

Abstract
Other than serving as building blocks for DNA and RNA, purines metabolites provide a cell with the necessary energy and cofactors to promote cell survival and proliferation. A renewed interest in how purine metabolism may fuel cancer progression has uncovered a new perspective into how a cell regulates purine need. Under cellular conditions of high purine demand, the de novo purine biosynthetic enzymes cluster near mitochondria and microtubules to form dynamic multi-enzyme complexes referred to as purinosomes. This review highlights the purinosome as a novel level of metabolic organization of enzymes in cells, its consequences for regulation of purine metabolism, and the extent that purine metabolism is being targeted for the treatment of cancers.
Keywords: purine metabolism, metabolon, purinosome
A historical perspective into purine metabolism
Cellular metabolism involves series of highly regulated sequential biochemical reactions aimed at generating the necessary substrates for basic cellular processes. One of the most abundant classes of metabolites within a mammalian cell are purines. In addition to the generation of DNA and RNA molecules, purine nucleotides such as adenosine 5′-triphosphate (ATP) and guanosine 5′-triphosphate (GTP) are crucial for providing cellular energy and intracellular signaling, respectively. Purines can also be incorporated into more complex biomolecules and serve as cofactors such as nicotinamide adenine dinucleotide (NAD) and coenzyme A.
The core of this review is attributable to the extensive studies on the sequential enzyme catalyzed reactions of the de novo purine biosynthetic pathway. This pathway was originally characterized with avian liver enzymes approximately six decades ago by Buchannan and Greenberg [1-3]. As the pathway enzymes were further characterized, a hypothesis emerged that the enzymes within the pathway must compartmentalize with one another to facilitate metabolic flux. Unfortunately, experimental evidence for such a cluster based on enzyme copurification with and without solution crosslinking and partial substrate channeling was not compelling [4, 5]. It was not until the advent of intracellular fluorescent imaging that the presence of a highly transient multi-enzyme complex, the purinosome, was discovered [6]. From there, our understanding of how purines are synthesized to meet cellular demand has broadened. This review highlights recent studies on purine biosynthesis and its regulation by viewing them into the context of a new level of metabolic enzyme organization – the purinosome. Consequently, the initial finding of this metabolic pathway is now subject to revision and expansion as we gain more insights into what drives purine metabolism in mammalian cells and how deregulation of these processes might contribute to human disease.
Purine metabolism
Purine levels in mammalian cells are maintained by a coordinated action of the salvage and de novo biosynthetic pathways (Figure 1). Under normal physiological conditions, most of the cellular purine pool is derived from the recycling of degraded bases via the salvage pathway [7-9]. Additionally, bases present in the extracellular matrix can be transported into the cell to generate the corresponding nucleotide. The salvage process uses hypoxanthine-guanine phosphoribosyltransferase (HPRT) to convert hypoxanthine and guanine to inosine 5′-monophosphate (IMP) and guanosine 5′-monophosphate (GMP), respectively (Figure 1). Adenine can also be combined with phosphoribosyl pyrophosphate (PRPP) to generate adenosine 5′-monophosphate (AMP) in a process catalyzed by adenine phosphoribosyltransferase (APRT). Under cellular conditions requiring higher purine levels, the intracellular purine demand is met by upregulating the de novo biosynthetic pathway [7, 9-11].
Figure 1. Purine metabolic pathways and their crosstalk with other metabolic processes.

The de novo purine biosynthetic pathway in humans consists of 10 highly conserved steps (green) that transforms phosphoribosylpyrophosphate (PRPP), generated through the pentose phosphate pathway (blue), into inosine 5′-monophosphate (IMP). Six enzymes catalyze these ten steps and include PRPP amidotransferase (PPAT, EC 2.4.2.14), trifunctional phosphoribosylglycinamide synthetase (GARS, EC 6.3.4.13)/phosphoribosylglycinamide formyltransferase (GAR Tfase, EC 2.1.2.2)/ phosphoribosylaminoimidazole synthetase (AIRS, EC 6.3.3.1) (GART), phosphoribosyl formylglycinamidine synthase (FGAMS, EC 6.3.5.3), bifunctional phosphoribosyl aminoimidazole carboxylase (CAIRS, EC 4.1.1.21)/phosphoribosyl aminoimidazole succinocarboxamide synthetase (SAICARS, EC 6.3.2.6) (PAICS), adenylosuccinate lyase (ADSL, EC 4.3.2.2), and bifunctional 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase (AICAR Tfase, EC 2.1.2.3)/IMP cyclohydrolase (IMPCH, EC 3.5.4.10) (ATIC). Pathway cofactor, 10-formyltetrahydrofolate (10-fTHF) is a product of one-carbon metabolism (denoted with a *, for biosynthetic pathway see Figure 3). Intermediate SAICAR was shown to allosterically activate pyruvate kinase isoform M2 (PKM2, EC 2.7.1.40) in glycolysis (red) [26-28]. AICAR is also a byproduct of histidine biosynthesis (purple). Downstream purine biosynthesis of IMP requires the use of IMP dehydrogenase (IMPDH, EC 1.1.1.205), GMP synthase (GMPS, EC 6.3.5.2) to make GMP whereas AMP can be generated by reactions catalyzed by adenylosuccinate synthase (ADSS, EC 6.3.4.4) and ADSL. Purine salvage (orange) can also be used to generate IMP and GMP using hypoxanthine-guanine phosphoribosyl transferase (HPRT, EC 2.4.2.8) and AMP from adenine phosphoribosyl transferase (APRT, EC 2.4.2.7).
