

Abstract
Ultraviolet (UV) radiation, including both UVB and UVA irradiation, is the major risk factor for causing skin cancer including melanoma. Recently, we have shown that Sesn2, a member of the evolutionarily conserved stress-inducible protein family Sestrins (Sesn), is upregulated in human melanomas as compared to melanocytes in normal human skin, suggesting an oncogenic role of Sesn2. However, the role of Sesn2 in UVB and UVA response is unknown. Here, we demonstrated that both UVB and UVA induce Sesn2 upregulation in melanocytes and melanoma cells. UVB induces Sesn2 expression through the p53 and AKT3 pathways. Sesn2 negatively regulates UVB-induced DNA damage repair. In comparison, UVA induces Sesn2 upregulation through mitochondria but not Nrf2. Sesn2 ablation increased UVA-induced Nrf2 induction and inhibits UVA-induced ROS production, indicating that Sesn2 acts as an upstream regulator of Nrf2. These findings suggest previously unrecognized mechanisms in melanocyte response to UVB and UVA irradiation and potentially in melanoma formation.
Introduction
Melanoma is one of the most deadly cancers in the United States. The incidence of melanoma continues to rise at an alarming rate each year, faster than any of the other common cancers. The major environmental risk factor for melanoma is ultraviolet radiation in sunlight and artificial tanning beds, including both UVB (280–315 nm) and UVA (315–400 nm) radiations (1). In the past decades, tremendous progress has been made in elucidating the mechanism of melanoma initiation and progression at the molecular, cellular and organismal levels (2). In mouse models, UVB and UVA induce melanoma through different mechanisms (3). UVA induces melanoma in association with oxidative damage depending on melanin, different from UVB-induced melanoma (3). However, our understanding of the molecular mechanism in UVB and UVA-induced melanoma development is far from complete.
Sestrins (Sesn) are a family of highly conserved stress-inducible proteins. Sestrins have multiple roles in physiology and pathology in different organisms. In drosophila, Sestrin protects cells and organisms from age-related physiological abnormalities through maintaining metabolic homeostasis (4). In C. elegans, Sestrin promotes health and life span and protects against life stressors (5). In contrast, mammalian species express three Sestrins, Sesn1, Sesn2 and Sesn3. They serve as antioxidant factors to suppress oxidative stress through their oxidoreductase activity, mTOR inhibition and Nrf2 activation (6–8). As stress-inducible proteins, Sesn2 are targets for the tumor suppressor p53 (9). Therefore, Sestrins are considered to have the potential to suppress tumors by properly responding to DNA damage, detoxifying reactive oxygen species, or inhibiting the oncogenic mTOR pathway (7,10–12).
Recently, we have found that Sesn2 is upregulated in human melanomas as compared with melanocytes in normal human skin (13), suggesting an oncogenic role of Sesn2 in melanoma formation. However, the role of Sesn2 in melanocyte response to UVB and UVA irradiation remains unknown. Here, we show that both UVB and UVA induce Sesn2 upregulation in melanocytes and melanoma cells. Sesn2 negatively regulates repair of UVB-induced DNA damage, while it promotes UVA-induced production of reactive oxygen species (ROS). Our findings suggest that Sesn2 is a molecular target for both UVB and UVA irradiation in melanoma development.
Materials and Methods
Cell culture
WT, Sesn2 KO mouse embryonic fibroblast (MEF) cells (14) and A375 (human amelanotic melanoma cells, ATCC) were maintained in a monolayer culture in 95% air/5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin and 100 μg mL−1 streptomycin (Invitrogen, Carlsbad, CA). iMC23 melanocytes were generated and cultured as described previously (15). Normal human epidermal melanocytes (NHEM) were obtained from Invitrogen and cultured according to the manufacturers' instructions. NHEM cells were cultured for less than four passages.
UVB and UVA radiation
UVB and UVA radiations were performed as described previously (16,17). Our UV radiation was monitored every other week to measure the exposure output and dose. Our UV system emits UVB radiation (51%) and UVA (49%) but does not emit UVC radiation. Our UVA radiation was monitored every other week to measure the exposure output and dose. DuPont Teijin Films (Melinex 453/400, cutoff 315 nm, DuPont) were used to remove UVB radiation.
siRNA transfection
A375 cells were transfected with negative control (NC) or siRNA (ON-TARGETplus SMARTpool, Dharmacon) targeting p53 or AKT3, using TransIT-siQUEST® Transfection Reagent (Madison, WI) according to the manufacturer's instructions.
