

Abstract
Objective:
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant adult-onset disease characterized by progressive ptosis, dysphagia, and proximal limb weakness. The genetic cause is an expanded (GCN)n mutation in the PABPN1 gene encoding for the polyadenylate-binding protein nuclear 1. We hypothesized a potential correlation between the size of the (GCN)n expansion and the severity of the phenotype. To do this, we characterized the distribution of the genotypes as well as their correlation with age at diagnosis and phenotypical features in a large cohort of heterozygous and homozygous patients with OPMD in France with a confirmed molecular diagnosis of PABPN1.
Methods:
We explored 354 unrelated index cases recruited between 1999 and 2014 in several neuromuscular centers in France.
Results:
This cohort allowed us to characterize the frequency of mutated alleles in the French population and to demonstrate a statistical correlation between the size of the expansion and the mean age at diagnosis. We also confirmed that homozygous patients present with a more severe disease.
Conclusions:
It has been difficult to establish phenotype–genotype correlations because of the rare nature of this disease. Our work demonstrates that patients with OPMD with longer PABPN1 expansion are on average diagnosed at an earlier age than patients with a shorter expansion, confirming that polyalanine expansion size plays a role in OPMD, with an effect on disease severity and progression.
Oculopharyngeal muscular dystrophy (OPMD) (MIM #164300) is an autosomal dominantly inherited muscle disease with a clinical onset usually in the fourth to sixth decade of life.1 OPMD is clinically characterized by ptosis and dysphagia. Proximal limb weakness may occur at later stages of the disease. OPMD is caused by an abnormal (GCN) triplet expansion within the PABPN1 gene (NG_008239) located on chromosome 14 (14q11.2-q13). While the wild-type PABPN1 gene contains 10 (GCN) repeats, the mutated form in OPMD is expanded to 11–18 repeats, adding 1–8 additional alanine residues at the N-terminus of the PABPN1 protein.2,3 OPMD shows a large worldwide distribution (described in more than 30 countries) and several studies describing the distribution of PABPN1 alleles on a large cohort have been published.4–11 In these studies, no correlation between the size of the expansion and the severity of the phenotype could clearly be evidenced, probably due to the relatively small size of genotyped patient cohorts. Here we performed a retrospective analysis on the French cohort diagnosed in neuromuscular reference centers between 1999 and 2014. We present 354 unrelated index patients genetically confirmed with a mutation in the PABPN1 gene. This large cohort allowed the evaluation of the distribution of genotypes and the frequency of mutated alleles in the French population. In addition, we searched for a potential correlation among disease severity, age at diagnosis, and number of repeats. We also added the clinical description of 3 specific groups of patients: patients carrying the (GCN)11 allele, compound heterozygous patients, and homozygous patients.
METHODS
Patients.
This collaborative study from neurology centers throughout France was conducted on 900 patients referred to the genetic laboratory of Pitié-Salpêtrière Hospital for the diagnosis of OPMD by genotyping the PABPN1 gene between 1999 and 2014. In 2/3 of the cases, the analysis was performed on patients who presented suggestive clinical features of OPMD, but in 1/3 of the cases, the analysis was done as a first exclusion of OPMD prior to invasive muscle biopsy or for the differential diagnosis of mitochondrial disease or hereditary myopathies. Out of these 900 patients, 354 unrelated index patients were found to have PABPN1 gene expansions, together with an additional group of 63 mutated relatives. In addition, control samples (n = 200) with no known family history of OPMD were also genotyped.
Standard protocol approvals, registrations, and patient consents.
Written informed consent and appropriate assent were obtained from each evaluated participant by a qualified investigator.
Clinical and paraclinical evaluation.
The referring clinicians were asked to provide detailed neurologic clinical data as well as the existence of a positive family history. Age at onset, evolution of disease symptoms (ptosis, dysphagia, muscle weakness, and cognitive functions for some of them), creatine kinase (CK) activity, and results of EMG were obtained when available. The great majority of patients were first tested for acetylcholine receptor anti-MuSK antibodies.
