

Abstract
Design, synthesis, and evaluation of α-methylene-γ-butyrolactone analogues and their evaluation as anticancer agents is described. SAR identified a spirocyclic analogue 19 that inhibited TNFα-induced NF-κB activity, cancer cell growth and tumor growth in an ovarian cancer model. A second iteration of synthesis and screening identified 29 which inhibited cancer cell growth with low-μM potency. Our data suggest that an isatin-derived spirocyclic α-methylene-γ-butyrolactone is a suitable core for optimization to identify novel anticancer agents.
INTRODUCTION
Twenty-one percent of currently marketed covalent drugs are used to treat cancer and several new covalent inhibitors are in clinical trials, suggesting the development of irreversible inhibitors as cancer therapeutics is making a strong comeback.1–3 Covalent inhibitors were not fully explored as their perceived risk-reward ratio was heavily biased by concerns regarding their potential idiosyncratic effects. The resurgence of covalent inhibitors as cancer therapeutics can be attributed to the successful development of currently marketed irreversible inhibitors of enzyme active sites.1 The current strategy for the development of covalent drugs for targeting oncogenic kinases is to append an electrophilic group to a reversible inhibitor. This electrophilic group on the reversible inhibitor then forms a covalent bond with the sulfhydryl group of a noncatalytic cysteine residue peripheral to the kinase active site.4 Here we report a biased approach for the identification of covalent inhibitors and their evaluation as anticancer agents. Nuclear factor kappa B (NF-κB) is a transcription factor that plays a key role in innate and adaptive immune responses, inflammation, cell growth, and apoptosis.5 In unstimulated cells, NF-κB is sequestered in the cytoplasm by its inhibitor, inhibitor of nuclear factor κB (IκBα). Upon stimulation with proinflammatory cytokines such as tumor necrosis factor alpha (TNFα), IκBα is phosphorylated by the IκB kinase β (IKKβ), ubiquitinated, and rapidly degraded, allowing NF-κB dimers to translocate to the nucleus and activate transcription.6 Immunohistochemistry (IHC) studies conducted with surgically resected tumor samples show that TNFα was found in ~50% of tumors, suggesting that the NF-κB pathway is constitutively activated in a variety of cancers including pancreatic, breast, and ovarian cancers and has been shown to contribute to proliferation, tumor progression, and chemoresistance.7
The key proteins in this pathway, i.e., kinase IKKβ and the transcription factor NF-κB, have surface exposed cysteine residues. Cys179 found in the activation loop of IKKβ is primed for targeting as it is between the serine residues 177 and 181. Phosphorylation of Ser177 and Ser181 results in the activation of IKKβ.8 Cys38 in NF-κB (p65 subunit) plays an important role in its translocation to the nucleus to activate gene expression.9 The sulfhydryl groups on Cys179 of IKKβ and Cys38 of NF-κB have been previously targeted using parthenolide, a sesquiterpene lactone natural product.10,11 In a cell-based assay, we recently showed that parthenolide inhibits TNFα-induced IKKβ-mediated NF-κB activity with low μM potency.12
Natural products with the α-methylene-γ-butyrolactone functionality exhibit a wide-range of biological activities including anticancer and anti-inflammatory effects.13–17 The available SAR with parthenolide analogues showed that the Michael acceptor in the α-methylene-γ-butyrolactone is critical for activity against the NF-κB pathway.11 The Colby lab synthesized fluorinated amino derivatives of parthenolide and screened them for antiproliferative activities.18,19 More recently, the Crooks lab generated a series of parthenolide and melampomagnolide-B analogues and screened them against a panel of 60 human cancer cell lines.20–22 The α-methylene-γ- butyrolactone functionality was appended to small molecules to covalently link them to their biological target.23,24 These compounds with α-methylene-γ-butyrolactone also show anticancer activities.25–27 In the studies presented here, we have expanded on this general theme via synthesis of α- methylene-γ-butyrolactone containing analogues and screened them to identify pathway specific inhibitors. Multiple proteins in the NF-κB pathway have surface exposed cysteine residues; therefore, we screened our analogues in a TNFα-induced IKKβ-mediated NF-κB reporter assay to identify covalent pathway specific inhibitors.
This exercise led to the identification of an isatin derived spirocyclic core with an α-methylene-γ-butyrolactone moiety (19) that inhibits the NF-κB pathway by covalently binding to IKKβ and NF-κB. This is the first report that identified a compound with spirocyclic α-methylene-γ-butyrolactone moiety as a NF-κB inhibitor. Analogue 19 inhibits cancer cell growth in vitro and tumor growth in an orthotopic ovarian cancer model. Analogue 19 is ~4-fold more stable in serum albumin when compared to parthenolide.
To explore this further, we generated seven analogues with substitutions at different positions on the isatin-derived spirocyclic core and evaluated their ability to inhibit cancer cell growth. This led to identification of analogue 29 with low μM potency and ~2–20-fold more active than parthenolide in a panel of cancer cell lines.
RESULTS AND DISCUSSION
We generated a biased library that features the α-methylene-γ- butyrolactone functionality using a reported two-step synthesis (Scheme 1)28 and screened analogues at 10 μM (Figure 1B) in a cell-based luciferase assay (A549) that specifically reports on the ability of the compound to inhibit TNFα-induced IKKβ- mediated NF-κB activity.12 ML-120B, a well-characterized IKKβ inhibitor, was used as a positive control.29
Scheme 1. Synthesis of α-Methylene-γ-butyrolactoneContaining Spiroisatin Analogue 20a.