Lentiviral production and infection
Lentiviral constructs expressing shNC (shLuc) and shSesn2 were generated as described previously (6,7). Negative control shRNA (shNC, kindly provided by Dr. Seungmin Hwang, University of Chicago), shPTEN1 (Plasmid #25638) and shPTEN2 (Plasmid #25639) were obtained from Addgene. Lentivirus was produced by cotransfection into 293T cells with lentiviral constructs together with the pCMVdelta8.2 packaging plasmid and pVSV-G envelope plasmid using X-Treme 9 (Roche) as described previously (18–20). Virus-containing supernatants were collected 24–48 h after transfection and used to infect recipients. Target cells were infected in the presence of Polybrene (8 μg mL−1, Sigma-Aldrich, St. Louis, MO) and selected with puromycin at 1 μg mL−1 for 6 days.
Western blotting
Protein concentration was determined using the BCA assay (Pierce, Rockford, IL). Western blotting was performed as described previously (21). Antibodies used included Sesn2 (Proteintech Group, Inc, Chicago, IL), GAPDH, p53, p21, PTEN, AKT, phosphor-AKT (p-AKT) (Cell Signaling Technology, Danvers, MA) and Nrf2 (Santa Cruz).
Real-time PCR
Total RNA was isolated using Qiagen's RNeasy plus mini kit. Quantitative real-time PCR assays were performed using ABI7300 (Applied Biosystems, Foster City, CA) in 96-well plates with the SYBR® Green PCR Master Mix (Applied Biosystems) as in our previous studies (22). The threshold cycle number (CT) for each sample was determined in triplicate. The CT for values for Sesn2 and PTEN was normalized against GAPDH as described previously (18,19,22). The sequences of the primers used were as follows: mouse Sesn2 gene, 5′-TAG CCTGCA GCC TCA CCT AT-3′ (forward) and 5′-TAT CTG ATG CCA AAG ACG CA-3′ (reverse); human Sesn2 gene, 5′-GAC CATGGC TAC TCG CTG AT-3′ (forward) and 5′-GCT GCC TGG AAC TTC TCA TC-3′ (reverse).
Determination of two major forms of UVB-induced DNA damage in genomic DNA by slot blot assay
Slot blot assays of CPD and 6-4PP were performed as described previously (23). Briefly, cells were collected at different time points post-UVB, and DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, 51304). The DNA concentration was calculated from the absorbance at 260 nm using NanoDrop 1000 (NanoDrop products, Wilmington, DE). The CPD and 6-4PP in DNA were quantified by slot blot (Bio-Rad) with antibodies (COSMO BIO Co., TDM-2 for CPD and 64M-2 for 6-4PP) as described previously (20,24-26). For examining repair kinetics, the percentage (%) of repair was calculated by comparing the optical density at the indicated time to that of the corresponding absorbance at time zero when there was no opportunity for repair and 100% of CPDs (or 6-4PPs) were present post-UVB.
ROS measurement
Cells were treated with or without UVB radiation and then incubated with CM-DCFH-DA (2 μm, Invitrogen) for 30 min at 37°C. Cells were washed with PBS three times, and oxidative stress was analyzed by flow cytometry, as described previously (19).
Statistical analyses
Statistical analyses were performed using Prism 5 (GraphPad software, San Diego, CA). Data were expressed as the mean of at least three independent experiments and analyzed by Student's t-test. Error bars indicate the standard error of the means (SE). A P-value of <0.05 was considered statistically significant.
Results
Role of p53 in UVB-induced Sesn2
Recently, we have shown that UVB induces Sesn2 upregulation in normal melanocytes and melanoma cells (13). To determine the mechanism in UVB-induced Sesn2 upregulation, we first analyzed UVB's regulation of Sesn2 at the transcriptional level. UVB increased the mRNA levels of Sesn2 in both A375 and MEF cells (Fig. 1A,B). The RNA synthesis inhibitor actinomycin D abolished UVB-induced Sesn2 mRNA upregulation (Fig. 1A,B). These data indicate that UVB does induce Sesn2 expression at the transcriptional level. As the tumor suppressor p53 has been shown to activate Sesn2 transcription (9), next we analyzed the role of p53 in UVB-induced Sesn2. UVB upregulated the protein levels of Sesn2, p53 and the well-studied p53 target p21 (Fig. 1C). Knockdown of p53 reduced but did not completely block UVB-induced Sesn2 expression at the protein and mRNA levels, while it completely blocked p21 expression (Fig. 1C,D). p53 knockdown also reduced the basal Sesn2 levels. These data indicate that p53 is important for basal and UVB-induced Sesn2 expression and that other factors may play active roles in UVB-induced Sesn2 expression.
Figure 1.