Genotyping method.
Blood samples were obtained after signing informed written consent according to French legislation and genomic DNA was extracted by standard methods.
The repeated sequence in exon 1 of the PABPN1 gene was amplified on genomic DNA by PCR using PABP forward primer (5′-CGCAGTGCCCCGCCTTAGA-3′) and PABP reverse primer (5′-ACAAGATGGCGCCGCCGCCCCGGC-3′) as described by Brais et al.2 The forward primer was fluorescent labelled with 6-FAM. Amplicons were then analyzed by a GeneScan analysis: the PCR products were separated according to their sizes by capillary electrophoresis on an Applied Biosystems (Foster City, CA) 3730 Genetic Analyzer,12 and the running files were analyzed by the GeneMapper 3.2 software. The PCR product of the normal allele containing 10 (GCN) repeats was 246 base pairs and the difference between each allele was 3 base pairs.
RESULTS
Distribution of pathogenic genotypes.
The group of 354 patients with genetically confirmed OPMD is composed of 173 female and 179 male patients. As OPMD is a dominant disease, most of the patients carried the normal (GCN)10 repeat on one allele associated with a pathogenic allele containing (GCN)11 to (GCN)17 repeats. Distribution of expanded genotypes in index patients was the following: (GCN)10/(GCN)12 was found in 33 patients (9.3%), (GCN)10/(GCN)13 in 176 patients (49.7%), (GCN)10/(GCN)14 in 55 patients (15.5%), (GCN)10/(GCN)15 in 56 patients (15.8%), (GCN)10/(GCN)16 in 4 patients (1%), and (GCN)10/(GCN)17 in 2 patients (0.6%) (figure 1A). In addition, 9 compound heterozygous (GCN)11/(GCN)12 patients (2.5%) and 13 homozygous patients, distributed as (GCN)11 (n = 9; 2.5%), (GCN)12 (n = 2; 0.6%), and (GCN)13 (n = 2; 0.6%) genotypes were also found (figure 1A). In addition, 6 (GCN)10/(GCN)11 patients (1.7%) presenting with clinical symptoms of OPMD were found, confirming our recent observation suggesting that the (GCN)11 allele—described until now as a recessive allele—could be a dominant allele.3 No patient with 18 expansions13 was found. This cohort allowed us to obtain the allelic distribution in France of the 7 mutant alleles from (CGN)11 to (CGN)17 of the 376 mutated alleles tested: 8.8% were (GCN)11, 12.2% were (GCN)12, 47.9% were (GCN)13, 14.6% were (GCN)14, 14.9% were (GCN)15, 1.1% were (GCN)16, and 0.5% were (GCN)17 (figure 1B).
Figure 1. Distribution and allelic frequency of PABPN1 genotypes.

(A) Distribution of PABPN1 genotypes in the 354 PABPN1 mutated patients. For clarity, genotype (GCN)x/(GCN)y is indicated as x/y on the horizontal axis. Red indicates heterozygous patients, green compound heterozygous patients, and blue homozygous patients. (B) Allelic frequency of the 354 expanded alleles of PABPN1. For clarity, only the expanded allele is presented, excluding the normal (GCN)10 allele.
Clinical presentation of classical expanded genotypes.