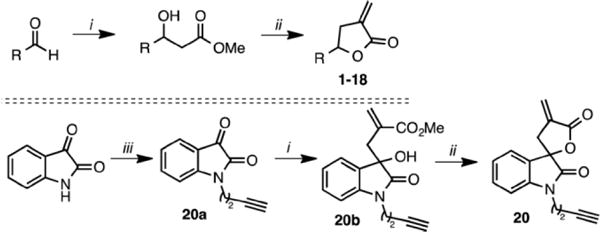
a(i) Methyl- 2-(bromomethyl)acrylate, In powder, NH4Cl, MeOH, 50 °C, 1 h; (ii) PTSA, CH2Cl2, 12 h; (iii) 4-bromo-1-butyne, Cs2CO3, CH3CN, 70 °C, 12 h.
Figure 1.
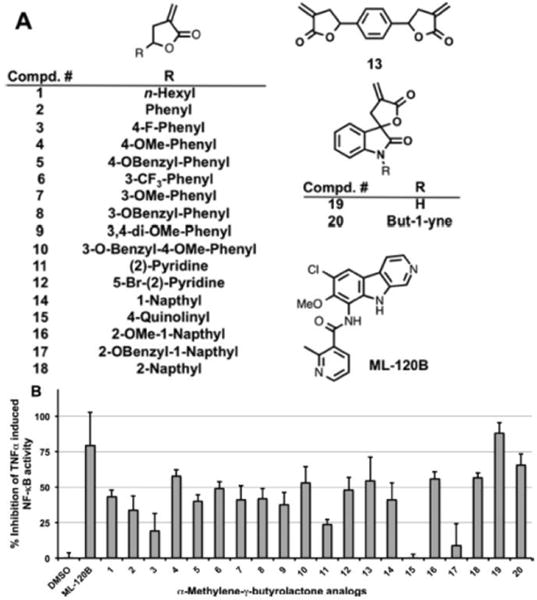
(A) Focused library of α-methylene-γ-butyrolactone analogues and the IKKβ inhibitor ML-120B. (B) Cell-based screen (A549) that reports on the ability of the inhibitors to specifically block TNFα-induced IKKβ-mediated NF-κB activity.
The unsubstituted, mono- and disubstituted phenyl compounds (2–10) had modest activity (25–50% inhibition) with no clear SAR. Pyridine substitution (11) resulted in decreased activity (<25% inhibition), while a bulky bromo group ortho to the nitrogen in 12 resulted in the recovery of activity (~50% inhibition). Interestingly, introducing a second α-methylene-γ- butyrolactone in 13 did not increase the activity. In the bicyclic fused aryl ring systems, unsubstituted 1- and 2-naphthyl substituted analogues (14 and 18) had modest activity (~50% inhibition) while the 4-quinolinyl substitution resulted in an inactive compound (15). The methoxy substitution at the 2-position of 1-naphthyl analogue (16) was tolerated, however a bigger benzyloxy substitution at the same position (17) resulted in reduced activity (<25% inhibition). The spirocyclic analogues (19 and 20), in which the α-methylene-γ- butyrolactone is rigid, had activities (~75% inhibition) comparable to the ML-120B, indicating that rigidification of the Michael acceptor is a favorable feature.
To determine if the Michael acceptor is critical for activity, we tested the reduced spirocyclic compound that lacks the Michael acceptor (21) and compared it with parthenolide and reduced parthenolide analogue (Red-P) (Figure 2A). At 10 μM, analogue 19 showed good activity (>70% inhibition) while reduced analogue 21 was inactive (~5% inhibition), demonstrating that the Michael acceptor in 19 is critical for NF-κB inhibitory activity. Parthenolide showed remarkable activity (>95% inhibition), while Red-P was ~4 fold less active (~20% inhibition) (Figure 2B).11 To confirm whether rigidification indeed resulted in increased NF-κB inhibition, the isatin derived acyclic analogue (22) was screened under identical conditions. Acyclic analogue 22 was ~2-fold less active than the cyclized version (19), suggesting that rigidification indeed increases activity against the NF-κB pathway proteins. In a dose–response study, analogue 19 had low μM (IC50 = 4 μM) inhibitory activity in the TNFα-induced IKKβ-mediated NF-κB activity assay (Figure 2C), which is comparable to parthenolide (IC50 = 4.7 ± 1.5 μM).12 To summarize, our synthesis and screening effort identified an isatin derived spirocyclic compound with the α-methylene-γ-butyrolactone as a potent NF-κB inhibitor. The acyclic analogue 22 adopts multiple conformations when compared to the rigidified analogue 19 and therefore could bind to additional targets, which explains the increased NF-κB inhibitory activity observed with 19.
Figure 2.