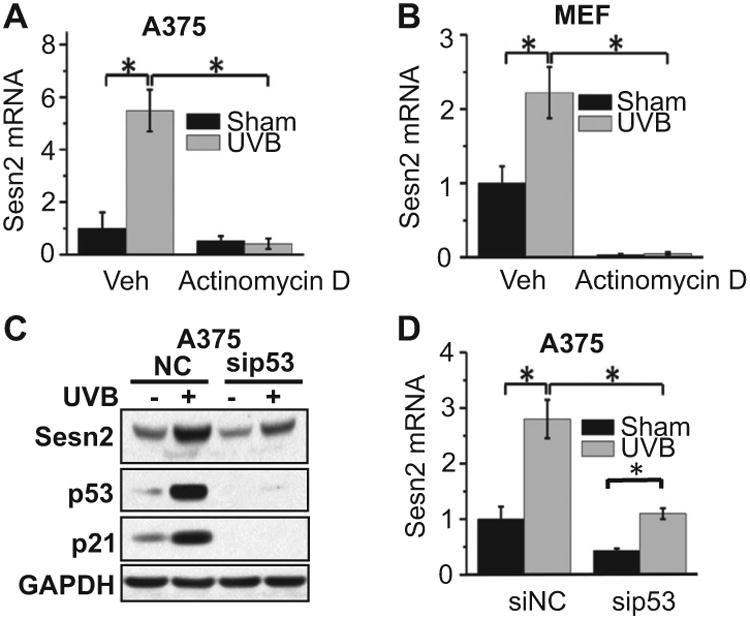
Role of p53 in UVB-induced Sesn2 expression. (A and B) Real-time PCR analysis of Sesn2 mRNA levels in A375 (A) and MEF (B) cells. (C) Immunoblot analysis of Sesn2, p53, p21 and GAPDH in A375 transfected with siRNA targeting p53 (sip53) or negative control siRNA (siNC) at 6 h post-UVB or post-sham irradiation. (D) Real-time PCR analysis of Sesn2 mRNA levels in A375 cells treated as in C. (A, B, D) *P < 0.05, Student's t-test.
Role of AKT in UVB-induced Sesn2
Next, we assessed the role of the AKT pathway in UVB-induced Sesn2 expression. In Drosophila, loss of PTEN, the key negative regulator for the PI3K/AKT pathway and a tumor suppressor in skin cancer (27), upregulates dSesn, the single ortholog of mammalian Sestrins (4). However, in mammalian cells, the role of PTEN/AKT in Sesn2 regulation remains unclear. We found that in A375 cells, knockdown of PTEN increased AKT activation and Sesn2 expression (Fig. 2A), indicating that PTEN loss upregulates Sesn2 in mammalian cells. Furthermore, the biochemical PI3K/AKT inhibitor LY294002 (LY) inhibited UVB-induced Sesn2 upregulation (Fig. 2B). Similarly, knockdown of AKT3, the predominant AKT isoform active in melanoma cells (28), inhibited basal Sesn2 expression and UVB-induced Sesn2 upregulation at the mRNA and protein levels (Fig. 2C,D). These findings demonstrated that the AKT pathways are critical for basal Sesn2 expression and UVB-induced Sesn2 upregulation.
Figure 2.
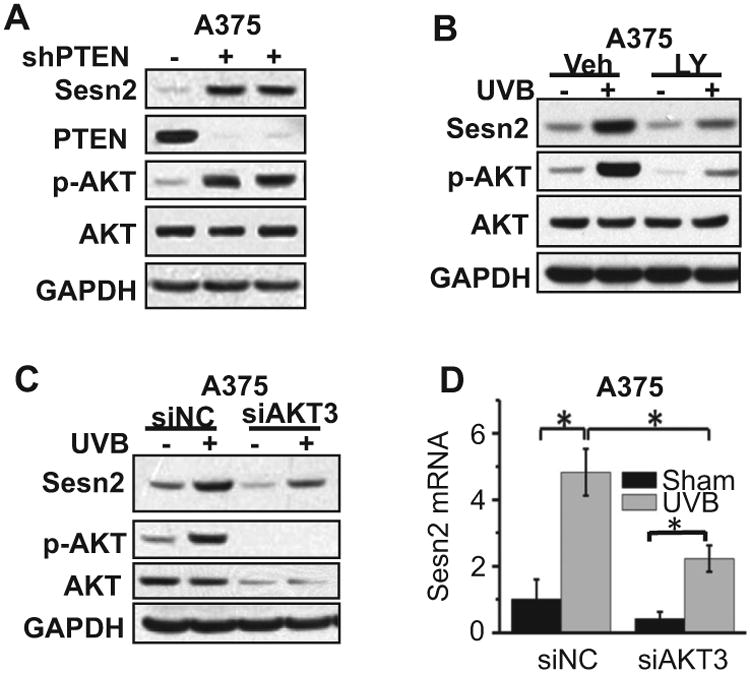
Role of AKT in UVB-induced Sesn2 expression. (A) Immunoblot analysis of Sesn2, PTEN, p-AKT, AKT and GAPDH in A375 cells infected with either a lentiviral vector expressing shRNA targeting PTEN (shPTEN1 and shPTEN2) or negative control (shNC). (B) Immunoblot analysis of Sesn2, p-AKT, AKT and GAPDH in A375 cells pretreated with the specific PI3K/AKT activation inhibitor LY294002 (LY, 10 μm) at 24 h post-UVB (20 mJ cm−2) or post-sham. (C) Immunoblot analysis of Sesn2, p-AKT and GAPDH in A375 cells transfected with siRNA targeting AKT3 (siAKT3) and negative control siRNA (NC) at 6 h post-UVB (20 mJ cm−2) or post-sham. (D) Real-time PCR analysis of Sesn2 mRNA levels in A375 treated as in G. For all of the graphs, data are shown as means ± SE for three independent experiments. *P < 0.05; t-test, compared with the control group.