In the 176 patients carrying one (GCN)13 allele, the mean age at diagnosis was 64 years, with a high dispersion (64 ± 10 [SD]). At onset of symptoms, half of the patients presented dysphagia and ptosis, whereas for the other half, only dysphagia was present and ptosis was delayed (table). In patients with expansions greater than (GCN)14, proximal weakness of the pelvic girdle was frequent at the time of diagnosis. Extension of muscle weakness to limbs was observed in half of patients and was linked to the age at examination. In addition, quantification of the CK biomarker showed levels 3 to 10 times higher (300 UI/L to 1,000 UI/L) than the normal range (<100 UI/L) (table). Patients carrying (GCN)16 (n = 4) and (GCN)17 (n = 2) repeats had an age at diagnosis ranging from 42 to 55 years. One of 2 patients carrying (GCN)17 presented with a severe phenotype characterized by a sudden bilateral ptosis at 50 years, which was treated by surgery. Seven years later, after a resuscitated cardiac arrest due to a myocardial infarction, the patient suddenly presented a loss of deglutition and a laryngeal palsy of unknown etiology needing tracheotomy and gastrostomy. As he complained of muscle weakness, neurologic examination showed a mild motor deficiency, but no diplopia and no diaphragmatic paralysis. EMG did not show any myopathic pattern. No cognitive symptoms were noted. His mother also presented a bilateral ptosis and complained of aspiration episodes.
Table.
Clinical phenotypes of patients with mutated oculopharyngeal muscular dystrophy
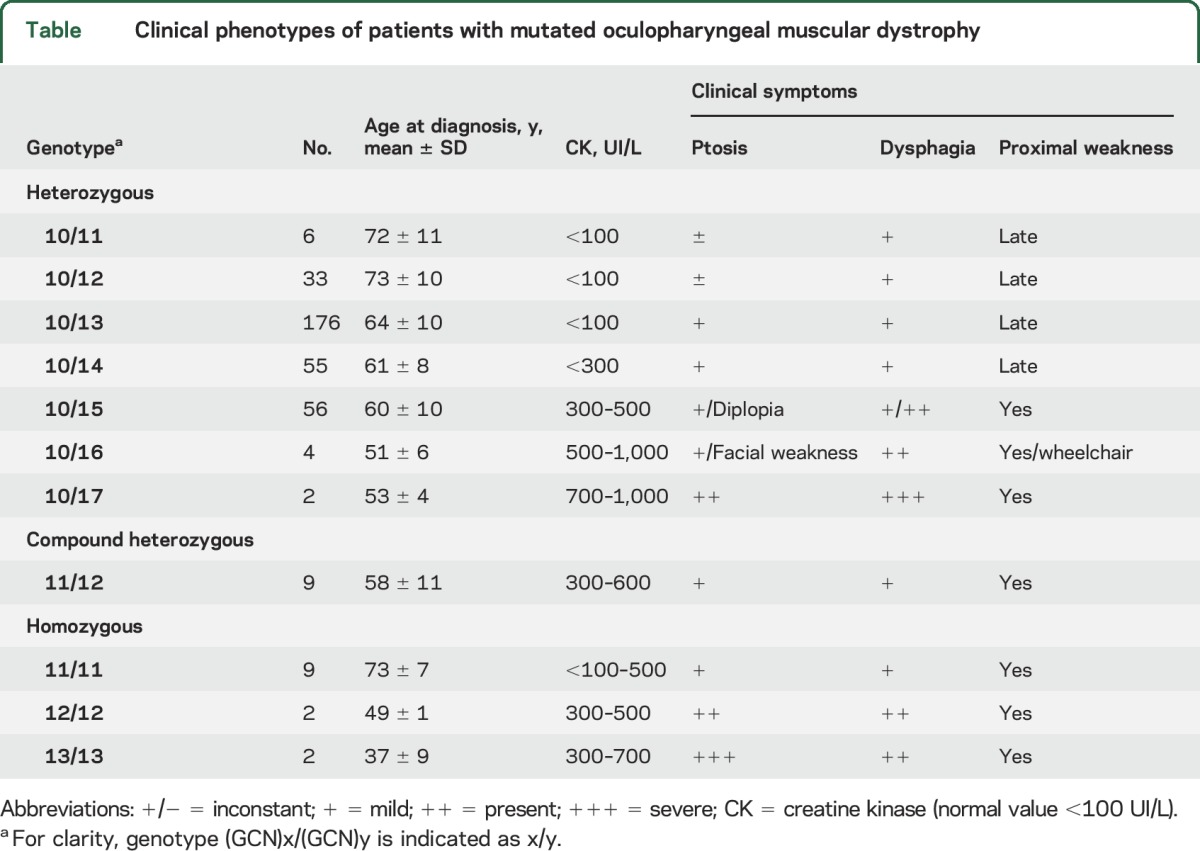
Six patients were found to be heterozygous for the (GCN)11 allele. They presented a late disease onset (mean age at diagnosis of 72 years) characterized by dysphagia with or without ptosis. Older patients could present a limb-girdle pattern of weakness although this was not confirmed to be directly linked to the disease. These patients confirmed that the (GCN)11 allele, formerly considered as a recessive allele, had in fact a dominant expression with a low penetrance.3 In accordance with UK patients,9 analysis of 200 control chromosomes showed that none of these controls carried the (GCN)11 allele; however, this allele was recently found with a 0.05% frequency in the Thai population.14
Clinical presentation and paraclinical examination of compound heterozygous and homozygous patients.