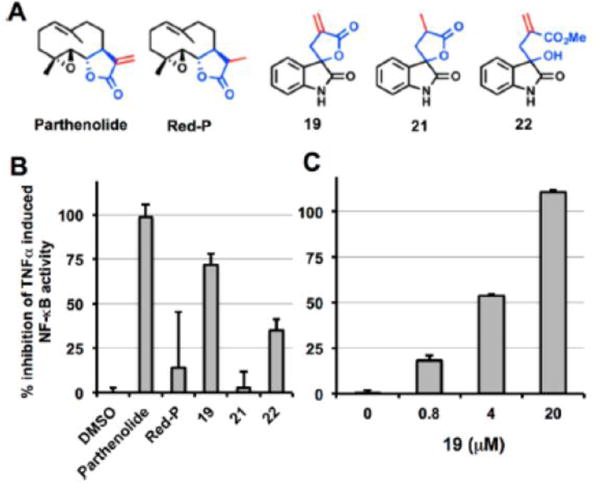
(A) Structure of parthenolide, reduced parthenolide, and isatin analogues. (B) Evaluation of inhibitors in TNFα-induced NF-κB activity assay. (C) Dose–response study with analogue 19.
One of the limitations with the use of parthenolide is the short half-life in serum. A recent study characterized the covalent binding of parthenolide through the α-methylene-γ- butyrolactone to the free cysteine34 residue of serum albumin by MS analyses. They also determined the half-life of parthenolide to be ~37 min.30 To determine the stability of our analogue 19, we conducted a head-to-head comparison of analogue 19 and parthenolide for serum albumin binding using HPLC (Supporting Information (SI) Figure S1). Our results show that half-life of 19 is 290 min as compared to 66 min for parthenolide, indicating the spirocyclic disposition in 19 as opposed to a fused disposition in parthenolide could contribute to the observed increased serum stability.
Next, we used click chemistry31 to determine if 19 indeed irreversibly binds to IKKβ and NF-κB. Recombinant IKKβ and NF-κB (p65) were incubated with analogue 20, which is analogue 19 with an alkyne linker, for 1 h at room temperature. Rhodamine azide and click reagents were added to the reaction mixture and incubated for an additional hour. At the end of the second hour, the mixture was subjected to SDS PAGE and the gel was imaged using the Typhoon 9410 variable mode imager, an imager that produces digital images of fluorescent samples. The data summarized in Figure 3A shows fluorescent bands at molecular weights that correspond to IKKβ and NF-κB (p65 protein truncated at C-terminus and has L159V, P180S, F309S, A439V, and V462 M mutations runs at ~50 kDa, accession no. AAA36408), demonstrating that analogue 20 is a covalent inhibitor of IKKβ and NF-κB. To determine if this is a Cys adduct, we conducted a HPLC study wherein Boc protected Cys or Lys amino acids were incubated with analogue 19 (data not shown). Our results showed that analogue 19 reacts with only Cys and not Lys, suggesting a Cys adduct.
Figure 3.
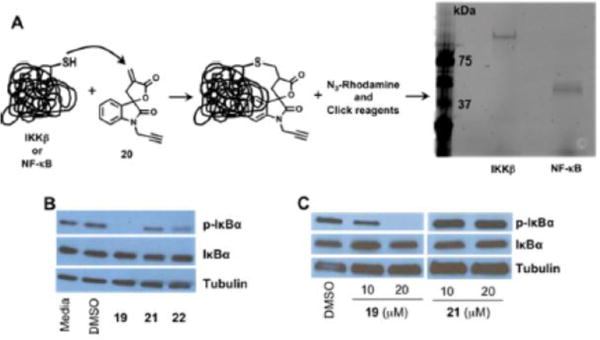
(A) Schematic of click chemistry used to demonstrate covalent binding (left) and covalent binding of 20 to IKKβ and p65 via click chemistry (right). (B) Inhibition of IKKβ kinase activity determined by Western blot analyses in MDA-MB-231 cells and (C) MiaPaCa2 cells.
To determine if covalent binding to IKKβ results in the inhibition of the kinase activity of IKKβ, cancer cells were incubated with analogues 19, 21, and 22 for 2 h. The cells were lysed and the proteins were separated on SDS PAGE, transferred to a membrane, and probed with total and phosphospecific IκBα antibodies. Because IκBα is a substrate of IKKβ, inhibition of the kinase activity of IKKβ should result in reduced IκBα phosphorylation. Indeed, we observed a complete inhibition of IκBα phosphorylation in cells treated with 19 but no inhibition of IκBα phosphorylation in cells treated with the reduced compound 21 that lacks the Michael acceptor (Figure 3B,C). Interestingly the acyclic analogue 22 showed partial inhibitory activity (Figure 3B), which indicates that rigidification of the Michael acceptor results in increased inhibition of IKKβ.
To determine if the IKKβ-NFκB inhibitory activity translatesto anticancer activity, we subjected a panel of cancer cell lines to 19, 21, and three previously reported IKKβ inhibitors 13–197,32 Bayer VIII, and TPCA1.33–36 Analogue 19 showed dose and time-dependent inhibition of cancer cell growth while analogue 21 did not (Figure 4A,B), further demonstrating that the Michael acceptor is critical for anticancer activity. Importantly, the growth inhibitory activity of 19 was comparable to known IKKβ inhibitors (SI Table S1).
Figure 4.