The de novo purine biosynthetic pathway is a highly conserved, energy intensive pathway that generates IMP from PRPP (Figure 1). In humans, this metabolic transformation is carried out in ten steps by sequential orchestration of six enzymes. The first reaction in the de novo purine biosynthetic pathway is the conversion of PRPP to 5-phosphoribosylamine (PRA) by PRPP amidotransferase (PPAT) and is presumed to be rate-limiting. Transformation of PRA to N-formylglycinamide ribonucleotide (FGAR) via glycine and formyl group addition is catalyzed by the phosphoribosylglycinamide synthetase (GARS) and phosphoribosylglycinamide formyltransferase (GAR Tfase) domains of GART (also referred to as TrifGART). GART carries out non-sequential steps in the de novo purine biosynthetic pathway. The intermediate FGAR is then converted to N-formylglycinamidine ribonucleotide (FGAM) by phosphoribosyl formylglycinamidine synthase (FGAMS or PFAS) in the fourth step of the pathway prior to formation of aminoimidazole ribonucleotide (AIR) by the phosphoribosylaminoimidazole synthetase (AIRS) domain of GART. Bifunctional phosphoribosyl aminoimidazole carboxylase (CAIRS)/phosphoribosyl aminoimidazole succinocarboxamide synthetase (SAICARS) (PAICS) utilizes AIR to generate N-succinocarboxyamide-5-aminoimidazole ribonucleotide (SAICAR) in two concerted steps. SAICAR then can be processed to aminoimidazole-4-carboxamide ribonucleotide (AICAR) in a reversible reaction catalyzed by adenylosuccinate lyase (ADSL). The last two steps in the pathway convert AICAR to IMP using bifunctional 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase (AICAR Tfase)/IMP cyclohydrolase (IMPCH) (ATIC).
The six enzymes within the de novo purine biosynthetic pathway also rely on numerous amino acid substrates and cofactors (Figure 1). For each molecule of IMP generated, five molecules of ATP, two molecules each of glutamine and formate, and one molecule each of glycine, aspartate, and carbon dioxide are needed. Glutamine and aspartate are generated from intermediates of the tricarboxylic acid cycle and are necessary for PPAT, FGAMS, and PAICS (SAICARS) activity. Glycine is a side product of serine-driven one-carbon metabolism in mitochondria and serves as a substrate for conversion of PRA to glycinamide ribonucleotide (GAR) by GART (GARS). Formate, a metabolite exported from mitochondria, is required for the biosynthesis of the 10-formyltetrahydrofolate cofactor, which in turn is needed for the transformylase activity of GART (GAR Tfase) and ATIC (AICAR Tfase) [12]. More recently, the knockdown of mitochondrial enzymes associated with generating one-carbon units was shown to activate production of cytosolic one-carbon units from extracellular serine and glycine sources [13]. However, despite the presence of analogous one-carbon metabolic pathways in both the cytosol and mitochondria, isotope labeling studies have shown that the formate used for the synthesis of cytoplasmic 10-formyltetrahydrofolate is largely mitochondria derived [14, 15].
Studies into the quaternary structures of these enzymes have implied that their oligomeric state may regulate its catalytic activity. Although structural data are still not available for human PPAT and FGAMS, homologous enzymes implicate a tetrameric and monomeric species, respectively. GART exists as a dimer, PAICS as an octamer, and ADSL as a tetramer. ATIC exists in a monomer-dimer equilibrium, where the dimer predominates when its substrate, AICAR and 10-formyltetrahydrofolate cofactor are present. The dimeric form of ATIC shows increased AICAR Tfase activity through sculpting of an active site favoring AICAR binding [16, 17].
We are only starting to gain an understanding of how the de novo purine biosynthetic pathway is being regulated. The presence of two nucleotide binding sites near the active site on PPAT allows for allosteric inhibition by downstream purine nucleotides through a feedback control mechanism [18-21]. Additionally, pathway intermediates may also serve to control other metabolic processes. AICAR, a key signaling intermediate within the pathway, is a byproduct of histidine biosynthesis (Figure 1). When AICAR levels are high, AMP-activated protein kinase (AMPK) is also allosterically activated to regulate the intracellular AMP:ATP ratio [23]. AICAR-mediated AMPK activation inhibits cell proliferation by blocking cell cycle progression via degradation of cdc25, the G2/M phosphatase [24]. Similarly, the activation of AMPK in HeLa ovarian carcinoma cells results in the formation of FGAMS granules, which is consistent with a mechanism to shutdown de novo purine biosynthesis [25]. Last, AICAR is also readily converted to SAICAR in the reverse reaction catalyzed by ADSL. This reaction is the only step within the pathway that has been shown to be reversible, suggesting that there may be a level of regulation yet to be explored. SAICAR was identified as an allosteric regulator of the cancer-specific pyruvate kinase isoform M2 under glucose limited conditions (Figure 1) [26-28]. These observations provide a glimpse into the interconnectivity of metabolic pathways in response to the bioenergetics and biosynthetic requirements of the cell.
Discovery of a metabolon in purine metabolism – the purinosome
A long-standing question in cellular metabolism is how metabolic enzymes in a given network organize within the cytosol, densely packed with myriad proteins and metabolites, to facilitate metabolic flux. One solution is through the formation of a macromolecular complex of enzymes termed a metabolon (Box 1). Enzymes in other metabolic pathways such as the tricarboxylic acid cycle and glycolysis have been found to form metabolons and well-orchestrated action of these components holds the key for efficient metabolite synthesis [29, 30]. The resulting microenvironment sequesters reactive intermediates to enhance their stability and avoid interferences by other cellular constituents. Given the number of enzymatic activities and the chemical instabilities of several pathway intermediates, a metabolon consisting of the pathway enzymes in de novo purine biosynthesis had been hypothesized for more than two decades. The rationale for compartmentalization has largely relied on kinetic arguments. Specifically, the product of the first reaction in the de novo purine biosynthetic pathway, PRA, has a very short solution half-life and may directly transfer to the subsequent enzyme, GART, through complexation of the two enzymes [4, 31, 32]. Similarly, GART catalyzes non-concerted steps within the same pathway (Figure 1) and requires the activity of FGAMS for the fourth step raising the possibility that FGAMS interacts with GART [33].
Box 1. Metabolons.
Paul A. Srere was the first to define a metabolon as a “supramolecular complex of sequential metabolic enzymes and cellular structural elements” [79]. His pivotal work focused on the enzymes within the tricarboxylic acid cycle and demonstrated five sequential enzymes including malate dehydrogenase, citrate synthase, and aconitase compartmentalized to the inner mitochondrial membrane for efficient conversion of fumarate to α-ketoglutarate [29, 57]. Since then, metabolons within other metabolic pathways have been observed. Representative examples of other reported metabolons include: 1) in glycolysis -- a binary complex of glycerol-3-phosphate dehydrogenase and fructose-bisphosphate aldolase [80-84] and a multienzyme complex of glyceraldehyde-3-phosphate dehydrogenase, phosphofructokinase, pyruvate kinase, and lactate dehydrogenase assembled on the human red blood cell membrane [30, 85]; 2) in pyrimidine biosynthesis -- the first three enzymes of the pathway (carbamoyl phosphate synthetase-2, aspartate transcarboamoylase, and dihydroorotase) form the CAD complex as reviewed by Evans and Guy [86]. CAD activity is stimulated through the phosphorylation of CAD Ser1859 by S6 kinase and shown to be sensitive to rapamycin treatment [87].