The clinical presentation of compound heterozygous patients for (GCN)11/(GCN)12 mutant alleles was typical of the OPMD phenotype but with an earlier mean age at diagnosis as compared with patients heterozygous for a single (GCN)12 allele (mean 58 and 73 years of age, respectively). These patients showed bilateral ptosis without diplopia and dysphagia appearing around 50–55 years of age. In half of them, a proximal weakness appeared around 60 years, affecting both lower (quadriceps, psoas) and upper (deltoid) limbs. The CK levels were slightly elevated from 300 to 650 UI/L.
A homozygous mutated genotype was found in 13 independent patients: 9 with (GCN)11, 2 with (GCN)12, and 2 with (GCN)13 alleles (figure 1). Patients carrying the homozygous (GCN)11 allele had a mean age at diagnosis of 73 years. They presented with a bilateral ptosis inconstantly associated with diplopia. All exhibited dysphagia, sometimes associated with dysphonia, and complications such as aspiration were noticed in 7 patients. Proximal and especially distal muscle weakness with myalgia and difficulty walking were observed in 4 patients older than 65. In 2 patients, an EMG was performed and showed a myogenic pattern. CK levels were normal or slightly elevated (300 UI/L). No cognitive decline was noticed although no specific tests were performed. The 2 patients homozygous for the (GCN)12 allele were from consanguineous families. The first patient was a 60-year-old man in whom the diagnosis of OPMD was first excluded on the basis of a transmission not clearly in favor of a dominant heredity although some cousins presented with ptosis both on the maternal side and on the paternal side. Upon clinical examination, the patient presented with ptosis and dysphagia associated with a proximal muscle weakness of lower limbs (walking difficulties, waddling gait), with hypertrophy of calves and quadriceps and a diffuse areflexia. The patient complained of walking difficulties since the age of 50 years. CK levels were slightly elevated, ranging from 300 to 500 UI/L. His older brother presented with the same phenotype but was never genotyped. Both underwent a surgery for their ptosis. The second patient was a man born in 1954 in an Indian family. The patient noticed bilateral ptosis and dysphagia around the age of 40 years. His older brother born in 1952 was heterozygous and presented with a moderate phenotype, whereas his younger brother born in 1966, who was genotyped homozygote for the mutant allele, exhibited at 45 years of age bilateral ptosis, dysphagia, ophthalmoparesis, and proximal and axial muscle weakness. Two patients were homozygous for the (GCN)13 repeat. The first one was a woman of Turkish origin. She presented with a bilateral ptosis noticed at 32 years of age and a progressive dysphagia from her 40s onward. At 48 years of age, she noticed proximal muscle weakness of the pelvic girdle, affecting both upper and lower limbs, with wasting and difficulties standing from a chair, walking, and climbing stairs. She underwent a cricopharyngeal myotomy at 51 years of age. Her CK levels were elevated (700 UI/L). The second patient was a 49-year-old woman originating from the North of France. She complained for many years about an asymmetric bilateral ptosis associated with dysphagia, dysphonia, and muscle weakness. The familial context was in favor of a dominant disease since both her mother and her 2 aunts also presented with a bilateral ptosis and dysphagia. The EMG disclosed a myogenic pattern in the lower limb muscles. At the age of 50 years, she noticed an exacerbation of muscle weakness of the 4 limbs, predominantly in the lower limbs, since the patient was unable to kneel.