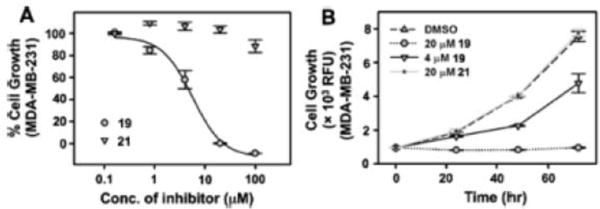
Dose-dependent (A) and time-dependent (B) effects on the growth of breast cancer cells by analogues 19 and 21. Cell viability was assessed using PrestoBlue dye after 3 days of treatment.
The ability of analogues 19 and 21 to inhibit colony formation was accessed using a clonogenic assay. Cells were sparsely plated and allowed to grow in the presence or absence of 19 or 21 for 7 days. Colonies were then stained using crystal violet and quantified (Figure 5A). In the plates treated with 19, we observed a dose-dependent decrease in the number of colonies while no such effect was observed with 21. This demonstrates that the Michael acceptor functionality is critical for 19 to inhibit colony formation.
Figure 5.
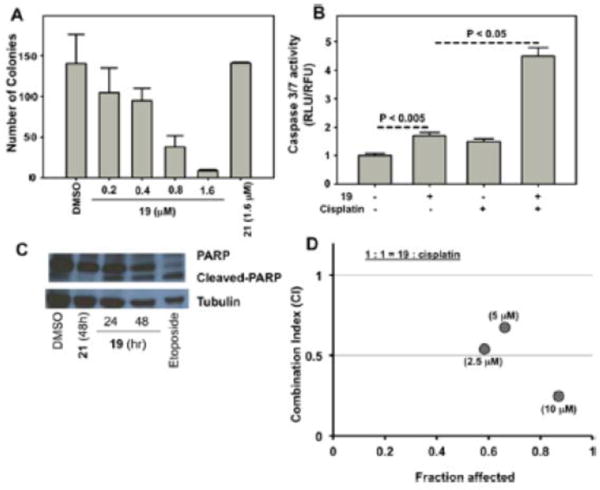
(A) Dose-dependent effects on colony formation of HeLa cells by analogues 19 and 21. (B) Caspase 3/7 activity induced by 19, cisplatin, and the combination in SKOV3 cells. (C) Effects of 19, 21, and Etoposide (positive control) on PARP cleavage in HeLa cells. (D) Combination index (CI) vs fraction affected plot, demonstrating synergistic growth inhibition with 19 and cisplatin. *p < 0.05 vs control.
Inhibition of the NF-κB pathway has been explored as a therapeutic strategy to sensitize cancers to current chemotherapeutics.37 The NF-κB inhibitor BAY 11–7085 that targets the NF-κB pathway proteins through covalent inhibition via its Michael acceptor sensitizes ovarian cancer cells to cisplatininduced apoptosis.38 Two key events during apoptosis are the activation of caspase 3/7 and poly(ADP-ribose) polymerase (PARP) cleavage. In cells treated with 19, we observed modest induction of caspase 3/7 activity by itself and synergistic induction in the presence of cisplatin (Figure 5B). This is consistent with reports that implicate activation of NF-κB in chemoresistance.39 We also observed decreased/cleaved PARP in cells treated with 19. Importantly, no such effects with 21 treated cells (Figure 5C and SI Figure S2) were observed. To determine if the observed synergistic induction of caspase 3/7 leads to growth inhibition, we subjected ovarian cancer cells to either 19 or cisplatin alone and their combination and monitored their effects on the cancer cell growth over a 3- day period (Figure 5D). The combination index (CI) values for the various combinations were derived from median effect plot and dose effect curves (SI Figure S3) using calcusyn (biosoft.com). CI < 1 indicates synergism, CI = 1 indicates additive effects, and CI > 1 indicates antagonism.40 Concentration combinations of 19 and cisplatin in the 1–10 μM range had CI < 1, indicating synergistic inhibition of ovarian cancer cell growth (Figure 5D). These studies demonstrate that 19 sensitizes cancer cells to cisplatin induced apoptosis and demonstrates synergism in the low μM ranges with cisplatin toward the inhibition of ovarian cancer cell growth.
Our in vitro studies clearly demonstrate the anticancer effects induced by 19 are dependent on the presence of the Michael acceptor. We next investigated if these effects translate to an ovarian cancer mouse model.41
Our first goal was to determine if analogue 19 has anticancer activity as a single agent and to define an optimal dose for both cisplatin and 19 in an orthotopic ovarian cancer model.42,43 Ovarian cancer cells (A2780) were injected into the peritoneal cavity of nude mice, and the tumors were allowed to establish. On day 3, mice were divided into four groups and the groups were treated with vehicle, 1, 2.5, or 5 mg/kg of 19, the dose and route of administration were selected based on literature reports with parthenolide.44–46 The mice were treated intraperitoneally 5 days a week for 4 weeks. At the end of the study, the mice were sacrificed and the tumor weights determined. None of the mice treated with 19 showed any overt toxicity. A dose-dependent effect on tumor growth (~10–40%) was observed in mice treated with 19. There was a ~40% reduction in tumor weights in mice treated with 19 at the highest dose (5 mg/kg). The ~10% reduction in tumor growth at 1 mg/kg is consistent with the effects observed with parthenolide at an equivalent dose in the prophylactic metastasis study.45 Consistent with our previous studies, we observed a ~ 25% reduction of tumor weights in mice treated with cisplatin (2 mg/kg) when compared to the controls (Figure 6A).41
Figure 6.
