-terminal subunits of the alpha and beta subunits of F1 (A and B subunits of V1) are shown in blue, orange, and green, respectively. The “bulge” domain of V1, which represents a new SCOPe superfamily in 2.06, is shown in red. Other subunits of F1 and V1 are shown in light grey. B) Four homologous domains from bacterial toxins are colored in a spectrum ranging from blue at the N-terminal end to red at the C-terminal end. Other domains from the structures are shown in grey. C) Two homologous domains from the PA14 superfamily are colored in a spectrum ranging from blue at the N-terminal end to red at the C-terminal end. Other domains from the structures are shown in grey. PA14 was part of the Anthrax Protective Antigen fold (the entire structure shown on the left) in versions of SCOP and SCOPe prior to SCOPe 2.04.
Example of superfamily merging
Van Itallie and colleagues solved the first structure of the C-terminal domain of Clostridium perfringens enterotoxin (CPE), a common cause of food poisoning [29]. They reported that their CPE structure, a 9-stranded β-sandwich (PDB code 2quo), revealed unexpected structural similarity to several other bacterial toxins: ColG collagenase from Clostridium histolyticum (PDB code 1nqd) and Cry4Ba toxin from Bacillus thuringiensis (PDB code 1w99). Of these structures, only 1nqd had been classified in SCOP, starting with version 1.65, in the “Collagen-binding domain superfamily,” sccs b.23.2 under the “CUB-like” fold, b.23. Although Cry4Ba had not been classified in SCOPe, other toxins from the Cry family are: for example, Cry3A from B. thuringiensis (PDB code 1dlc) has been classified in SCOP since the earliest version with stable identifiers, with its C-terminal β-sandwich domain in the “Galactose-binding domain superfamily,” sccs b.18.1, under the “Galactose-binding domain-like” fold, b.18. All four structures are shown in Fig 2B.
Three pieces of evidence from the Van Itallie study convinced us that the superfamilies b.23.2 and b.18.1 were in fact homologous, despite having originally been classified in separate SCOP folds. First, the authors of the CPE study showed that when the CPE, ColG, and Cry4Ba structures are aligned, analogous positions in the core β-strands have similar sequences (albeit insufficiently significant to be identified without the benefit of structural alignment). Second, all four structures have identical β-sheet topologies, and are more similar to other structures in the b.18 fold (where most structures have 9 β-strands) than to structures in the b.23 fold (where most structures have 10 β-strands, with several of the strands typically being longer, or distorted). Third, the proteins have similar functional roles, as bacterial toxins. We therefore merged the Collagen-binding domain superfamily b.23.2 into the Galactose-binding domain superfamily b.18.1 in SCOPe 2.05, making it into a new family under the existing superfamily. CPE C-terminal domain-like proteins were classified as another new family within the same superfamily.
Example of domain splitting
The Anthrax Protective Antigen (APA, pdb code 1acc) is a multi-domain protein that has been classified in its own fold (f.11, “Anthrax protective antigen”) in SCOP since the earliest version with stable identifiers. Although described in the SCOP curator's comments as having four domains, no homologs of the other domains were ever classified in SCOP, since no structures of these domains in other contexts were available prior to the last release of SCOP. However, the N-terminal domain of APA, called Protective Antigen 14 (PA14), has been observed in a wide variety of bacterial toxins, enzymes, adhesins, and signaling molecules [30]; some of these have recently been structurally characterized. In building SCOPe 2.04, we classified several members of the GLEYA domain family (Pfam PF10528), which are homologous to PA14. We therefore split the APA entries into two parts: the N-terminal domain, and the remaining C-terminal domains. The N-terminal fold contains the PA14 superfamily, which in turn contains PA14 and GLEYA families. Structures of several PA14 representatives are shown in Fig 2C. The remaining C-terminal domains of APA still do not have structurally characterized homologs classified in SCOPe, but would be split in the future should that occur.
Artifact Removal
We moved cloning artifacts (e.g., expression tags) that we could identify to a new class (l: Artifacts) in order to separate them from the homology-based curations in the rest of the SCOPe hierarchy. Including such artifacts can result in spurious similarity between non-homologous protein sequences. We identified 21,876 tags that were experimental observed in protein structures, with lengths ranging from 1 to 28 residues, and separated them from the SCOPe domains to which they had originally been attached. Where possible, we kept the same stable identifiers for the trimmed domains.