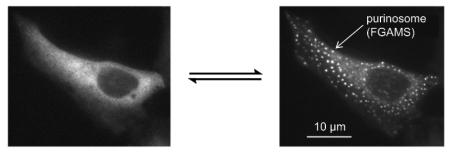
The first evidence that GART and FGAMS might interact within cells was made by confocal microscopy of HeLa cells using chimeric constructs of these enzymes under cellular conditions that stimulated de novo purine biosynthesis [6]. In these studies, co-clustering of the enzymes FGAMS and GART was observed in the cytoplasm. Probing with the remaining four pathway enzymes also showed co-clustering with FGAMS [6]. Immunofluorescence has demonstrated purinosome formation on the endogenous level and provided evidence that the observed compartmentalization is not a consequence of overexpression due to transient transfection [6, 34-36]. Additionally, particle characterization in transient transfected models demonstrated that these enzyme clusters are distinct in size and cell density from processing bodies (P-bodies), stress granules, and aggresomes (Box 2) [37]. Using G3BP as a stress granule marker and gp250 and gp170* as aggresome markers, the purinosome enzyme cluster did not colocalize [38]. Finally, Western blot analysis found the levels of all six enzymes remained unchanged as the composition of the culturing media was switched [37]. Together, these conclusions further define the purinosome as a unique cellular body and not an artifact of transient transfection. Unlike more traditional static metabolons, purinosome formation appears to be a reversible process as suggested by the finding that purinosome formation in HeLa cells can be controlled by altering the composition of the incubation media (Table 1). With purine-depleted media, purinosomes assemble and upon purine supplementation, disassemble [6]. Hence, we may infer that purinosomes are formed as a response to depleted cellular purine levels and higher metabolic demands.
Box 2. Comparison of physical parameters between cytoplasmic cellular bodies.
The identification of metabolons in living cells has greatly benefitted from advancements in fluorescence microscopy. However, caution must be given when characterizing metabolon formation in transient transfected models. Metabolons are distinct cellular bodies from the other commonly observed aggresome, processing body (P-body), and stress granule foci. Aggresomes are formed when the capacity of the proteasome to degrade proteins has been exceeded whereas P-bodies and stress granules are a result of mRNA compartmentalization either for degradation or storage. A listing of the physical parameters associated with specific cellular bodies observed in the cytoplasm and nucleus has been compiled by David Spector, and these criteria used to distinguish the purinosome as a unique subcellular assembly as shown in Table I [88].
Table 1.
Experimental Conditions Affecting Purinosome Formation.
| |
|
conditions that result in purinosome
disassembly |
conditions that result in purinosome
assembly |
| purine supplementation [6] | purine depletion [6] |
| Hsp90 inhibitors (17-AAG, NVP-AUY922) [38] |
CK2 inhibitors (DMAT, TBI) [48] |
| microtubule polymerization inhibitor (nocodazole) [60] |
GPCR agonist (oxymetazoline) [51] |
| ADSL and ATIC mutations [35] | HPRT deficiency [55] |
| electron transport by respiratory chain inhibitor (antimycin A) [61] |
|
| oxidative phosphorylation inhibitor (oligomycin) [61] |
|
Purinosome composition
Characterization of the proximity and the determination of the diffusion coefficient of individual proteins within the purinosome revealed that the purinosome is comprised of a core scaffolding protein assembly and peripheral proteins (with weaker inter-protein interactions) (Figure 2) [39]. The core scaffolding proteins include the first three enzymes in the pathway: PPAT, GART, and FGAMS. The peripheral proteins are PAICS, ADSL, and ATIC. Additional proteomic studies have verified interactions between purinosome enzymes as shown in Figure 2C [33, 40-42]. Recently, mutations in ADSL and ATIC observed in skin fibroblasts from patients with AICAR-ribosiduria and ADSL deficiency showed decrease purinosome formation suggesting that activity of these periphery purinosome enzymes impact complex stability (Table 1) [35]. Likewise, HeLa cell lines deficient in specific pathway enzymes resulted either in a complete loss of purinosomes (GART, ADSL, ATIC) or a significant reduction (FGAMS, PAICS) compared to normal HeLa cells [34]. Measurement of the diffusion coefficients of these enzymes in Hs578T breast carcinoma cells using fluorescence recovery after photobleaching substantiated the spatial model of core and peripheral enzymes that assemble stepwise (Figure 2B) [43]. Additionally, adenylsuccinate synthase (ADSS) and inosine monophosphate dehydrogenase (IMPDH) were identified as members of the purinosome [36].
Figure 2. Purinosome assembly in cells is proposed as a stepwise process.

While the exact triggers for purinosome formation are not known, diffusion and protein-protein interaction studies have provided insight into how purinosomes may assemble in cells under purine-depleted growth conditions. (B) The first three enzymes in the de novo purine biosynthetic pathway (PPAT, GART, and FGAMS) form the core of the purinosome and assemble first before secondary complexes of [PAICS·ADSL] and [ATIC] interact with the core. (C) Several proteomic studies have uncovered interactions between the enzymes in the de novo purine biosynthetic pathway [33, 39-43]. Captured here is the oligomeric state of the active form of each enzyme in the de novo purine biosynthetic pathway and those protein-protein interactions reported between them where the interaction strength (edge thickness) corresponds to the number of studies referencing the interaction. Downstream enzymes of IMP, IMPDH and ADSS, were also shown to be part of the purinosome; however, no protein-protein interactions between these enzymes and the de novo purine biosynthetic enzymes have been reported [36]. Likewise, Hsp90 and CK2 have been demonstrated to influence purinosome formation in cells by unknown mechanisms [38, 48].