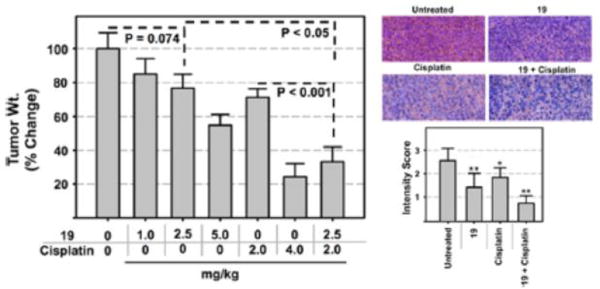
Dose-dependent effects on ovarian (A2780) tumor growth by analogue 19, cisplatin, and the combination in an orthotopic model of ovarian cancer (left) and NF-κB(p65) staining of the excised tumors (right top) and quantification (right bottom) (n = 10). **p < 0.01, *p < 0.05.
To determine if 19 can chemosensitize ovarian tumors to cisplatin, we performed a follow up study in which mice were treated with 2.5 mg/kg of 19 (resulted in ~21% reduction in tumor growth bar no. 3 Figure 6A) and 2 mg/kg of cisplatin (resulted in ~30% reduction in tumor growth bar no. 5 Figure 6A). Therefore, a reduction of >51% in tumor growth with the combination of 2.5 mg/kg of 19 and 2.0 mg/kg of cisplatin will indicate synergism. Indeed, the combination was synergistic with ~65% (bar no. 7 Figure 6A) reduction in tumor weights compared to the controls. The reduction of tumor weights by treatment with 19 + cisplatin is significant compared to treatment with either drug alone (One-Way ANOVA, P < 0.001). These studies clearly demonstrate that analogue 19 has anticancer activity as a single agent and also demonstrates the ability to chemosensitize ovarian tumors to cisplatin (Figure 6A).
To determine if 19 affects NF-κB (p65) levels and NF-κB regulated proteins (Mcl-1) in the tumors, we conducted IHC studies with p65 and Mcl-1 antibodies with the excised tumor tissue. IHC studies showed reduced p65 staining in tumors of animals treated with 19 and cisplatin individually and the combination (Figure 6B). Similar effects were observed in Mcl- 1 levels (SI Figure S4). These studies suggest that the antitumor effects of 19 in vivo are mediated by the inhibition of the NF-κB pathway.
Because in vitro and in vivo studies clearly demonstrate the anticancer effects of 19, we functionalized the spirocyclic oxindole core with substitutions at various positions to generate seven additional analogues (Figure 7). We screened these for inhibition of cancer cell growth in A2780 cells (Table 1). In this cell line, we observed a ~4-fold higher potency in cell growth inhibition with 19 compared to parthenolide which correlates with ~4-fold higher serum stability.
Figure 7.
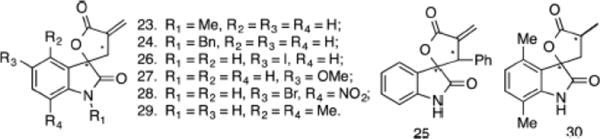
Focused library of substituted α-methylene-γ-butyrolactone–oxindole analogues.
Table 1.
Inhibition of A2780 cancer Cell Growth by Substituted α-Methylene-γ-butyrolactone–Oxindoles
| analogues | EC50 values (μΜ) | analogues | EC50 values (μΜ) |
|---|---|---|---|
| 20 | 5.5 ± 0.58 | 19 | 2.3 ± 1.4 |
| 23 | 3.9 ± 0.45 | 29 | 1.0 ± 0.16 |
| 24 | 2.7 ± 023 | 30 | >100 |
| 25 | 3.8 ± 0.21 | ibrutinib | 20.1 ± 1.26 |
| 26 | 2.2 ± 0.24 | Red-I | 15.2 ± 1.11 |
| 27 | 3.1 ± 0.32 | parthenolide | 12.9 ± 0.72 |
| 28 | 2.0 ± 0.12 | Red-P | >100 |
Alkylation (20, 23, 24) of the nitrogen atom on the oxindole had modest effects on the growth inhibitory activity. Analogue 25 with a phenyl substitution on the lactone ring did not have a significant effect on the activity. Likewise, substitutions at 4–7 positions (26–29) on the phenyl ring of the oxindole had modest effects when compared to 19 on the growth of A2780 cells. Only methyl substitutions at the both 4- and 7-positions of the oxindole (29) showed >2-fold improvement in the growth inhibitory effects when compared to analogue 19. As expected, the corresponding reduced spirocyclic analogue 30 was inactive (IC50 > 100 μM). Importantly, under our assay conditions, the growth inhibitory activity of 29 was ~13-fold better (IC50 values 1.0 vs 12.9 μM) than parthenolide and ~20- fold better than ibrutinib. Surprisingly, reducing the Michael acceptor in ibrutinib (Red-I) did not alter the growth inhibitory effects. However, reduction of the Michael acceptor in parthenolide resulted in loss of activity (Table 1, SI Figure S5). Although limited in number, this preliminary study clearly demonstrates that the spirocyclic oxindole core with the α- methylene-γ-butyrolactone core can be functionalized to improve biological activity and possible drug-like properties.