N-terminal and C-terminal tags were primarily identified using PDB metadata (SEQADV records) referring to cloning or expression tags at the beginning or end of each chain; a full list of these tags is available on the help page of our website. We annotated additional tags using exact sequence matches to these tagged chains, and to terminal tag sequences at least 5 residues long that were not otherwise annotated in the PDB metadata (DBREF records) as belonging to the reference protein sequence associated with the PDB chain.
We also generated a new set of full-length ASTRAL chain sequences based on PDB SEQRES records, with tags removed, as well as nonredundant subsets of this set. The removal of tags also resulted in changes to all nonredundant sets that were built using fixed E-value or % identity thresholds: in some cases, removal of a tag caused pairwise sequence similarity to fall below the threshold, while in other cases, removal of dissimilar tags caused similarity of the “natural” parts of the proteins to increase. For example, among the sets of PDB chain sequences we created for ASTRAL 2.06 with a 95% sequence identity threshold (see [8] for details), the tagless set contains 25,631 representatives (out of 180,206 total PDB chains in ASTRAL 2.06), while the representative set with tags contains 25,917 representatives. 518 chains from the tag-containing representative set are not present in the tagless set, while 232 other chains were added.
Automated Classification Protocol
Our automated classification algorithm and benchmarking protocol are described in detail in a previous manuscript [5]. We previously found that the error rate for manually classified entries in SCOP was as low as 0.08%, mostly as the result of typos in entering domain boundaries [5]. We have undertaken studies to demonstrate that we can liberalize some parameters of our automated protocol while retaining the same accuracy. We introduced these changes starting with SCOPe 2.04 in order to classify more PDB entries. As with our prior algorithm, we have validated our current automated classification method against all manually curated versions of SCOP, finding no cases in which the superfamily was predicted incorrectly, or any predicted domain boundary differed from the correct boundary by more than 10 residues. While our current pipeline is very accurate, these strict requirements still limit its application to about 50% of newly solved structures. Changes made since our previous publication include:
Removing prohibitions against automatically classifying low-resolution, NMR, and ribosomal structures (low-resolution and ribosomal structures are still limited to being classified in the applicable sections of the SCOPe hierarchy).
Allowing PDB chains with any number of domains to be classified (was previously limited to two domains).
Increasing the number of residues by which we extend BLAST annotations to chain ends or gaps, from 10 to 15 residues.
Removing the requirement that multiple BLAST hits from a query PDB chain being automatically classified must be to different target SCOPe domains.
New Website
In order to improve the usability of the SCOPe website on tablet and mobile phone browsers, we rebuilt the front end using Bootstrap (http://getbootstrap.com). The new site is “responsive,” meaning that the layout and navigation controls automatically adjust based on the browser size, making the site convenient to use on wide range of devices, from desktop computers to tablets and phones. The website also supports SSL (i.e., encrypted connections using the HTTPS protocol). Like our previous website, the new SCOPe website can display data from all versions of SCOPe, SCOP, and ASTRAL since release 1.55. All data are stored in a relational (MySQL) database back end, which is also available for download.
Research Highlights.
SCOPe is a backwards-compatible extension of the SCOP (version 1) database
SCOPe 2.06 includes manually curated structures from 126 Pfam families not in SCOP
SCOPe 2.06 separates cloning artifacts from the rest of the protein structure
SCOPe now contains double the number of structures as SCOP
Acknowledgments
We would like to thank all of the other SCOP authors: Alexey G. Murzin, Antonina Andreeva, Dave Howorth, Loredana Lo Conte, Bartlett G. Ailey, Tim J. P. Hubbard, and Cyrus Chothia.