The transient nature of the purinosome suggests that other auxillary proteins may be involved in regulating enzyme recruitment and activity. Initial identification of such auxiliaries have revealed that the purinosome is a cellular macromolecular complex of unusual dimensions not limited to the pathway enzymes or membrane bound. Immunoprecipitation of FGAMS under purinosome forming conditions resulted in a list of potential direct interactions with ADSL, PAICS, and numerous components of the Hsp70/Hsp90 chaperone machinery [38]. Colocalization of FGAMS with Hsp90 and Hsp70 implies that molecular chaperones might be involved in purinosome formation. Inhibition of Hsp90 with NVP-AUY922 or 17-AAG caused purinosome disassembly on the same time scale as purinosome disassembly triggered by purine supplementation (Table 1) [38]. Moreover, removal of Hsp90 inhibitors from purine-depleted growth medium resulted in the resurgence of purinosomes [38].
Insights into purinosome assembly
For proliferating cells, purine metabolism must be upregulated during G1- and S-phases of the cell cycle to meet nucleotide demand. Consequently, as HCT116 human colon carcinoma cells progress from mid-G1-phase to S-phase, a 5-fold increase in de novo purine biosynthesis was observed [44]. In addition, a 3-fold increase in the intracellular PRPP concentration accompanied this same cell cycle stage transition suggesting that the increased flux through the pathway is due in part to the increased concentrations of the pathway’s input substrate. Using time-lapse fluorescence microscopy, the percentage of purinosome-positive HeLa cells was found to be highest in G1-phase with a 3.8-fold increase in the purinosome containing cells arrested in the G1-phase by dibutyryl-cAMP [37]. As HeLa cells then progress through S-phase onto G2/M-phases, dramatic decreases in the purinosome content among the cell population were observed.
Various signaling pathways have been attributed to purinosome formation. An in vitro survey of potential proteome-wide casein kinase II (CK2) phosphorylation sites revealed that core scaffolding proteins PPAT, GART, and FGAMS might serve as substrates for CK2, and this phosphorylation event might alter purinosome formation [45]. CK2 is a pleiotropic serine/threonine kinase that has been implicated in a wide variety of cellular processes, complicating our understanding of how CK2 exactly affects de novo purine biosynthesis [46, 47]. Insights into the contribution of CK2 on de novo purine biosynthesis and purinosome formation have relied on the use of three classic CK2 inhibitors: DMAT (2-dimethylamino- 4,5,6,7-tetrabromo-1H-benzimidazole), TBB (4,5,6,7-tetrabromo-1H-benzotriazole), and TBI (4,5,6,7-tetrabromo-1H-benzimidazole). DMAT and TBI showed increased formation of purinosomes in HeLa cells grown under normal growth conditions whereas TBB resulted in a biphasic response to purinosome formation (Table 1) [48]. As the concentration of TBB increased, purinosome dissociation was observed suggesting that TBB is acting on purinosome formation differently than the rest of the CK2 inhibitors. It is worth noting that TBB has been shown to be more selective for CK2 compared to DMAT and its parent compound, TBI, raising the question of whether off target (or indirect) effects of CK2 inhibitors are present and have a greater effect on purinosome formation than CK2 inhibition alone [49, 50].
The effect of these CK2 inhibitors in modulating purinosome formation was validated using a label-free mass redistribution assay [51]. The label-free mass redistribution assay uses a resonant waveguide grating biosensor to monitor real-time changes in the local refractive index due to changes in biomass of living cells near the surface of the sensor. The mass redistribution assay further demonstrated that G-protein coupled receptor (GPCR) signaling could trigger purinosome formation in HeLa cells [54]. Using a library of GPCR agonists, activation of Gαi-coupled receptors correlated with purinosome formation, suggesting that purinosome formation is a downstream event of GPCR signaling [51].
Purinosome function
The classic rationale for metabolon formation is to increase metabolic flux through enzyme proximity, pathway intermediate sequestration, and enzyme activation. For a transient complex, the flux through the pathway must be adaptable to cellular needs. Under cellular conditions that promote purinosome formation, a 50% enhancement of metabolic flux was observed leading to a 3-fold increase in IMP formation compared to normal growth conditions [36]. Since the cells in these studies were asynchronous, we anticipate significant increases in the pathway flux during the G1 phase of the cell cycle, where the number of purinosomes in a cell is highest [37]. Moreover, fibroblasts with loss-of-function mutations in HPRT1, impeding IMP production by salvage and solely relying on the de novo pathway, showed a 25% increase in purinosome formation (Table 1) [55].
What can spatial organization tell us about the function of the purinosome?
Similar to the tricarboxylic acid metabolon, we anticipated that purine biosynthetic enzyme activity in the purinosome could be enhanced by concentrating the proteins within a smaller volume to better sense metabolite levels and coordinate their cellular movements [56-59]. These precedents led us to investigate the locus of the purinosome within a cell. Spatial control of purinosome assembly in HeLa cells was found to be microtubule assisted, shown by fluorescent live cell imaging [60]. Nocodazole disruption of the microtubule network decreased the purinosome-microtubule colocalization and was accompanied by a marked decrease in the metabolic flux through the de novo purine biosynthetic pathway under purinosome forming conditions (Table 1) [60].
In addition to the colocalization with microtubules, purinosomes were found to colocalize with mitochondria through application of super-resolution fluorescence microscopy [61]. Figure 3A-B shows the colocalization of purinosomes with mitochondria. A 2-fold increase in the number of purinosome containing cells was observed after disruption of mitochondrial processes such as electron transport by the respiratory chain and oxidative phosphorylation by antimycin A and oligomycin, respectively (Table 1) [61]. Furthermore, increased intracellular malate production was observed under conditions known to initiate purinosome formation, suggesting that there must be a functional link between mitochondria and purinosomes [61].
Figure 3. mTOR is involved in purinosome localization near mitochondria likely through ATF-directed MTHFD2 expression.

(A) Using FGAMS-mEos2 as a purinosome marker, the colocalization of purinosomes (red) with mitochondria (green) was determined in HeLa cells under purine-depleted conditions using Stochastic Optical Reconstruction Microscopy (STORM). Scale bar = 5 μM. (B) Those purinosomes deemed to be colocalized with mitochondria shown in magenta. Details of how colocalization was determined can be found in French, J.B. et al. [61]. Images courtesy of Sara A. Jones and Xiaowei Zhuang. (C) Our working hypothesis on how mTOR likely controls purinosome-mitochondria colocalization. mTOR regulates expression of MTHFD2, which encodes the mitochondrial dehydrogenase (MTHFD2) responsible for formate release into the cytoplasm [64]. Formate is readily converted into 10-formyltetrahydrofolate (10-fTHF) by the enzyme MTHFD1 where it is used as a cofactor for GAR and AICAR Tfase reactions within the purinosome. Inhibition of mTOR with rapamycin, causing a decrease in purinosome-mitochondria colocalization, results in a decrease in MTHFD2 expression and formate release suggesting that one reason for the observed colocalization is to compartmentalize the enzymes near areas of high formate concentration [89].