We also compared the efficacy of analogue 29, its reduced analogue 30, parthenolide, reduced parthenolide (Red-P), ibrutinib, and reduced ibrutinib (Red-I) in a panel of cancer cell lines (Table 2, SI Figure S6). 29 was ~2–10-fold more potent than parthenolide and ~5–20-fold more potent than ibrutinib. Reduced compounds 30 and Red-P resulted in >10- fold loss of activity. However, reduced ibrutinib (Red-I) showed modest improvement in the growth inhibitory activity when compared to ibrutinib in all the lines. At the present time, we do not have an explanation for the results observed with ibrutinib and its reduced analogue. On the other hand, the cell based activity of both parthenolide and 29 are largely dependent on the presence of the Michael acceptor.
Table 2.
Inhibition of HeLaGFP, MiaPaCa-2, and SW480 Cancer Cell Growth
| inhibitors | IC50 ± SEM (μΜ)
|
||
|---|---|---|---|
| HeLaGFP | MiaPaCa-2 | SW480 | |
| ibrutinib | 16.8 ± 1.7 | 16.6 ± 2.4 | 25.6 ± 0.3 |
| Red-I | 15.1 ± 0.1 | 8.2 ± 1.4 | 13.4 ± 0.2 |
| parthenolide | 5.7 ± 3.3 | 9.8 ± 0.05 | 15.3 ± 0.1 |
| Red-P | 66.1 ± 1.9 | 84.3 ± 11.1 | >100 |
| 29 | 32 ± 0.03 | 0.9 ± 0.1 | 1.2 ± 0.2 |
| 30 | >100 | >100 | >100 |
CONCLUSION
In conclusion, a cell-based pathway screen with a focused library of α-methylene-γ-butyrolactone containing analogues led to the identification of the isatin derived spirocyclic analogue 19 as a potent inhibitor of TNFα-induced IKKβ- mediated NF-κB activation. SAR studies revealed that rigidification of the α-methylene-γ-butyrolactone and the Michael acceptor in the spirocyclic system are critical features required for activity. Changing the context of the α-methylene-γ- butyrolactone from a fused to a spirocyclic system could explain the increased stability of 19 in serum when compared to parthenolide. Using click chemistry, we show that the inhibition of TNFα-induced IKKβ-mediated NF-κB activation is due to covalent binding of 19 to IKKβ and NF-κB. Analogue 19 inhibited the phosphorylation of IκBα in cancer cells and exhibited anticancer activities that was comparable to known IKKβ inhibitors. On the other hand, analogue 21, a reduced form of 19, was inactive in all the assays. Analogue 19 inhibits ovarian tumor growth as a single agent and sensitizes ovarian tumors to cisplatin in an orthotopic model. Our studies clearly demonstrate that α-methylene-γ-butyrolactone containing spirocyclic oxindole analogue 19 phenocopies the effects of natural product parthenolide. A second iteration of synthesis and screening led to the identification of a dimethyl analogue 29, which inhibited cancer cell growth with low μM potency. Depending on cell lines, analogue 29 was ~2–20-fold better than parthenolide. Studies to expand the SAR of α-methylene- γ-butyrolactone containing spirocyclic oxindole analogues for the identification of suitable pretherapeutic lead compounds with anticancer effects are currently underway, and the results from these studies will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported in part by CA182820, CA127297, Nebraska Research Initiative to A.N., CA009476 to E.C.B. (training grant), LB506 to P.R., and DOD to R.R. We thank Ed Ezell for NMR, Krishna Kattel for mass spectrometry, and the Natarajan lab members for helpful discussions. We are grateful for a Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727) for the NMR core facility.
ABBREVIATIONS USED
- SAR
structure–activity relationship
- TNFα
tumor necrosis factor alpha
- NF-κB
nuclear factor kappa B
- IκBα
inhibitor of nuclear factor κB
- IKKβ
IκB kinase β
- PARP
poly(ADP ribose) polymerase
- IHC
immunohistochemistry
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem. 6b00400. Experimental for Western blotting, cell viability assay, kB-luciferase assay, click chemistry, PARP cleavage, caspase 3/7 assay, colony formation assay, mouse studies, immunohistochemistry, synthetic procedures, and NMR spectra (PDF) Molecular formula strings (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 2.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discovery. 2012;7:561–581. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 4.Dou D, Park JG, Rana S, Madden BJ, Jiang H, Pang YP. Novel selective and irreversible mosquito acetylcholinesterase inhibitors for controlling malaria and other mosquito-borne diseases. Sci Rep. 2013;3:1068. doi: 10.1038/srep01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246:239–253. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 8.Byun MS, Choi J, Jue DM. Cysteine-179 of IkappaB kinase beta plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp Mol Med. 2006;38:546–552. doi: 10.1038/emm.2006.64. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Pineres AJ, Lindenmeyer MT, Merfort I. Role of cysteine residues of p65/NF-kappaB on the inhibition by the sesquiterpene lactone parthenolide and N-ethyl maleimide, and on its transactivating potential. Life Sci. 2004;75:841–856. doi: 10.1016/j.lfs.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 10.Govindachari TR, Joshi BS, Kamat VN. Structure of parthenolide Tetrahedron. 1965;21:1509–1519. [Google Scholar]
