

Abstract
Electrophilic small molecules are an important class of chemical probes and drugs that produce biological effects by irreversibly modifying proteins. Examples of electrophilic drugs include covalent kinase inhibitors that are used to treat cancer and the multiple sclerosis drug dimethyl fumarate. Optimized covalent drugs typically inactivate their protein targets rapidly in cells, but ensuing time-dependent, off-target protein modification can erode selectivity and diminish the utility of reactive small molecules as chemical probes and therapeutics. Here, we describe an approach to confer kinetic selectivity to electrophilic drugs. We show that an analogue of the covalent Bruton’s tyrosine kinase (BTK) inhibitor Ibrutinib bearing a fumarate ester electrophile is vulnerable to enzymatic metabolism on a time-scale that preserves rapid and sustained BTK inhibition, while thwarting more slowly accumulating off-target reactivity in cell and animal models. These findings demonstrate that metabolically labile electrophilic groups can endow covalent drugs with kinetic selectivity to enable perturbation of proteins and biochemical pathways with greater precision.
TOC image

Covalent small molecules are valuable tools for interrogating biological processes and promising therapeutics for treating human disease.1 By reacting irreversibly with protein targets, covalent small molecules can produce more complete and sustained pharmacological effects compared to traditional reversible compounds.1–3 Covalent small molecule-protein adducts also provide a convenient handle for visualizing and quantifying target engagement and selectivity in biological systems.3–5 Activity-based protein profiling (ABPP) and related chemical proteomic methods have accordingly been utilized to assess the proteome-wide reactivity of electrophilic small molecules, facilitating optimization of on-target activity while minimizing off-target interactions.3
Many electrophilic small molecules act by modifying cysteine residues in proteins, and we, and others, have shown that broad-spectrum cysteine-reactive chemical probes can be used to globally map the targets of such electrophilic drugs in native biological systems.6 Chemical proteomic studies have also revealed that electrophilic drugs often react rapidly with their intended targets in cells, but then show substantial time-dependent increases in proteome-wide reactivity.4 Minimizing this cross-reactivity, which can confound the interpretation of drug action in biological systems and jeopardize drug safety in humans,1 presents a major challenge. One potential solution is the use of hyper-electrophilic drugs that bind to proteins in a covalent, reversible manner.7 Here, we describe an alternative and complementary strategy that achieves kinetic selectivity, where irreversible on-target engagement is preserved and time-dependent proteomic cross-reactivity minimized by endowing covalent small molecules with metabolically labile electrophilic groups.
We recently generated a chemical proteomic map of cysteine residues targeted by the immunomodulatory drug dimethyl fumarate (DMF) in human T cells.8a In this study, we found that the hydrolytic product of DMF – monomethyl fumarate – showed negligible reactivity with proteinaceous cysteines. A methyl fumarate-bearing analog of the opioid receptor antagonist naltrexone has also been shown to be thiol-reactive.8b We were inspired by these results to consider the fumarate ester as a metabolically labile switch for controlling electrophilic drug activity. In this kinetic selectivity model, treating cells with a fumarate ester drug would produce rapid engagement of the intended drug target(s) on a time scale that outcompetes esterolysis by cellular carboxylesterases (CESs), which would then inactivate excess free drug to prevent slower off-target reactivity (Fig. 1A). As a proof-of-concept for achieving kinetic selectivity for irreversible inhibitors, we generated a fumarate ester analogue of the Bruton’s tyrosine kinase (BTK) inhibitor Ibrutinib (1), which reacts with an active-site cysteine via a terminal acrylamide (Fig. 1B).4,9 Ibrutinib and its fumarate ester analogue (2) were further modified with alkyne handles to furnish probes 3 and 4, respectively.
Figure 1.