Correlation between mean age at diagnosis and number of repeats.
With the large size of our cohort (n = 354), we were able to search for a possible correlation between the number of repeats and the mean age at diagnosis, as an indicator of disease severity. The mean age at diagnosis for the different groups of heterozygous patients decreased gradually from 72 years for (GCN)11 down to 53 years for (GCN)17 with a high dispersion on each genotype. Statistical analysis done on the mean age at diagnosis for these heterozygous patients clearly showed a strong negative correlation between the length of the triplet expansion and the mean age at diagnosis (linear regression, r2 = 0.9148, p = 0.0007, figure 2). The group of 9 compound heterozygous carriers of the (GCN)11/(GCN)12 repeats was genetically diagnosed at a mean age of 58 years, which was comparable to the mean age of diagnosis of heterozygous (GCN)14–15 patients (61 and 60 years, respectively). The analysis of the 13 homozygous patients further illustrated a correlation between expansion size and disease severity: homozygous (GCN)11 mean age at diagnosis was 73 years; (GCN)12, 49 years; and (GCN)13, 37 years.
Figure 2. Correlation between mean age at diagnosis and number of repeats.

A significant correlation (r2 = 0.9148; p = 0.0007) was found between the mean age at diagnosis and the number of repeats for heterozygous patients with oculopharyngeal muscular dystrophy. Homozygous and compounds heterozygous patients are also indicated. Red indicates heterozygous patients, green compound heterozygous patients, and blue homozygous patients. Each patient is represented by a black diamond. Mean and SEM are indicated for each genotype. Linear regression of the mean age at diagnosis for the heterozygous compounds is represented on the graph and was obtained using Prism GraphPad software. The relationship between mean age at diagnosis and number of repeats is indicated on the bottom of the graph. For clarity, genotype (GCN)x/(GCN)y is indicated as x/y on the horizontal axis.
DISCUSSION
Among the triplet expansion diseases, OPMD is one of the few caused by a very short and meiotically stable trinucleotide-repeat expansion with no anticipation observed within families.2 It has been difficult to establish phenotype–genotype correlations because of the rare nature of this disease, particularly for the rarest genotypes. Prior to this study, this important issue had not been confirmed, probably due to the relatively small size of genotyped cohorts ranging from 17 to 86 patients.4–11 Nevertheless, it seemed that phenotypes including early onset, severe ptosis and dysphagia, proximal muscle weakness, and loss of ambulation were observed in patients with the largest PABPN1 heterozygous expansion repeats, as well as in compound heterozygous and homozygous patients.2,13,15,16 Cognitive decline and psychological disorders were described in homozygous patients17 and in 2 heterozygous patients harboring 18 expansions.13 In addition, a negative correlation between repeat size and neuropsychological scores was observed in 11 heterozygous patients with OPMD.18 Our large cohort of 354 unrelated patients with OPMD allowed us to determine the distribution of the expanded alleles in France and more importantly to demonstrate that there is a negative correlation between the mean age at diagnosis and the number of repeats. Within each genotype, the ages at diagnosis were dispersed (with an SD ranging from 1 to 11 years) but the large number of patients in this cohort allowed us to have an accurate determination of the mean age at diagnosis for each group. The linear regression of these means highlighted that longer repeat expansions resulted in earlier diagnosis in OPMD. Because of the insidious nature of the disease, it is often difficult to pinpoint an accurate dating of disease onset, thus, we choose to indicate the age at diagnosis, which was available for all 354 patients. When available, the delay between onset and diagnosis of OMPD ranged usually from 3 to 6 years. These ages at diagnosis and onset are directly related to the severity of the clinical symptoms. Among the 900 patients referred to our center from 1999 