- 11.Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IkappaB kinase. Chem Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 12.Bryant VC, Kishore Kumar GD, Nyong AM, Natarajan A. Synthesis and evaluation of macrocyclic diarylether heptanoid natural products and their analogs. Bioorg Med Chem Lett. 2012;22:245–248. doi: 10.1016/j.bmcl.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandran PV, Yip-Schneider M, Schmidt CM. Natural and synthetic alpha,beta-unsaturated carbonyls for NF-kappaB inhibition. Future Med Chem. 2009;1:179–200. doi: 10.4155/fmc.09.15. [DOI] [PubMed] [Google Scholar]
- 14.Kempema AM, Widen JC, Hexum JK, Andrews TE, Wang D, Rathe SK, Meece FA, Noble KE, Sachs Z, Largaespada DA, Harki DA. Synthesis and antileukemic activities of C1-C10-modified parthenolide analogues. Bioorg Med Chem. 2015;23:4737–4745. doi: 10.1016/j.bmc.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khazir J, Riley DL, Chashoo G, Mir BA, Liles D, Islam MA, Singh SK, Vishwakarma RA, Pilcher LA. Design, synthesis and anticancer activity of Michael-type thiol adducts of alpha-santonin analogue with exocyclic methylene. Eur J Med Chem. 2015;101:769–779. doi: 10.1016/j.ejmech.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 16.Qin XY, Chen BY, Fu JJ, Shan L, Lei XG, Zhang WD. Synthesis, cytotoxicity and inhibition of NO production of ivangustin enantiomer analogues. Eur J Med Chem. 2015;102:256–265. doi: 10.1016/j.ejmech.2015.07.051. [DOI] [PubMed] [Google Scholar]
- 17.Xu YZ, Gu XY, Peng SJ, Fang JG, Zhang YM, Huang DJ, Chen JJ, Gao K. Design, synthesis and biological evaluation of novel sesquiterpene mustards as potential anticancer agents. Eur J Med Chem. 2015;94:284–297. doi: 10.1016/j.ejmech.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Woods JR, Mo H, Bieberich AA, Alavanja T, Colby DA. Fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide, as (19)f NMR probes in deuterium-free environments. J Med Chem. 2011;54:7934–7941. doi: 10.1021/jm201114t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woods JR, Mo H, Bieberich AA, Alavanja T, Colby DA. Amino-derivatives of the sesquiterpene lactone class of natural products as prodrugs. MedChemComm. 2013;4:27–33. [Google Scholar]
- 20.Janganati V, Penthala NR, Madadi NR, Chen Z, Crooks PA. Anti-cancer activity of carbamate derivatives of melampomagnolide B. Bioorg Med Chem Lett. 2014;24:3499–3502. doi: 10.1016/j.bmcl.2014.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janganati V, Ponder J, Jordan CT, Borrelli MJ, Penthala NR, Crooks PA. Dimers of Melampomagnolide B Exhibit Potent Anticancer Activity against Hematological and Solid Tumor Cells. J Med Chem. 2015;58:8896–8906. doi: 10.1021/acs.jmedchem.5b01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penthala NR, Bommagani S, Janganati V, MacNicol KB, Cragle CE, Madadi NR, Hardy LL, MacNicol AM, Crooks PA. Heck products of parthenolide and melampomagnolide-B as anticancer modulators that modify cell cycle progression. Eur J Med Chem. 2014;85:517–525. doi: 10.1016/j.ejmech.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baraldi PG, Del Carmen Nunez M, Tabrizi MA, De Clercq E, Balzarini J, Bermejo J, Estevez F, Romagnoli R. Design, synthesis, and biological evaluation of hybrid molecules containing alpha-methylene-gamma-butyrolactones and polypyrrole minor groove binders. J Med Chem. 2004;47:2877–2886. doi: 10.1021/jm031104y. [DOI] [PubMed] [Google Scholar]
- 24.Ropp S, Guy J, Berl V, Bischoff P, Lepoittevin JP. Synthesis and photocytotoxic activity of new alpha-methylene-gammabutyrolactone- psoralen heterodimers. Bioorg Med Chem. 2004;12:3619–3625. doi: 10.1016/j.bmc.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Ramachandran PV, Pratihar D, Nair HN, Walters M, Smith S, Yip-Schneider MT, Wu H, Schmidt CM. Tailored alpha-methylene-gamma-butyrolactones and their effects on growth suppression in pancreatic carcinoma cells. Bioorg Med Chem Lett. 2010;20:6620–6623. doi: 10.1016/j.bmcl.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 26.Jun-Tao F, De-Long W, Yong-Ling W, He Y, Xing Z. New antifungal scaffold derived from a natural pharmacophore: synthesis of alpha-methylene-gamma-butyrolactone derivatives and their antifungal activity against Colletotrichum lagenarium. Bioorg Med Chem Lett. 2013;23:4393–4397. doi: 10.1016/j.bmcl.2013.05.073. [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran PV, Nicponski DR, Nair HN, Helppi MA, Gagare PD, Schmidt CM, Yip-Schneider MT. Synthetic alpha-(aminomethyl)-gamma-butyrolactones and their anti-pancreatic cancer activities. Bioorg Med Chem Lett. 2013;23:6911–6914. doi: 10.1016/j.bmcl.2013.09.065. [DOI] [PubMed] [Google Scholar]