A kinetic selectivity model for covalent small molecules and its application to Ibrutinib. A, Standard covalent inhibitor (CI). Fast on-target (green arrow) and slower off-target reactivity (red arrow). Kinetically-selective CI. Fast on-target (green arrow) and slower off-target reactivity (red arrow), with an intermediary rate of hydrolysis of the electrophilic fumarate ester to unreactive free acid (orange arrow). B, Ibrutinib-based compounds and probes. C, 2 is hydrolyzed to inactive 5 by hCES1-, but not hCES2- or control protein (MetAP2)-transfected HEK293T cells. Cells were treated with 2 (10 μM, 1 h) prior to extraction and LC-MS analysis to quantify relative amounts of 2 and 5.
We confirmed concentration-dependent labeling of BTK by 3 and 4 in Ramos cell lysates using ABPP involving copper-catalyzed azide-alkyne cycloaddition (CuAAC)10 of probe-labeled proteins to a fluorescent tag followed by SDS-PAGE (Fig. S1A).4 Probe 4 exhibited greater in vitro proteomic reactivity than probe 3, and we also found that 4 reacted more rapidly with cysteine as a model nucleophile (Fig. S1B). We next incubated 2 with HEK293T cells expressing human carboxylesterase-1 (hCES1), carboxylesterase-2 (hCES2) or a control protein (methionine aminopeptidase 2, MetAP2; Fig. S2A), and found that hCES1-, but not hCES2- or MetAP2-expressing cells converted 2 to the corresponding carboxylic acid (5, Fig. 1C). In contrast, Ibrutinib (1) was unaffected by either CES (Fig. S2B).
We had previously found that tumor xenografts express high CES activity originating mainly from stromal/host cells.11 We attempted to mimic this endogenous environment using a dual-cell culture system, where Ramos cells were co-cultured with HEK293T cells stably expressing hCES1 (Fig. S3). Using a 6:1 ratio of Ramos and HEK293T cells expressing either hCES1 or MetAP2, we found that hCES1, but not MetAP2, produced a marked reduction in the in situ proteome-wide reactivity of 4, while only modestly reducing the potency (~10-fold) of this probe for BTK (Fig. S4). In contrast, hCES1 had no effect on the proteome-wide reactivity of 3. Time course studies verified these findings, where the initial engagement of BTK by 3 or 4 was followed by substantial proteome-wide reactivity that increased over 24 h, except under conditions where 4 was incubated with Ramos-hCES1-HEK293T co-cultures, which instead furnished rapid and sustained labeling of BTK with negligible increases in background proteome cross-reactivity (Figs. 2 and S5). These results, taken together, support a model where hCES1 imparts kinetic selectivity to 3. Notably, the heightened reactivity of 4 compared to 3 seen in our in vitro studies (Fig. S1) was not observed in situ, suggesting that some basal level of fumarate ester metabolism in human cells, independent of exogenous hCES1 expression, may serve to normalize the proteomic reactivity of 3 and 4.
Figure 2.

hCES1 suppresses the proteome-wide reactivity of probe 4. Ramos cells co-cultured with HEK293T cells expressing hCES1, Met-AP2, or mock-transfected (−) were treated with 3 or 4 (1 μM, 0–24 h). Gel-based ABPP revealed rapid BTK engagement and time-dependent increases in proteome-wide reactivity for both probes, except for 4 in the presence of CES1, where proteome-wide reactivity was blocked.