to 2014 for PABPN1 genotyping, only patients who displayed the typical features of OPMD with dysphagia or ptosis were genetically proven to carry expanded (GCN)n repeats. The presence of proximal limb weakness was a more variable feature depending on the age of patients and the size of the expansion. Patients in whom the genotyping failed to detect any PABPN1 triplet expansion often display atypical features and this request of genotyping was often an exclusion diagnosis before considering a mitochondrial myopathy. In the vast majority of patients, dysphagia preceded or occurred simultaneously with ptosis, and proximal limb muscle weakness tended to follow. In patients at the beginning of the disease, with only ocular and pharyngeal symptoms, EMG examination showed no abnormal pattern. When motor weakness was observed, EMG appeared myogenic. When motor weakness was mild, myogenic signs were focalized on proximal muscles, whereas in severe motor weakness cases, both proximal and distal muscles were affected. In our patients, no obvious cognitive impairments were observed, but this aspect should be more extensively explored in the future with specific tests.18 CK levels were normal in patients with a repeat number <14, while this level increased with the number of repeats and in homozygote patients. This observation further reinforced the correlation between the severity of the disease and the number of repeat expansions. An interesting finding concerned the (GCN)11 allele, which was initially described as a polymorphism8,9 then as a recessive allele19,20 and finally recently as a dominant allele in our previous case report.3 In 1998, it was suggested that 20% of individuals with more severe dominant OPMD have inherited both a dominant allele in the (GCN)12–17 range and a polymorphism in the other PABPN1 allele that causes the insertion of one extra GCN triplet, thus producing the (GCN)11 recessive allele.2 Here examination of patients carrying this (GCN)11 allele led us to suggest that this allele is both associated with a mild phenotype in heterozygous patients with the normal allele in trans and also considered as a disease-modifying allele by worsening the phenotype when associated with another expanded allele such as allele (GCN)12. This was illustrated by the mean age at diagnosis in compound heterozygous patients (GCN)11/12, which was the same as in patients carrying the (GCN)10/(GCN)14 or (GCN)10/15 alleles and significantly lower than in (GCN)10/(GCN)12 patients. Simple heterozygous patients frequently had no ptosis but presented with bulbar signs (that could be attributed to age) leading to an underdiagnosis or misdiagnosis of these heterozygous patients. The risk of misdiagnosis with a mitochondrial myopathy is high. Such a difficult situation requires an adapted strategy consisting of a genotyping test for PAPBN1 expansion and a muscle biopsy. A mitochondrial accumulation may be confusing, since mitochondrial dysfunction is frequently found in OPMD.21 The hallmark of OPMD being the presence of mutated PABPN1 aggregated in the form of insoluble filamentous intranuclear inclusions in skeletal muscle fibers,22 the proper OPMD diagnosis will be ascertained by detection of these inclusions by electron microscopy or immunostaining.3,22,23 The underdiagnosis of patients with the (GCN)11 allele is demonstrated by the fact that this allele is underrepresented at the heterozygous status in our cohort (only 6 heterozygous patients [CGN]10/[CGN]11) as compared with homozygous patients (9 homozygous patients [CGN]11/[CGN]11), whereas testing of 200 normal chromosomes failed to detect it in the French population. A study of 33 patients with OPMD from France concluded that the geographic dispersion of OPMD did not suggest the existence of a recent founder effect,24 in contrast to what is observed in the French-Canadian community.25 The distribution of repeat lengths in our cohort and the large prevalence of the (GCN)13 allele suggests that it is likely that there is a common founder effect despite the dispersion of patients on the territorial area, although this hypothesis should be confirmed by haplotype analysis of all the (GCN)10/(GCN)13 patients.