The molecular details of the mitochondria-purinosome interaction remain to be explored; however, several hypotheses can be generated for why this colocalization is important for efficient production of purine nucleotides (Figure 3C). First, the de novo purine biosynthetic pathway is energy intensive. For every molecule of IMP generated from PRPP, five molecules of ATP are consumed. Localization of these enzymes near mitochondria would position these enzymes in areas of high ATP concentrations and promote forward flux through the pathway. Second, the utilization of ATP would likewise generate pools of high ADP concentrations. Mitochondria contains carrier proteins that facilitate the rapid exchange of ADP for ATP. Third, in addition to transport proteins shuttling ADP and ATP across the mitochondrial membrane, amino acid substrates and formate, the precursor to 10-formyltetrahydrofolate needed for the transformylase enzymes, GART and ATIC, are exported from mitochondria into the cytosol (Figure 3C).
The strong link between the mitochondria and the level of the 10-formyltetrahydrofolate cofactor is further revealed by examination of mitochondrial dysfunction. Genetic defects in mitochondria DNA replication (mitochondrial myopathy or spinocerebellar ataxia) show tissue-specific induction of the mitochondrial and cytosolic folate cycle [62]. Likewise, depletion of mitochondria DNA to induce respiratory chain deficiency impaired the production of formate needed for folate cofactor biosynthesis [63]. Therefore, the positioning of purinosomes near mitochondria provide a significant advantage for reaching optimal enzymatic activity.
How does mTOR regulate purinosome-mitochondria colocalization?
Our initial studies into understanding the link between purinosomes and mitochondria led us to perform a short hairpin RNA (shRNA) loss-of-function kinome high-throughput screen [61]. In this screen, HeLa cells grown under purine-depleted conditions were treated with shRNA resulting in the silencing of one of the 673 target kinase genes targeted. Changes in the biomass due to purinosome assembly or disassembly were monitored using a label-free mass redistribution assay to identify those kinases that might be involved in regulating purinosome formation. Among the kinases identified in this series was the mechanistic target of rapamycin (mTOR), whose activity modulates mitochondrial physiology and associated endoplasmic reticulum membranes [61]. Increasing doses of the mTOR inhibitor, rapamycin, did not show a decrease in the number of purinosome positive cells; however, it decreased the fractional colocalization between purinosomes and mitochondria [61].
The projected participation of mTOR in purinosome localization was strengthened by the discovery that the expression of a key mitochondrial enzyme, methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is associated with mTORC1 signaling both in normal and cancer cells (Figure 3C) [64]. A link between MTHFD2 expression and purine synthesis was shown through the mTORC1-mediated activation of transcription factor 4 (ATF4) that in turn increased the translation of MTHFD2 [64]. The net result in the study suggests that the degree of formate release from mitochondria into the cytosol is governed by MTHFD2 expression [64]. Once in the cytosol, the formate is eventually incorporated into tetrahydrofolate by MTHFD1 to produce the 10-formyltetrahydrofolate cofactor needed for GART and ATIC Tfase activity and subsequent IMP production (Figure 3C). Decreased transcription of MTHFD2 upon depletion of mitochondrial DNA also resulted in less serine being processed by one-carbon metabolism [63]. Moreover, mTOR has been shown to promote the expression of genes associated with the pentose phosphate pathway that leads to the biosynthesis of PRPP, the substrate for the first reaction in the de novo purine biosynthetic pathway (Figure 1) [65]. Combined these studies are assigning mTOR an important regulatory role in the de novo purine biosynthetic pathway. Additional studies are warranted to understand the exact manner in which purinosome-mitochondria localization are directed by mTOR.
Leveraging purine metabolism as a therapeutic strategy
A characteristic feature in numerous diseases, including cancer, is the reprogramming of metabolic processes, often resulting from genetic mutations [66]. A recent framework explains the role of altered cellular metabolism in tumorigenesis and outlines the bioenergetics and biomolecular needs of cancer cells [67]. These characteristics include an increase in the demand for nitrogen sources due to the deregulation of nutrient uptake, use of glycolytic and citric acid cycle intermediates for NAPDH production, and changes in metabolite-driven gene regulation. The ability to impede metabolic reprogramming has renewed the scrutiny of purine anti-metabolites for treatment of cancers such as acute leukemias [68, 69].
Therapeutics that target purine metabolism have long been used in the clinic. The first to target purine metabolism was 6-mercaptopurine, a purine anti-metabolite, for the treatment of acute lymphocytic leukemia and is still widely used [70]. Since then, the number of purine anti-metabolites steadily increased with several granted FDA approval [71]. Purine anti-metabolites (thiopurine and deoxypurine analogs) often are prodrugs and upon intracellular conversion to the active drug molecule, stall DNA replication through inhibition of replicative enzymes or integrate into DNA/RNA causing DNA damage, gene silencing, and/or the induction of apoptosis [71]. The use of anti-folates (methotrexate, premetrexed) have also been powerful therapeutics in the clinic by inhibiting the production of 10-formyltetrahydrofolate as well as dihydrofolate reductase [72]. Likewise, anti-folates such as lometrexol have been developed to target GAR and AICAR Tfase activity and inhibiting de novo purine biosynthesis [73]. However, many of these therapeutics have undesired toxicities, so there is a need to identify new levels of regulation within purine metabolism as novel ways to inhibit tumorigenesis.
One strategy may arise from disrupting the purinosome itself. Combining Hsp90 inhibitors, shown to inhibit purinosome formation, with an anti-folate such as methotrexate, increased the efficacy of methotrexate in HeLa cells [38]. A second approach may emerge by targeting protein-protein interactions of a single enzyme within the pathway. Elevated expression levels of PAICS and ATIC in numerous cancers is suggestive of the importance of these enzymes and the role of purinosomes in tumorigenesis [74]. Recently, a peptide-based inhibitor of ATIC was shown to disrupt homodimerization, and impair its activity, resulted in a 40% decrease in MCF-7 breast cancer cells proliferation [75]. Consistent with the inhibitor’s proposed mechanism of action, these cells showed a 3-fold increase in its substrate, AICAR, 24 hours post-treatment and activated AMPKα through a dose-dependent increase in phosphorylation of AMPKα residue Thr172 [23].