We next evaluated high-occupancy targets of 1 and 2 by performing competition experiments. Co-cultures of Ramos and hCES1- or MetAP2-HEK293T cells were treated with 1 or 2 (1 nM – 10 μM, 1 h) followed by treatment with 3 (200 nM, 1 h). Gel-based ABPP revealed complete blockade of BTK by both 1 and 2, with 1 showing ~10-fold greater potency (Figs. S6A, B). The potency of BTK blockade by 2 was only marginally affected in the presence of hCES1 (Figs. S6A, B), further supporting that engagement of this kinase occurs at a rate that exceeds CES-mediated metabolism of fumarate ester analogues of 2. We also analyzed competition experiments using quantitative mass spectrometry (MS)-based proteomics (ABPP-SILAC (stable isotope labeling with amino acids in cell culture)12), as described previously.4 Isotopically labeled co-cultures of Ramos cells with hCES1- or MetAP2-HEK293T cells were treated with DMSO or inhibitor (1 or 2; 10 μM, 1 h) prior to addition of 3 (1 μM, 1 h). Conjugation of 3-labeled proteins to an azidebiotin tag, followed by streptavidin enrichment and LC-MS-based proteomics identified high-occupancy targets of 1 that matched those reported previously4 (Fig S6C and Table S1). None of these targets was affected by hCES1 expression. Inhibitor 2 showed only three high-occupancy targets, two of which (BTK and TEC) were hCES1-insensitive, while a third (BLK) showed markedly reduced inhibition by 2 in the presence of hCES1 (Fig. S6C). The fewer high-occupancy off-targets for 2 compared to 1 indicates the fumarate reactive group imparts improved selectivity to the Ibrutinib scaffold (as has been observed for other beta-substitutions to the acrylamide of this inhibitor).4 The limited number of high-occupancy targets for 1 and 2 further suggested that the substantial concentration- and time-dependent proteome-wide cross-reactivity observed for the corresponding probes 3 and 4 (Figs. 2 and S4–S5) likely reflected low-stoichiometry interactions. We set out to identify these proteins and assess the impact of CES expression on their probe reactivity by ABPP-SILAC. We catalogued proteins that reacted with 3 and/or 4 by performing probe (1 μM, 24 h) vs no-probe (DMSO) experiments, which identified ~30–40 proteins that showed high probe/DMSO ratios (> 4) in 3 or 4-treated cells (Fig. 3A and Table S1). The majority of these targets showed greatly reduced reactivity with 4 in co-cultures of Ramos with hCES1- versus MetAP2-HEK293T cells (Figs. 3A and B). Exceptions were BTK and TEC (another high potency target of Ibrutinib),9 which reacted with 4 in a CES-insensitive manner (Figs. 3A, 3B and Table S1). In contrast, hCES1 had a negligible effect on the reactivity of targets with 3 (Figs. 3A, 3B and Table S1).
Figure 3.
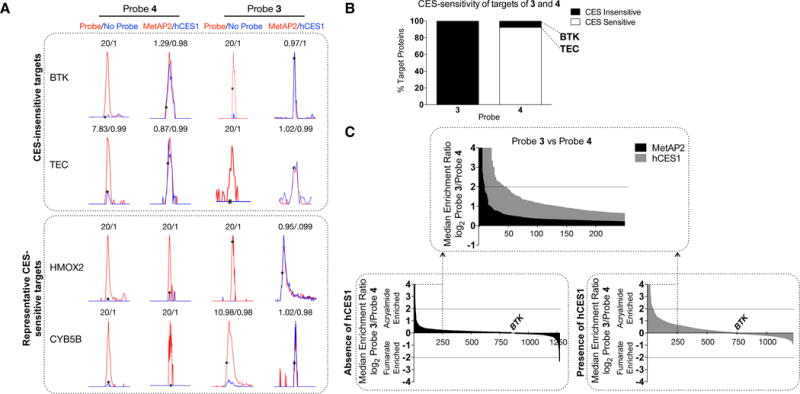
Characterization and hCES1-dependency of targets of 3 and 4. A, Representative MS1 spectra for probe targets. Ratios > 20 are assigned as 20 values. B, The hCES1-sensitivity of targets of 3 and 4. Proteins showing ratios of ≥ 2.5 in MetAP2/hCES1 ABPP-SILAC experiments were assigned as CES-sensitive. C. Comparison of the reactivity of 3 and 4. Co-cultures of Ramos and HEK293T cells expressing hCES1 or MetAP2 were treated with 3 or 4 (1 μM, 24 h). Average SILAC ratios for proteins from three experiments are shown.