- 28.Rana S, Natarajan A. Face selective reduction of the exocyclic double bond in isatin derived spirocyclic lactones. Org Biomol Chem. 2013;11:244–247. doi: 10.1039/c2ob27008k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen D, Nong Y, Morgan JG, Gangurde P, Bielecki A, Dasilva J, Keaveney M, Cheng H, Fraser C, Schopf L, Hepperle M, Harriman G, Jaffee BD, Ocain TD, Xu Y. A selective small molecule IkappaB Kinase beta inhibitor blocks nuclear factor kappaBmediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. J Pharmacol Exp Ther. 2006;317:989–1001. doi: 10.1124/jpet.105.097584. [DOI] [PubMed] [Google Scholar]
- 30.Ploger M, Sendker J, Langer K, Schmidt TJ. Covalent modification of human serum albumin by the natural sesquiterpene lactone parthenolide. Molecules. 2015;20:6211–6223. doi: 10.3390/molecules20046211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bateman LA, Zaro BW, Miller SM, Pratt MR. An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J Am Chem Soc. 2013;135:14568–14573. doi: 10.1021/ja408322b. [DOI] [PubMed] [Google Scholar]
- 32.Chen Q, Bryant VC, Lopez H, Kelly DL, Luo X, Natarajan A. 2,3-Substituted quinoxalin-6-amine analogs as antiproliferatives: a structure-activity relationship study. Bioorg Med Chem Lett. 2011;21:1929–1932. doi: 10.1016/j.bmcl.2011.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajule R, Bryant VC, Lopez H, Luo X, Natarajan A. Perturbing pro-survival proteins using quinoxaline derivatives: a structure-activity relationship study. Bioorg Med Chem. 2012;20:2227–2234. doi: 10.1016/j.bmc.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaturvedi NK, Rajule RN, Shukla A, Radhakrishnan P, Todd GL, Natarajan A, Vose JM, Joshi SS. Novel treatment for mantle cell lymphoma including therapy-resistant tumor by NFkappaB and mTOR dual-targeting approach. Mol Cancer Ther. 2013;12:2006–2017. doi: 10.1158/1535-7163.MCT-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gautam N, Bathena SP, Chen Q, Natarajan A, Alnouti Y. Pharmacokinetics, protein binding and metabolism of a quinoxaline urea analog as an NF-kappaB inhibitor in mice and rats by LC-MS/MS. Biomed Chromatogr. 2013;27:900–909. doi: 10.1002/bmc.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radhakrishnan P, Bryant VC, Blowers EC, Rajule RN, Gautam N, Anwar MM, Mohr AM, Grandgenett PM, Bunt SK, Arnst JL, Lele SM, Alnouti Y, Hollingsworth MA, Natarajan A. Targeting the NF-kappaB and mTOR pathways with a quinoxaline urea analog that inhibits IKKbeta for pancreas cancer therapy. Clin Cancer Res. 2013;19:2025–2035. doi: 10.1158/1078-0432.CCR-12-2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao L, Wu L, Zhou D. Sensitization of tumor cells to cancer therapy by molecularly targeted inhibition of the inhibitor of nuclear factor kappaB kinase. Transl Cancer Res. 2012;1:100–108. doi: 10.3978/j.issn.2218-676X.2012.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K, Murata Y. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279:23477–23485. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- 39.Veiby OP, Read MA. Chemoresistance: impact of nuclear factor (NF)-kappaB inhibition by small interfering RNA. Commentary re J. Guo et al., Enhanced chemosensitivity to irinotecan by RNA interference-mediated down-regulation of the NF-kappaB p65 subunit. Clin Cancer Res. 2004;10:3262–3264. doi: 10.1158/1078-0432.CCR-04-0703. [DOI] [PubMed] [Google Scholar]
- 40.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 41.Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13:483–491. doi: 10.1593/neo.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Athanassiou AE, Bafaloukos D, Pectasides D, Dimitriadis M, Varthalitis I, Barbounis V. First line combination chemotherapy with cisplatin and etoposide in advanced ovarian cancer. Br J Cancer. 1989;60:755–758. doi: 10.1038/bjc.1989.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng F, Sun G, Zhong M, Yu Y, Brewer MA. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br J Cancer. 2013;108:579–586. doi: 10.1038/bjc.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweeney CJ, Mehrotra S, Sadaria MR, Kumar S, Shortle NH, Roman Y, Sheridan C, Campbell RA, Murry DJ, Badve S, Nakshatri H. The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol Cancer Ther. 2005;4:1004–1012. doi: 10.1158/1535-7163.MCT-05-0030. [DOI] [PubMed] [Google Scholar]
- 45.Kishida Y, Yoshikawa H, Myoui A. Parthenolide, a natural inhibitor of Nuclear Factor-kappaB, inhibits lung colonization of murine osteosarcoma cells. Clin Cancer Res. 2007;13:59–67. doi: 10.1158/1078-0432.CCR-06-1559. [DOI] [PubMed] [Google Scholar]
- 46.Curry EA, III, Murry DJ, Yoder C, Fife K, Armstrong V, Nakshatri H, O’Connell M, Sweeney CJ. Phase I dose escalation trial of feverfew with standardized doses of parthenolide in patients with cancer. Invest New Drugs. 2004;22:299–305. doi: 10.1023/B:DRUG.0000026256.38560.be. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.