Our current hypothesis suggests that de novo purine biosynthesis is likely most efficient when purinosomes are located near mitochondria. One can imagine that the development of a molecule disrupting the purinosome juxtaposition to the mitochondria could lead to the development of a novel therapeutic. A large scale study of mRNA profiling 1,454 metabolic enzymes in 1,981 tumors representing 19 different cancer types identified MTHFD2 mRNA and protein overexpression in many cancers, with increasing expression correlating with poor prognosis in breast cancers [74]. The findings of this study are consistent with the importance of one-carbon metabolism in the biochemistry studies mentioned and drives home the importance that mitochondrial reprogramming may have in tumorigenesis [14, 76, 77].
Concluding perspectives
Despite its importance in cellular proliferation, the biochemical mechanisms that regulate purine metabolism are still not well understood. The purinosome presents itself as a new level of metabolic enzyme organization and regulation in purine biosynthesis and is largely contributed to the advancements in in-cell enzymology by fluorescence microscopy [78]. The emergence of super-resolution imaging techniques such as Stochastic Optical Reconstruction Microscopy (STORM) in characterizing the subcellular localization of de novo purine biosynthetic enzymes has renewed the importance mitochondria play in the control of purine metabolism [61]. Such an association would enable the exchange of cofactors, substrates, and products facilitating purine production. Therefore, the ability to leverage advancements in fluorescence imaging to link current regulatory mechanisms of purine metabolism with the spatio-temporal properties of the purinosome is critical to further understand the importance of such a complex in meeting a cell’s purine demand.
The discovery and characterization of the purinosome opens up the general question as to whether transient metabolon formation is a commonly employed regulatory strategy for metabolic pathways as a whole (see Outstanding Questions). Cellular advantages for forming metabolons within cells include the increase in metabolite production efficiency and the sequestration of substrates to avoid toxicity or unwanted activation of regulatory nucleases. The purinosome through localization of the pathway enzymes has the potential to achieve flux values rivaling those enzymes that channel pathway intermediates.
These protein assemblies might represent a novel means to pharmacologically control cellular metabolism and could emerge as an important new class of drug targets. Recent studies have shown that deficiency in enzymatic activity of metabolic enzymes impact metabolon formation [34, 35]. These data argue that enzymatic activity of the individual components are important for complex stability; however, enzymatic activity is not likely the only criterion for complex formation. So the question remains as to the best means to target enzyme compartmentalization. As new tools and probes are developed to study the functions, interactions, and dynamics of transient metabolons, new directions into targeting cellular metabolism are likely to emerge.
Table I.
Comparison of Purinosomes with Other Reported Cytosolic Cellular Bodies.
| cellular body | size of body (μm) | number of bodies per cell |
cellular marker used to compare cellular body to purinosome |
|---|---|---|---|
| purinosome | 0.2 – 0.9 (average: 0.56 ± 0.16) [37] |
50 – 1000 (median: 278) [37] |
FGAMS-GFP [37] |
| processing body (P- body) |
0.1 – 1.0 | 0 – 30 | - |
| stress granule | 0.4 – 5.0 | 5 – 30 | G3BP [38] |
| aggresome | 2 – 10 | 1 | gp250, gp170* [38] |
Outstanding Questions.
To what extent is subcellular localization important for purine biosynthesis? Do enzymes sense localized areas of high metabolite concentration for efficient energy production? How do these enzymes and enzyme clusters traffic to these hot spots within a cell?
Besides enzyme proximity, what other biochemical features promote enzyme activity and pathway efficiency (post-translational modifications, allosteric regulation, etc.)?
How exactly does a cell sense changes in intracellular purine levels? What signaling events occur that result in the switch between salvage and de novo biosynthetic pathways?
Trends Box.
The discovery of the purinosome presents itself as a new form of enzyme organization within de novo purine biosynthesis.
Mechanisms surrounding purinosome assembly and disassembly have provided insights into how purine metabolism is regulated within cells.
The classification of the purinosome as a metabolon brings to light whether enzyme clustering is a commonly employed regulatory mechanism used by cells to meet their bioenergetics and biomolecular demands.
Elucidating the mechanisms in how purine metabolism is deregulated in diseases, such as cancer, has brought forth the potential to develop new therapeutic strategies.
Acknowledgments
The authors thank Vidhi Pareek for careful reading of the manuscript, and Sara A. Jones and Xiaowei Zhuang for permission to use the STORM images presented in Figure 3. Financial support is provided by The National Institutes of Health (NIH GM024129, SJB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buchanan JM, Hartman SC. Enzymic Reactions in the Synthesis of the Purines. In: Ford FF, editor. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley & Sons, Inc; 1959. pp. 199–261. [Google Scholar]
- 2.Greenberg GR, Jaenicke L. On the Activation of the One-Carbon Unit for the Biosynthesis of Purine Nucleotides. In: Wolstenholme GEW, O'Connor CM, editors. Ciba Foundation Symposium - Chemistry and Biology of Purines. John Wiley & Sons, Ltd; 1957. pp. 204–232. [Google Scholar]
- 3.Hartman SC, Buchanan JM. Nucleic acids, purines, pyrimidines (nucleotide synthesis) Annual review of biochemistry. 1959;28:365–410. doi: 10.1146/annurev.bi.28.070159.002053. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph J, Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995;34:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- 5.Smith GK, et al. Characterization of the enzyme complex involving the folate-requiring enzymes of de novo purine biosynthesis. Biochemistry. 1980;19:4313–4321. doi: 10.1021/bi00559a026. [DOI] [PubMed] [Google Scholar]
- 6.An S, et al. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 7.Henderson JF, Khoo KY. On the Mechanism of Feedback Inhibition of Purine Biosynthesis De Novo in Ehrlich Ascites Tumor Cells in Vitro. The Journal of biological chemistry. 1965;240:3104–3109. [PubMed] [Google Scholar]
- 8.Murray AW. The biological significance of purine salvage. Annual review of biochemistry. 1971;40:811–826. doi: 10.1146/annurev.bi.40.070171.004115. [DOI] [PubMed] [Google Scholar]
- 9.Yamaoka T, et al. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. The Journal of biological chemistry. 1997;272:17719–17725. doi: 10.1074/jbc.272.28.17719. [DOI] [PubMed] [Google Scholar]