We next directly compared the proteomic reactivities of 3 and 4 (1 μM each, 24 h). In control Ramos-MetAP2-HEK293T co-cultures, 3 and 4 showed comparable reactivity with a handful of proteins being preferentially labeled by one or the other probe (Fig 3C and Table S1). In contrast, in Ramos-hCES1-HEK293T co-cultures, 3 showed much greater proteomic reactivity than 4 (Fig 3C and Table S1), consistent with CES-mediated attenuation of 4 reactivity.
Having established that 4 exhibits kinetic selectivity in cell models expressing hCES1, we wondered whether this concept applied in vivo. Rodents express an elaborate network of CES enzymes compared to humans,13 so we also tested an O-isopropyl fumarate analogue of Ibrutinib (6) (Fig. 1B) to determine if it showed different CES-sensitivity in mice to compared to 4. Probe 6, as well as the O-ethyl analogue 7 (Fig. 1B) reacted similarly with BTK compared to 4 (Figure S7). We treated mice with 3, 4, and 6 or vehicle (20 mg/kg) for 2 h and visualized probe-reactive proteins in tissues by gel-based ABPP. All probes reacted with BTK in the spleen, and these labeling events were blocked by pretreatment with 1 (20 mg/kg, 2 h) (Fig 4A). Importantly, 4 and 6 exhibited much less off-target reactivity compared to 3 in tissue proteomes (Figs. 4A, B and S8), indicating that the fumarate ester probes were metabolized by mouse CESs in vivo. Also consistent with this conclusion, we found that 4 and 6 were rapidly metabolized in mouse plasma with half-lives of 0.751 min and 1.90 min, respectively (Fig. S9A), and these half-lives were substantially extended (25.5 and 352 min, respectively) by pretreatment with a CES inhibitor JZL18415 (10 μM, 1 h) (Fig. S9B). In contrast, probe 3 was stable in mouse plasma even in the absence of the CES inhibitor (half-life of 168 min) (Fig. S9A). Finally, we should note that, while 4 and 6 showed substantial reactivity with BTK in vivo, the extent of BTK engagement appeared consistently lower than that of probe 3, possibly reflecting the reduced potency displayed by fumarate ester analogues of Ibrutinib for BTK or that CES metabolism is sufficiently high in mice to compete with full labeling of BTK by these probes.
Figure 4.
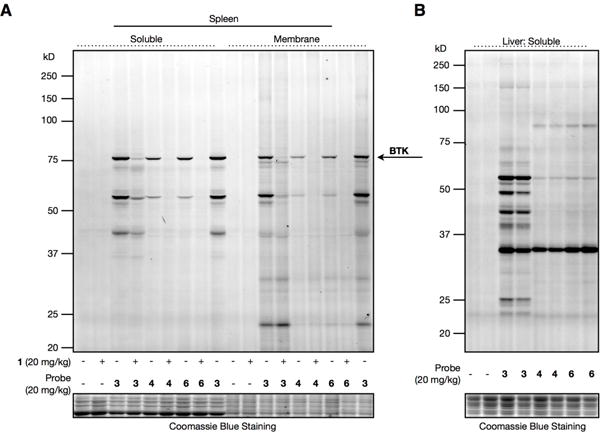
Characterization of probe reactivity in vivo. A. Gel- based ABPP of spleens (A) or livers (B) from mice treated intraperitoneally with probes 3, 4, or 6 (20 mg/kg, 2 h). For A, animals mice were pretreated with vehicle or Ibrutinib (20 mg/kg, 2 h).