Role of Sesn2 in UVB-induced DNA damage repair
To determine the function of Sesn2 in UVB response, we analyzed the effect of Sesn2 overexpression or knockdown in repair of UVB-induced DNA damage, an essential mechanism to prevent tumorigenic transformation. We measured the function of Sesn2 in repair of UVB-induced cyclobutane pyrimidine dimers (CPD), a major DNA damage lesion that causes tumorigenesis (29). Overexpressing Sesn2 in A375 melanoma cells reduced CPD repair (Fig. 3A,B), while knockdown of Sesn2 increased CPD repair (Fig. 3C,D). These results indicate that Sesn2 negatively regulates UVB-induced DNA damage repair.
Figure 3.
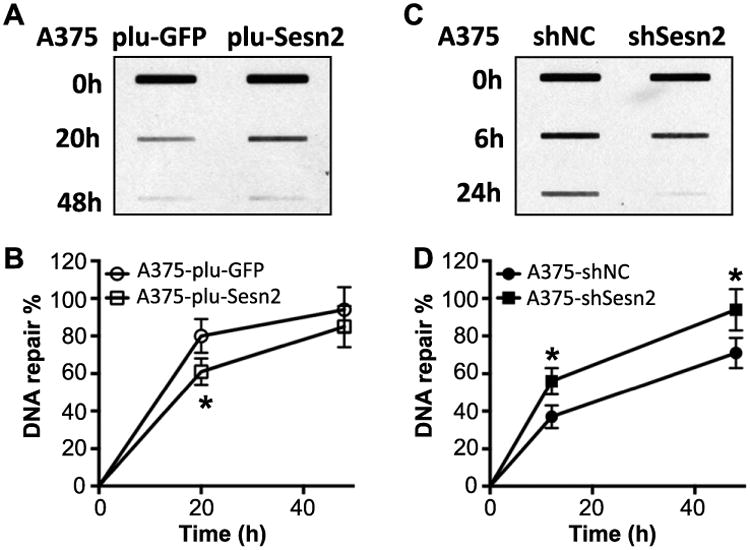
Role of Sesn2 in UVB-induced DNA damage repair. (A) Slot blot analysis of CPD levels in A375 cells stably infected with plu-GFP (control) or plu-Sesn2 at 0, 20 h or 48 h post-UVB irradiation. (B) Quantification of A. (C) Slot blot analysis of CPD levels in A375 cells stably infected with shNC (control) and shSesn2 at 0, 6 or 24 h post-UVB irradiation. (D) Quantification of C. *P < 0.05 as compared with their control group (Student's t-test).
Effect of UVA on Sesn2
UVA and UVB regulate shared and distinct molecular pathways. To determine the effect of UVA on Sesn2 expression, we analyzed the difference in Sesn2 protein levels in sham and UVA-irradiated normal melanocytes, melanoma cells and MEF cells. UVA irradiation induced Sesn2 upregulation in a dose-dependent manner in immortalized iMC23 melanocytes (Fig. 4A). UVA induced Sesn2 upregulation at 6 h, while it had no effect at 1.5 h post-UVA irradiation (Fig. 4B). In normal human epidermal melanocytes (NHEM), UVA induced Sesn2 upregulation at 6 and 24 h (Fig. 4C). Furthermore, UVA induced Sesn2 upregulation at 6 h postirradiation in A375 cells and MEF cells. These results indicate that UVA upregulates Sesn2 in melanocytes, melanoma cells and MEF cells.