This large cohort allowed us to demonstrate that in heterozygous and homozygous patients with OPMD, the mean age at diagnosis and the severity of the clinical symptoms correlate to the number of (GCN) repeats. Homozygous patients showed the worse phenotype, suggesting a gene–dose effect in addition to the repeat number expansion. Altogether this suggests that the determination of the size of the triplet expansion is an important test to perform in patients with OPMD, notably in aged patients with bulbar signs, after exclusion of autoimmune myasthenia gravis and amyotrophic lateral sclerosis.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the neurologists who referred patients with OPMD for genetic analysis, the patients and their families, and Laurence Demay and Valérie Jobic for their help with genotyping of patients.
GLOSSARY
- CK
creatine kinase
- OPMD
oculopharyngeal muscular dystrophy
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
P.R., C.T., and B.E. conceived and designed the study. P.R., C.T., T.S., A.d.B., S.P., J.P., and B.E. acquired and analyzed the data. P.R., C.T., T.S., A.d.B., S.P., J.P., and B.E. wrote the manuscript and figures. The following neurologists of French Neuromuscular Reference Centers CORNEMUS and FILNEMUS contributed to data acquisition: Pascal Laforêt, Anthony Behin, Aleksandra Nadaj-Pakleza, Rémi Bellance, Nina Canki, Arnaud Lacour, Claude Desnuelle, Françoise Chapon, Philippe Petiot, Pascal Cintas, Andoni Urtizberea, Emmanuelle Campana-Salort, Xavier Ferrer-Monasterio, and Jean Lacau St Guily.
STUDY FUNDING
This work was supported by the Assistance Publique -Hôpitaux de Paris (APHP), the Centre National pour la Recherche Scientifique (CNRS), the Association Française contre les Myopathies (AFM), the University Paris VI Pierre et Marie Curie (UPMC), and the Institut National de la Santé et de la Recherche Médicale (INSERM).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bouchard JP, Brais B, Brunet D, Gould PV, Rouleau GA. Recent studies on oculopharyngeal muscular dystrophy in Quebec. Neuromuscul Disord 1997;7(1 suppl):S22–S29. [DOI] [PubMed] [Google Scholar]
- 2.Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 1998;18:164–167. [DOI] [PubMed] [Google Scholar]
- 3.Richard P, Trollet C, Gidaro T, et al. PABPN1 (GCN)11 as a dominant allele in oculopharyngeal muscular dystrophy: consequences in clinical diagnosis and genetic counselling. J Neuromuscul Dis 2015;2:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tondo M, Gamez J, Gutierrez-Rivas E, Medel-Jimenez R, Martorell L. Genotype and phenotype study of 34 Spanish patients diagnosed with oculopharyngeal muscular dystrophy. J Neurol 2012;259:1546–1552. [DOI] [PubMed] [Google Scholar]
- 5.Shan J, Chen B, Lin P, et al. Oculopharyngeal muscular dystrophy: phenotypic and genotypic studies in a Chinese population. Neuromolecular Med 2014;16:782–786. [DOI] [PubMed] [Google Scholar]
- 6.Robinson DO, Hammans SR, Read SP, Sillibourne J. Oculopharyngeal muscular dystrophy (OPMD): analysis of the PABPN1 gene expansion sequence in 86 patients reveals 13 different expansion types and further evidence for unequal recombination as the mutational mechanism. Hum Genet 2005;116:267–271. [DOI] [PubMed] [Google Scholar]