- 10.Mayer D, et al. Expression of key enzymes of purine and pyrimidine metabolism in a hepatocyte-derived cell line at different phases of the growth cycle. Journal of cancer research and clinical oncology. 1990;116:251–258. doi: 10.1007/BF01612899. [DOI] [PubMed] [Google Scholar]
- 11.Natsumeda Y, et al. Enzymic capacities of purine de Novo and salvage pathways for nucleotide synthesis in normal and neoplastic tissues. Cancer research. 1984;44:2475–2479. [PubMed] [Google Scholar]
- 12.Fan J, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ducker GS, et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell metabolism. 2016 doi: 10.1016/j.cmet.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer & metabolism. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis CA, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Molecular cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greasley SE, et al. Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nature structural biology. 2001;8:402–406. doi: 10.1038/87555. [DOI] [PubMed] [Google Scholar]
- 17.Vergis JM, et al. Human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine 5'-monophosphate cyclohydrolase. A bifunctional protein requiring dimerization for transformylase activity but not for cyclohydrolase activity. The Journal of biological chemistry. 2001;276:7727–7733. doi: 10.1074/jbc.M009940200. [DOI] [PubMed] [Google Scholar]
- 18.Holmes EW, et al. Human glutamine phosphoribosylpyrophosphate amidotransferase. Kinetic and regulatory properties. The Journal of biological chemistry. 1973;248:144–150. [PubMed] [Google Scholar]
- 19.Smith JL. Glutamine PRPP amidotransferase: snapshots of an enzyme in action. Current opinion in structural biology. 1998;8:686–694. doi: 10.1016/s0959-440x(98)80087-0. [DOI] [PubMed] [Google Scholar]
- 20.Yamaoka T, et al. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. The Journal of biological chemistry. 2001;276:21285–21291. doi: 10.1074/jbc.M011103200. [DOI] [PubMed] [Google Scholar]
- 21.Zhou G, et al. Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. The Journal of biological chemistry. 1994;269:6784–6789. [PubMed] [Google Scholar]
- 22.Kim PB, et al. An ancient riboswitch class in bacteria regulates purine biosynthesis and one-carbon metabolism. Molecular cell. 2015;57:317–328. doi: 10.1016/j.molcel.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asby DJ, et al. AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chemistry & biology. 2015;22:838–848. doi: 10.1016/j.chembiol.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E435–444. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt DL, et al. Sequestration-Mediated Downregulation of de Novo Purine Biosynthesis by AMPK. ACS chemical biology. 2016 doi: 10.1021/acschembio.6b00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller KE, et al. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Molecular cell. 2014;53:700–709. doi: 10.1016/j.molcel.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keller KE, et al. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012;338:1069–1072. doi: 10.1126/science.1224409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan M, et al. Succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5'-Phosphate (SAICAR) Activates Pyruvate Kinase Isoform M2 (PKM2) in Its Dimeric Form. Biochemistry. 2016;55:4731–4736. doi: 10.1021/acs.biochem.6b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes SJ, Weitzman PD. Organization of citric acid cycle enzymes into a multienzyme cluster. FEBS letters. 1986;201:267–270. doi: 10.1016/0014-5793(86)80621-4. [DOI] [PubMed] [Google Scholar]
- 30.Campanella ME, et al. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schendel FJ, et al. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 32.Antle VD, et al. Substrate specificity of glycinamide ribonucleotide synthetase from chicken liver. The Journal of biological chemistry. 1996;271:8192–8195. doi: 10.1074/jbc.271.14.8192. [DOI] [PubMed] [Google Scholar]
- 33.Zhang QC, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baresova V, et al. CRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation. Molecular genetics and metabolism. 2016 doi: 10.1016/j.ymgme.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Baresova V, et al. Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Human molecular genetics. 2012;21:1534–1543. doi: 10.1093/hmg/ddr591. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, et al. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. The Journal of biological chemistry. 2015;290:6705–6713. doi: 10.1074/jbc.M114.628701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan CY, et al. Purinosome formation as a function of the cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1368–1373. doi: 10.1073/pnas.1423009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.French JB, et al. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2528–2533. doi: 10.1073/pnas.1300173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng Y, et al. Mapping protein-protein proximity in the purinosome. The Journal of biological chemistry. 2012;287:36201–36207. doi: 10.1074/jbc.M112.407056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Havugimana PC, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kristensen AR, et al. A high-throughput approach for measuring temporal changes in the interactome. Nature methods. 2012;9:907–909. doi: 10.1038/nmeth.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan C, et al. Panorama of ancient metazoan macromolecular complexes. Nature. 2015;525:339–344. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyoung M, et al. Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry. 2015;54:870–880. doi: 10.1021/bi501480d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fridman A, et al. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. The Biochemical journal. 2013;454:91–99. doi: 10.1042/BJ20130153. [DOI] [PubMed] [Google Scholar]
- 45.Allen JJ. Chemistry and Chemical Biology. University of California; San Francisco: 2008. Development and Application of Technologies to Study Individual Kinase Substrate Relationships. [Google Scholar]
- 46.Salvi M, et al. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochimica et biophysica acta. 2009;1793:847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 47.Venerando A, et al. Casein kinase: the triple meaning of a misnomer. The Biochemical journal. 2014;460:141–156. doi: 10.1042/BJ20140178. [DOI] [PubMed] [Google Scholar]
- 48.An S, et al. Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. The Journal of biological chemistry. 2010;285:11093–11099. doi: 10.1074/jbc.M110.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duncan JS, et al. An unbiased evaluation of CK2 inhibitors by chemoproteomics: characterization of inhibitor effects on CK2 and identification of novel inhibitor targets. Molecular & cellular proteomics : MCP. 2008;7:1077–1088. doi: 10.1074/mcp.M700559-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Pagano MA, et al. The selectivity of inhibitors of protein kinase CK2: an update. The Biochemical journal. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 51.Verrier F, et al. GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nature chemical biology. 2011;7:909–915. doi: 10.1038/nchembio.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang Y. Label-free drug discovery. Frontiers in pharmacology. 2014;5:52. doi: 10.3389/fphar.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrie AM, et al. Label-free functional selectivity assays. Methods in molecular biology. 2015;1272:227–246. doi: 10.1007/978-1-4939-2336-6_16. [DOI] [PubMed] [Google Scholar]