Figure 4.
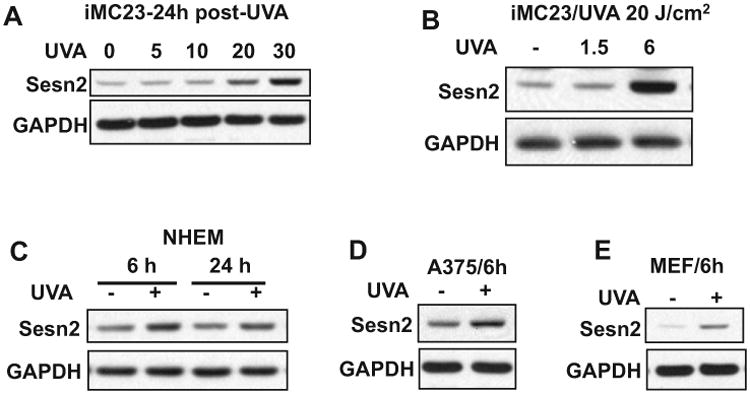
Effect of UVA radiation on Sesn2. (A) Immunoblot analysis of Sesn2 and GAPDH in iMC23 cells at 24 h post-UVA irradiation at different doses (0, 5, 10, 20 and 30 J cm−2). (B) Immunoblot analysis of Sesn2 and GAPDH in iMC23 cells at 1.5 or 6 h post-UVA irradiation (0 or 20 J cm−2). (C) Immunoblot analysis of Sesn2 and GAPDH in NHEM cells at 6 or 24 h post-UVA irradiation (0 or 20 J cm−2). (D) Immunoblot analysis of Sesn2 and GAPDH in A375 cells at 6 h post-UVA irradiation (0 or 20 J cm−2). (E) Immunoblot analysis of Sesn2 and GAPDH in MEF cells at 6 h post-UVA irradiation (0 or 20 J cm−2).
Role of mitochondria in UVA-induced Sesn2 upregulation
To determine the mechanism in UVA-induced Sesn2 upregulation, we first assessed the role of Nrf2, a transcription factor that regulates expression of numerous antioxidant genes (30,31) and is activated by UVA irradiation (32,33). UVA irradiation induced Nrf2 accumulation in A375 and iMC23 cells (Fig. 5A,B). Knockdown of Nrf2 had no effect on UVA-induced Sesn2 upregulation in A375 cells (Fig. 5A). Treatment of cells with mitochondria-targeted antioxidant Mito-carboxy proxyl (Mito-CP) prevented UVA-induced Nrf2 accumulation and Sesn2 upregulation (Fig. 5B). These results indicate that mitochondria have an active role in UVA-induced Sesn2 upregulation independent of Nrf2.
Figure 5.
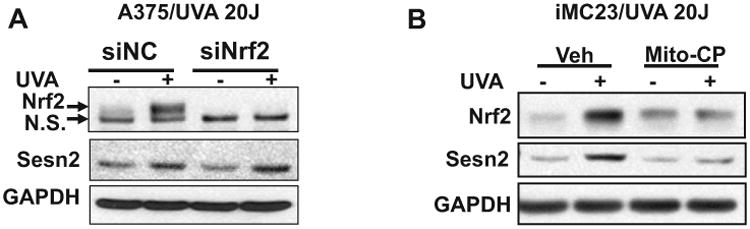
Role of mitochondrial function in UVA-induced Sesn2 upregulation. (A) Immunoblot analysis of Nrf2, Sesn2 and GAPDH in A375 cells transfected with siNC or siNrf2 at 6 h post-UVA irradiation (0 or 20 J cm−2). n.s., nonspecific band. (B) Immunoblot analysis of Sesn2 and GAPDH in iMC23 cells at 6 h post-UVA irradiation (0 or 20 J cm−2). The cells were pretreated with vehicle or Mito-CP (1 μm) one hour prior to and after UVA irradiation.
Role of Sesn2 in UVA-induced oxidative stress
To determine the function of Sesn2 in UVA response, we assessed the role of Sesn2 in UVA-induced oxidative stress, a distinct mechanism from UVB response. In MEF cells, Sesn2 deletion increased UVA-induced Nrf2 accumulation (Fig. 6A). UVA increased the levels of reactive oxygen species (ROS) as measured by DCFH-DA (Fig. 6B). Sesn2 ablation inhibited UVA-induced ROS generation (Fig. 6B). These results demonstrate that Sesn2 inhibition protects against UVA-induced ROS production in association with increased Nrf2 accumulation.