- 7.Muller T, Deschauer M, Kolbe-Fehr F, Zierz S. Genetic heterogeneity in 30 German patients with oculopharyngeal muscular dystrophy. J Neurol 2006;253:892–895. [DOI] [PubMed] [Google Scholar]
- 8.Mirabella M, Silvestri G, de Rosa G, et al. GCG genetic expansions in Italian patients with oculopharyngeal muscular dystrophy. Neurology 2000;54:608–614. [DOI] [PubMed] [Google Scholar]
- 9.Hill ME, Creed GA, McMullan TF, et al. Oculopharyngeal muscular dystrophy: phenotypic and genotypic studies in a UK population. Brain 2001;124:522–526. [DOI] [PubMed] [Google Scholar]
- 10.Becher MW, Morrison L, Davis LE, et al. Oculopharyngeal muscular dystrophy in Hispanic New Mexicans. JAMA 2001;286:2437–2440. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal PK, Mansfield DC, Mechan D, et al. Delayed diagnosis of oculopharyngeal muscular dystrophy in Scotland. Br J Ophthalmol 2012;96:281–283. [DOI] [PubMed] [Google Scholar]
- 12.You P, Ma Q, Tao T. Gene diagnosis of oculopharyngeal muscular dystrophy in a Chinese family by a GeneScan method. J Clin Lab Anal 2010;24:422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jouan L, Rocheford D, Szuto A, et al. An 18 alanine repeat in a severe form of oculopharyngeal muscular dystrophy. Can J Neurol Sci 2014;41:508–511. [DOI] [PubMed] [Google Scholar]
- 14.Pulkes T, Papsing C, Busabaratana M, Dejthevaporn C, Witoonpanich R. Mutation and haplotype analysis of oculopharyngeal muscular dystrophy in Thai patients. J Clin Neurosci 2011;18:674–677. [DOI] [PubMed] [Google Scholar]
- 15.Blumen SC, Sadeh M, Korczyn AD, et al. Intranuclear inclusions in oculopharyngeal muscular dystrophy among Bukhara Jews. Neurology 1996;46:1324–1328. [DOI] [PubMed] [Google Scholar]
- 16.Blumen SC, Brais B, Korczyn AD, et al. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann Neurol 1999;46:115–118. [PubMed] [Google Scholar]
- 17.Blumen SC, Bouchard JP, Brais B, et al. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology 2009;73:596–601. [DOI] [PubMed] [Google Scholar]
- 18.Dubbioso R, Moretta P, Manganelli F, et al. Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophy. J Neurol 2012;259:833–837. [DOI] [PubMed] [Google Scholar]
- 19.Semmler A, Kress W, Vielhaber S, Schroder R, Kornblum C. Variability of the recessive oculopharyngeal muscular dystrophy phenotype. Muscle Nerve 2007;35:681–684. [DOI] [PubMed] [Google Scholar]
- 20.Garibaldi M, Pennisi EM, Bruttini M, et al. Dropped-head in recessive oculopharyngeal muscular dystrophy. Neuromuscul Disord 2015;25:869–872. [DOI] [PubMed] [Google Scholar]
- 21.Chartier A, Klein P, Pierson S, et al. Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis. PLoS Genet 2015;11:e1005092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tome FM, Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol 1980;49:85–87. [DOI] [PubMed] [Google Scholar]
- 23.Gidaro T, Negroni E, Perie S, et al. Atrophy, fibrosis, and increased PAX7-positive cells in pharyngeal muscles of oculopharyngeal muscular dystrophy patients. J Neuropathol Exp Neurol 2013;72:234–243. [DOI] [PubMed] [Google Scholar]
- 24.Brunet G, Tome FM, Eymard B, Robert JM, Fardeau M. Genealogical study of oculopharyngeal muscular dystrophy in France. Neuromuscul Disord 1997;7(1 suppl):S34–S37. [DOI] [PubMed] [Google Scholar]
- 25.Scriver CR. Human genetics: lessons from Quebec populations. Annu Rev Genomics Hum Genet 2001;2:69–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.