- 54.Fang Y, et al. G-protein-coupled receptor regulation of de novo purine biosynthesis: a novel druggable mechanism. Biotechnology & genetic engineering reviews. 2013;29:31–48. doi: 10.1080/02648725.2013.801237. [DOI] [PubMed] [Google Scholar]
- 55.Fu R, et al. Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Molecular genetics and metabolism. 2015;114:55–61. doi: 10.1016/j.ymgme.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beeckmans S, Kanarek L. Demonstration of physical interactions between consecutive enzymes of the citric acid cycle and of the aspartate-malate shuttle. A study involving fumarase, malate dehydrogenase, citrate synthesis and aspartate aminotransferase. European journal of biochemistry / FEBS. 1981;117:527–535. doi: 10.1111/j.1432-1033.1981.tb06369.x. [DOI] [PubMed] [Google Scholar]
- 57.Velot C, et al. Model of a quinary structure between Krebs TCA cycle enzymes: a model for the metabolon. Biochemistry. 1997;36:14271–14276. doi: 10.1021/bi972011j. [DOI] [PubMed] [Google Scholar]
- 58.Wu F, Minteer S. Krebs cycle metabolon: structural evidence of substrate channeling revealed by cross-linking and mass spectrometry. Angewandte Chemie. 2015;54:1851–1854. doi: 10.1002/anie.201409336. [DOI] [PubMed] [Google Scholar]
- 59.Wu F, et al. Krebs cycle metabolon formation: metabolite concentration gradient enhanced compartmentation of sequential enzymes. Chemical communications. 2015;51:1244–1247. doi: 10.1039/c4cc08702j. [DOI] [PubMed] [Google Scholar]
- 60.An S, et al. Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12872–12876. doi: 10.1073/pnas.1008451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.French JB, et al. Spatial colocalization and functional link of purinosomes with mitochondria. Science. 2016;351:733–737. doi: 10.1126/science.aac6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nikkanen J, et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell metabolism. 2016;23:635–648. doi: 10.1016/j.cmet.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 63.Bao XR, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife. 2016;5:e10575. doi: 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ben-Sahra I, et al. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell metabolism. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robak P, Robak T. Older and new purine nucleoside analogs for patients with acute leukemias. Cancer treatment reviews. 2013;39:851–861. doi: 10.1016/j.ctrv.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. Journal of experimental & clinical cancer research : CR. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sahasranaman S, et al. Clinical pharmacology and pharmacogenetics of thiopurines. European journal of clinical pharmacology. 2008;64:753–767. doi: 10.1007/s00228-008-0478-6. [DOI] [PubMed] [Google Scholar]
- 71.Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chemical reviews. 2009;109:2880–2893. doi: 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allegra CJ, et al. Evidence for direct inhibition of de novo purine synthesis in human MCF-7 breast cells as a principal mode of metabolic inhibition by methotrexate. The Journal of biological chemistry. 1987;262:13520–13526. [PubMed] [Google Scholar]
- 73.Christopherson RI, et al. Inhibitors of de novo nucleotide biosynthesis as drugs. Accounts Chem Res. 2002;35:961–971. doi: 10.1021/ar0000509. [DOI] [PubMed] [Google Scholar]
- 74.Nilsson R, et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nature communications. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spurr IB, et al. Targeting tumour proliferation with a small-molecule inhibitor of AICAR transformylase homodimerization. Chembiochem : a European journal of chemical biology. 2012;13:1628–1634. doi: 10.1002/cbic.201200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caino MC, Altieri DC. Disabling mitochondrial reprogramming in cancer. Pharmacological research. 2015;102:42–45. doi: 10.1016/j.phrs.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caino MC, Altieri DC. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:540–545. doi: 10.1158/1078-0432.CCR-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kohnhorst CL, et al. Subcellular functions of proteins under fluorescence single-cell microscopy. Biochimica et biophysica acta. 2016;1864:77–84. doi: 10.1016/j.bbapap.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srere PA. The metabolon. Trends in Biochemical Sciences. 1985;10:109–110. [Google Scholar]
- 80.Batke J, et al. Substrate-induced dissociation of glycerol-3-phosphate dehydrogenase and its complex formation with fructose-bisphosphate aldolase. European journal of biochemistry / FEBS. 1980;107:389–394. doi: 10.1111/j.1432-1033.1980.tb06041.x. [DOI] [PubMed] [Google Scholar]
- 81.Ovadi J, et al. Kinetic pathways of formation and dissociation of the glycerol-3-phosphate dehydrogenase-fructose-1,6-bisphosphate aldolase complex. The Biochemical journal. 1985;229:57–62. doi: 10.1042/bj2290057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ovadi J, et al. Interaction of the dissociable glycerol-3-phosphate dehydrogenase and fructose-1,6-bisphosphate aldolase. Quantitative analysis by an extrinsic fluorescence probe. European journal of biochemistry / FEBS. 1983;133:433–437. doi: 10.1111/j.1432-1033.1983.tb07482.x. [DOI] [PubMed] [Google Scholar]
- 83.Vertessy B, Ovadi J. A simple approach to detect active-site-directed enzyme-enzyme interactions. The aldolase/glycerol-phosphate-dehydrogenase enzyme system. European journal of biochemistry / FEBS. 1987;164:655–659. doi: 10.1111/j.1432-1033.1987.tb11176.x. [DOI] [PubMed] [Google Scholar]
- 84.Vertessy BG, et al. Modulation of the interaction between aldolase and glycerol-phosphate dehydrogenase by fructose phosphates. Biochimica et biophysica acta. 1991;1078:236–242. doi: 10.1016/0167-4838(91)90564-g. [DOI] [PubMed] [Google Scholar]
- 85.Puchulu-Campanella E, et al. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. The Journal of biological chemistry. 2013;288:848–858. doi: 10.1074/jbc.M112.428573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. The Journal of biological chemistry. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 87.Robitaille AM, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 88.Spector DL. SnapShot: Cellular bodies. Cell. 2006;127:1071. doi: 10.1016/j.cell.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 89.Ma EH, Jones RG. CELL GROWTH. (TORC)ing up purine biosynthesis. Science. 2016;351:670–671. doi: 10.1126/science.aaf1929. [DOI] [PubMed] [Google Scholar]