Figure 6.
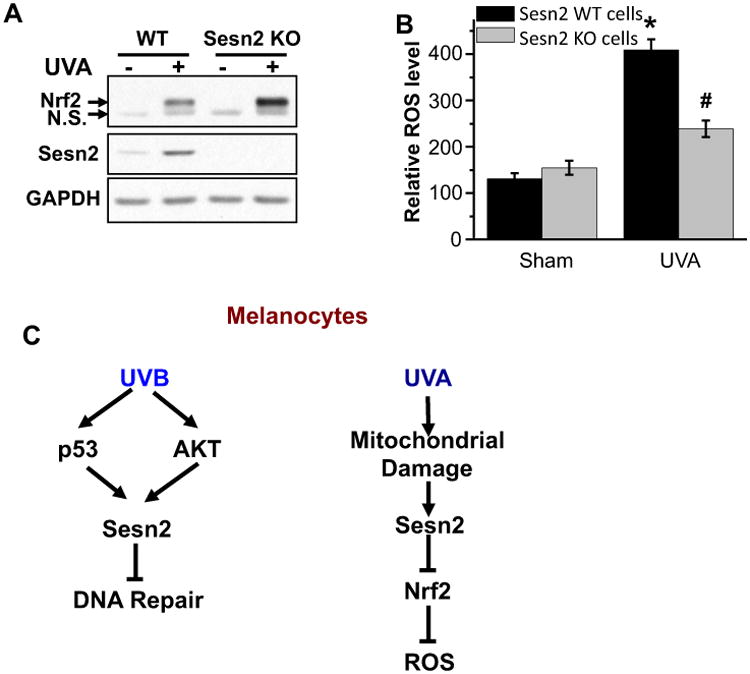
Role of Sesn2 in UVA-induced oxidative stress. (A) Immunoblot analysis of Nrf2, Sesn2 and GAPDH in WT or Sesn2 KO MEF cells at 6 h post-UVA irradiation (0 or 20 J cm−2). n.s., nonspecific band. (B) DCFH-DA analysis of ROS generation in WT or Sesn2 KO MEF cells at 6 h post-UVA irradiation (0 or 20 J cm−2). *P < 0.05 as compared with sham group; #P < 0.05 as compared with UVA-irradiated WT group (Student's t-test). (C) A schematic for the distinct role of Sesn2 in response to UVB and UVA radiation in melanocytes.
Discussion
Sestrins (Sesn) are a family of evolutionarily conserved stress-inducible proteins (34). Recently, we have shown that Sesn2 promotes AKT activation in both SCC and melanoma cells to promote cell survival (13). Sesn2 is upregulated in human melanomas as compared to normal human skin melanocytes, suggesting an oncogenic role of Sesn2 (13). However, the role of Sesn2 in UVB and UVA response is unknown. Here, we demonstrated that both UVB and UVA induce Sesn2 upregulation in melanocytes and melanoma cells (Fig. 6C). UVB induces Sesn2 expression through the p53 and AKT3 pathways. Sesn2 negatively regulates UVB-induced DNA damage repair. In comparison, UVA induces Sesn2 upregulation through mitochondria ROS but in a way that is independent of Nrf2. Sesn2 ablation inhibits UVA-induced ROS production in association with increased Nrf2 induction. These findings suggest a previously unrecognized mechanism in melanocyte response to UVB and UVA irradiation and potentially in melanocyte transformation and melanoma formation.
Expression of Sestrins is inducible in response to a number of cellular stresses such as DNA damage, hypoxia, energy starvation and oxidative damage (34). UVB and UVA induce Sesn2 upregulation through different mechanisms. It is known that the major molecular target for UVB is DNA, causing DNA damage, and thus, a major mechanism is the activation of DNA damage response pathways including p53. We found that UVB induces Sesn2 upregulation through the p53 pathway. In addition, AKT3, the major AKT pathway in melanoma cells, also plays a role in UVB-induced Sesn2 upregulation. In contrast, UVA causes the formation of ROS, possibly through multiple mechanisms including mitochondrial dysfunction. We found that UVA-induced Sesn2 upregulation is not through the Nrf2 pathway, while it is blocked by the mitochondria-targeted antioxidant Mito-CP. Instead, Sesn2 acts as an upstream suppressor of Nrf2 in response to UVA radiation. These data suggest that UVA induces Sesn2 through mitochondrial damage, either ROS formation or energetics. Further investigation is needed to elucidate the mechanism in UVA-induced Sesn2 upregulation. In addition, it requires more studies on how UVB and UVA regulate Sesn2 activity. This will require further characterization of the enzymatic activity of Sesn2 in stress response.