Funding: This work was supported by the National Institutes of Health (R01-GM073109) through the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 2.Lo Conte L, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res. 2002;30:264–267. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox NK, Brenner SE, Chandonia JM. SCOPe: Structural Classification of Proteins--extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014;42:D304–309. doi: 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose PW, Prlić A, Bi C, Bluhm WF, Christie CH, Dutta S, Green RK, Goodsell DS, Westbrook JD, Woo J, Young J, Zardecki C, Berman HM, Bourne PE, Burley SK. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner SE, Koehl P, Levitt M. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandonia JM, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. ASTRAL compendium enhancements. Nucleic Acids Res. 2002;30:260–263. doi: 10.1093/nar/30.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandonia JM, Hon G, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. The ASTRAL Compendium in 2004. Nucleic Acids Res. 2004;32:D189–192. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox NK, Brenner SE, Chandonia JM. The value of protein structure classification information—Surveying the scientific literature. Proteins Struct Funct Bioinforma. 2015;83:2025–2038. doi: 10.1002/prot.24915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheek S, Qi Y, Krishna SS, Kinch LN, Grishin NV. SCOPmap: Automated assignment of protein structures to evolutionary superfamilies. BMC Bioinformatics. 2004;5:197. doi: 10.1186/1471-2105-5-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YJ, Patel JM. A framework for protein structure classification and identification of novel protein structures. BMC Bioinformatics. 2006;7:456. doi: 10.1186/1471-2105-7-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lima Morais DA, Fang H, Rackham OJL, Wilson D, Pethica R, Chothia C, Gough J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011;39:D427–434. doi: 10.1093/nar/gkq1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox NK, Brenner SE, Chandonia JM. SCOPe: Structural Classification of Proteins--extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014;42:D304–309. doi: 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH – a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1109. doi: 10.1016/S0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 17.Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH, Grishin NV. ECOD: An Evolutionary Classification of Protein Domains. PLoS Comput Biol. 2014;10:e1003926. doi: 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Day R, Beck DAC, Armen RS, Daggett V. A consensus view of fold space: Combining SCOP, CATH, and the Dali Domain Dictionary. Protein Sci Publ Protein Soc. 2003;12:2150–2160. doi: 10.1110/ps.0306803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadley C, Jones DT. A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Struct Lond Engl 1993. 1999;7:1099–1112. doi: 10.1016/s0969-2126(99)80177-4. [DOI] [PubMed] [Google Scholar]
- 20.Lewis TE, Sillitoe I, Andreeva A, Blundell TL, Buchan DW, Chothia C, Cuff A, Dana JM, Filippis I, Gough J. Genome3D: a UK collaborative project to annotate genomic sequences with predicted 3D structures based on SCOP and CATH domains. Nucleic Acids Res. 2013;41:D499–D507. doi: 10.1093/nar/gks1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan C, Hang J, Wan R, Huang M, Wong CCL, Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182–1191. doi: 10.1126/science.aac7629. [DOI] [PubMed] [Google Scholar]
- 24.Brenner SE, Chothia C, Hubbard TJ, Murzin AG. Understanding protein structure: using scop for fold interpretation. Methods Enzymol. 1996;266:635–643. doi: 10.1016/s0076-6879(96)66039-x. [DOI] [PubMed] [Google Scholar]
- 25.Brenner SE, Levitt M. Expectations from structural genomics. Protein Sci Publ Protein Soc. 2000;9:197–200. doi: 10.1110/ps.9.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 27.Maher MJ, Akimoto S, Iwata M, Nagata K, Hori Y, Yoshida M, Yokoyama S, Iwata S, Yokoyama K. Crystal structure of A3B3 complex of V-ATPase from Thermus thermophilus. EMBO J. 2009;28:3771–3779. doi: 10.1038/emboj.2009.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maegawa Y, Morita H, Iyaguchi D, Yao M, Watanabe N, Tanaka I. Structure of the catalytic nucleotide-binding subunit A of A-type ATP synthase from Pyrococcus horikoshii reveals a novel domain related to the peripheral stalk. Acta Crystallogr D Biol Crystallogr. 2006;62:483–488. doi: 10.1107/S0907444906006329. [DOI] [PubMed] [Google Scholar]
- 29.Van Itallie CM, Betts L, Smedley JG, McClane BA, Anderson JM. Structure of the claudin-binding domain of Clostridium perfringens enterotoxin. J Biol Chem. 2008;283:268–274. doi: 10.1074/jbc.M708066200. [DOI] [PubMed] [Google Scholar]
- 30.Rigden DJ, Mello LV, Galperin MY. The PA14 domain, a conserved all-beta domain in bacterial toxins, enzymes, adhesins and signaling molecules. Trends Biochem Sci. 2004;29:335–339. doi: 10.1016/j.tibs.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Andreeva A, Howorth D, Chothia C, Kulesha E, Murzin AG. SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 2014;42:D310–D314. doi: 10.1093/nar/gkt1242. [DOI] [PMC free article] [PubMed] [Google Scholar]