Under stress conditions, Sestrin induction is critical for adaptation by maintaining redox balance and metabolic homeostasis (34,35). Previously DNA damage has been shown to induce Sesn1 and Sesn2 expression and thus activate AMPK to inhibit the mTOR pathway (7,36). Therefore, Sestrins have been proposed as tumor suppressors. In contrast, recently, AKT-mediated but p53-independent Sesn2 induction has been shown to promote cell survival under energetic stress (37). Consistent with these results, our recent studies demonstrated that Sesn2 is upregulated in human SCC and melanoma as compared with normal human skin and that Sesn2 promotes the oncogenic AKT pathway to enhance cell survival (13), suggesting an oncogenic function of Sesn2. Furthermore, here, we show that Sesn2 plays an active role in response to UVB and UVA-induced damage. Sesn2 inhibits the repair of UVB-induced DNA damage and promotes UVA-induced oxidative stress, further supporting an oncogenic role of Sesn2 in melanoma formation. Our results differ from previous studies showing that Sesn1 and Sesn2 promote Nrf2 activation in liver in response to fasting (8). These findings suggest a context-dependent role of Sesn2 in oxidative stress response. Future investigation will elucidate the molecular mechanism by which Sesn2 modulates DNA repair and oxidative damage in melanocytes and melanoma cells.
In summary, our findings demonstrate that both UVB and UVA irradiations upregulate Sesn2 in normal melanocytes, immortalized melanocytes and melanoma cells. Sesn2 inhibits UVB-induced DNA damage repair and promotes UVA-induced ROS generation. Our results can add new mechanistic insights into UVB- and UVA-induced melanoma formation and may facilitate developing better preventative and therapeutic strategies to reduce the melanoma burden.
Acknowledgments
This work was supported by the NIH/NIEHS grant ES024373 and ES016936 (YYH), the American Cancer Society (ACS) grant RSG-13-078-01 (YYH), the University of Chicago Cancer Research Center (P30 CA014599), the CTSA (UL1 TR000430) and the University of Chicago Friends of Dermatology Endowment Fund. We thank Drs Balaraman Kalyanaraman (Medical College of Wisconsin) for providing the Mito-CP and Ann Motten for a critical reading of the manuscript.
Abbreviations
- CPD
cyclobutane pyrimidine dimers
- iMC23
immortalized melanocyte cell line
- KO
knockout
- MEF
mouse embryonic fibroblast
- Sesn2
Sestrin 2
- Sesn
Sestrins
- shRNA
small hairpin RNA
- shSesn2
shRNA targeting Sesn2
- UVA
ultraviolet A (315–400 nm)
- UVB
ultraviolet B (280–415 nm)
- UV
ultraviolet radiation
- WT
wild type
Footnotes
This article is part of the Special Issue highlighting Dr. Azis Sancar's outstanding contributions to the field of DNA photodamage repair in honor of his recent Nobel Prize in Chemistry.
References
- 1.D'Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. UV radiation and the skin. Int J Mol Sci. 2013;14:12222–12248. doi: 10.3390/ijms140612222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1131–1155. doi: 10.1101/gad.191999.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noonan FP, Zaidi MR, Wolnicka-Glubisz A, Anver MR, Bahn J, Wielgus A, Cadet J, Douki T, Mouret S, Tucker MA, Popratiloff A, Merlino G, De Fabo EC. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat Commun. 2012;3:884. doi: 10.1038/ncomms1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, Karin M. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang YL, Loh KS, Liou BY, Chu IH, Kuo CJ, Chen HD, Chen CS. SESN-1 is a positive regulator of lifespan in Caenorhabditis elegans. Exp Gerontol. 2013;48:371–379. doi: 10.1016/j.exger.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 7.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013;17:73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, Skaliter R, Gudkov AV, Chumakov PM, Feinstein E. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- 10.Budanov AV. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Anti-oxid Redox Signal. 2011;15:1679–1690. doi: 10.1089/ars.2010.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cam M, Bid HK, Xiao L, Zambetti GP, Houghton PJ, Cam H. p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J Biol Chem. 2014;289:4083–4094. doi: 10.1074/jbc.M113.530303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao B, Shah P, Budanov AV, Qiang L, Ming M, Aplin A, Sims DM, He YY. Sestrin2 protein positively regulates AKT enzyme signaling and survival in human squamous cell carcinoma and melanoma cells. J Biol Chem. 2014;289:35806–35814. doi: 10.1074/jbc.M114.595397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, Wolfe AM, Perkins GA, Ellisman MH, Bier E, Scadeng M, Foretz M, Viollet B, Olefsky J, Karin M. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012;16:311–321. doi: 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang K, Chen J, Jiang W, Huang E, Cui J, Kim SH, Hu N, Liu H, Zhang W, Li R, Chen X, Kong Y, Zhang J, Wang J, Wang L, Shen J, Luu HH, Haydon RC, Lian X, Yang T, He TC. Conditional immortalization establishes a repertoire of mouse melanocyte progenitors with distinct melanogenic differentiation potential. J Invest Dermatol. 2012;132:2479–2483. doi: 10.1038/jid.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ming M, Han W, Maddox J, Soltani K, Shea CR, Freeman DM, He YY. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene. 2010;29:492–502. doi: 10.1038/onc.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao B, Ming M, He YY. Suppression of PTEN transcription by UVA. J Biochem Mol Toxicol. 2013;27:184–191. doi: 10.1002/jbt.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ming M, Shea CR, Guo X, Li X, Soltani K, Han W, He YY. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci USA. 2010;107:22623–22628. doi: 10.1073/pnas.1010377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han W, Ming M, Zhao R, Pi J, Wu C, He YY. Nrf1 CNC-bZIP protein promotes cell survival and nucleotide excision repair through maintaining glutathione homeostasis. J Biol Chem. 2012;287:18788–18795. doi: 10.1074/jbc.M112.363614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu CL, Qiang L, Han W, Ming M, Viollet B, He YY. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene. 2013;32:2682–2689. doi: 10.1038/onc.2012.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiang L, Yang Y, Ma YJ, Chen FH, Zhang LB, Liu W, Qi Q, Lu N, Tao L, Wang XT, You QD, Guo QL. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009;279:13–21. doi: 10.1016/j.canlet.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Han W, Ming M, He TC, He YY. Immunosuppressive cyclosporin A activates AKT in keratinocytes through PTEN suppression: implications in skin carcinogenesis. J Biol Chem. 2010;285:11369–11377. doi: 10.1074/jbc.M109.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ming M, Soltani K, Shea CR, Li X, He YY. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene. 2015;34:357–363. doi: 10.1038/onc.2013.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda T, Chua PP, Chong MT, Sim AB, Nikaido O, Tron VA. Nucleotide excision repair genes are upregulated by low-dose artificial ultraviolet B: evidence of a photoprotective SOS response? J Invest Dermatol. 2001;117:1490–1497. doi: 10.1046/j.0022-202x.2001.01562.x. [DOI] [PubMed] [Google Scholar]
- 25.Qiang L, Zhao B, Shah P, Sample A, Yang S, He YY. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy. 2016;12:357–368. doi: 10.1080/15548627.2015.1110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiang L, Shah P, Barcellos-Hoff MH, He YY. TGF-beta signaling links E-cadherin loss to suppression of nucleotide excision repair. Oncogene. 2016;35:3293–3302. doi: 10.1038/onc.2015.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ming M, He YY. PTEN: new insights into its regulation and function in skin cancer. J Invest Dermatol. 2009;129:2109–2112. doi: 10.1038/jid.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L, Robertson GP. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 29.Jans J, Schul W, Sert YG, Rijksen Y, Rebel H, Eker AP, Nakajima S, van Steeg H, de Gruijl FR, Yasui A, Hoeijmakers JH, van der Horst GT. Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol. 2005;15:105–115. doi: 10.1016/j.cub.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 31.Pi J, Freeman ML, Yamamoto M. Nrf2 in toxicology and pharmacology: the good, the bad and the ugly? Toxicol Appl Pharmacol. 2010;244:1–3. doi: 10.1016/j.taap.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marrot L, Jones C, Perez P, Meunier JR. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment Cell Melanoma Res. 2008;21:79–88. doi: 10.1111/j.1755-148X.2007.00424.x. [DOI] [PubMed] [Google Scholar]
- 33.Hirota A, Kawachi Y, Itoh K, Nakamura Y, Xu X, Banno T, Takahashi T, Yamamoto M, Otsuka F. Ultraviolet A irradiation induces NF-E2-related factor 2 activation in dermal fibroblasts: protective role in UVA-induced apoptosis. J Invest Dermatol. 2005;124:825–832. doi: 10.1111/j.0022-202X.2005.23670.x. [DOI] [PubMed] [Google Scholar]
- 34.Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013;18:792–801. doi: 10.1016/j.cmet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO Mol Med. 2010;2:388–400. doi: 10.1002/emmm.201000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 2012;7:e32035. doi: 10.1371/journal.pone.0032035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A, Tanti JF, Bost F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013;20:611–619. doi: 10.1038/cdd.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]