Our results, taken together, demonstrate that incorporating a fumarate ester electrophile into the Ibrutinib scaffold furnishes an irreversible inhibitor with striking kinetic selectivity for BTK in cell and animal models due to CES-dependent metabolic inactivation. The time scale for CES-mediated hydrolysis of probe 4 appears appropriately positioned to proceed more slowly than probe reactivity with the preferred target BTK, but faster than the proteome-wide cross-reactivity observed for this probe in the absence of hCES1. Importantly, the kinetic selectivity of the fumarate ester probes persisted in cell models over the entire 24 h time-period in the presence of hCES1, which contrasted with the continuous, time-dependent increases in proteome-wide reactivity observed for the acrylamide probe 3. Considering that covalent inhibitors are often used in pharmacological studies that require one or more days of treatment (e.g., to assess cytotoxicity),4,14 the ability to impart kinetic selectivity upon probes should improve interpretability of such experiments by minimizing confounding time-dependent off-target reactions. We should note that some protein targets of terminal acrylamides may not accommodate fumarate ester analogues without substantial reductions in potency, and future work will be required to determine the generality and extent to which such reactive groups can be interchanged. It may alternatively be possible to achieve kinetic selectivity with other metabolically-vulnerable electrophilic groups such as acrylates and thioacrylates. Additionally, proteins with short half-lives may be less suitable for targeting by kinetic selectivity. Regardless, our data should encourage the consideration of fumarate esters as starting points for the development of covalent inhibitors with potentially improved selectivity profiles in living systems. Indeed, one could speculate that DMF itself exploits the principle of kinetic selectivity to produce immunosuppression with limited side effects in humans.
Supplementary Material
Acknowledgments
We thank M. Hayward and A. Gilbert (Pfizer), K. Backus for helpful discussions, and K. Masuda for technical assistance. We thank M. Cameron (Scripps Florida) for performing plasma stability studies.
Funding Sources: No competing financial interests are declared. This work was supported by NIH (CA087660), American Cancer Society (B.W.Z., PF-15-142-01-CDD), and Pfizer.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
Author Contributions: B.W.Z. and B.F.C. designed experiments and interpreted results. B.W.Z. performed experiments and synthesized compounds. L.R.W. provided reagents and synthesized compounds. K.M.L. assisted with proteomics analysis. B.W.Z. and B.F.C. wrote the manuscript.
References
- 1.a Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Chem Biol. 2013;20:146. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Mah R, Thomas JR, Shafer CM. Bioorg Med Chem Lett. 2014;24:33. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Singh J, Petter RC, Baillie TA, Whitty A. Nat Rev Drug Discov. 2011;10:307. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 4.Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, Roberts LR, Schnute ME, Wang C, Hulce JJ, Wei B, Whiteley LO, Hayward MM, Cravatt BF. Nat Chem Biol. 2014;10:760. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DS, Weerapana E, Cravatt BF. Future Med Chem. 2010;2:949. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Nature. 2016;534:570. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Medina-Cleghorn D, Bateman LA, Ford B, Heslin A, Fisher KJ, Dalvie ED, Nomura DK. Chem Biol. 2015;22:1394. doi: 10.1016/j.chembiol.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Cancer Discov. 2016;6:316. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 7.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, din MFO, Taunton J. Nat Chem Biol. 2012;8:471. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, Cravatt BF. Sci Signal. 2016;9:rs10. doi: 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Larson DL, Hua M, Takemori AE, Portoghese PS. J Med Chem. 1993;36(23):3669. doi: 10.1021/jm00075a023. [DOI] [PubMed] [Google Scholar]
- 9.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. Proc Natl Acad Sci USA. 2010;107:13075. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, Mueller BM, Cravatt BF. Proc Natl Acad Sci USA. 2004;101:13756. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann M. Nat Rev Mol Cell Biol. 2006;7:952. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 13.Holmes RS, Wright MW, Laulederkind SJF, Cox LA, Hosokawa M, Imai T, Ishibashi S, Lehner R, Miyazaki M, Perkins EJ, Potter PM, Redinbo MR, Robert J, Satoh T, Yamashita T, Yan B, Yokoi T, Zechner R, Maltais LJ. Mamm Genome. 2010;21:427. doi: 10.1007/s00335-010-9284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Ji X, Peng T, Zhang X, Li J, Yang W, Tong L, Qu R, Jiang H, Ding J, Xie H, Liu H. Bioorg Med Chem. 2014;22:2366. doi: 10.1016/j.bmc.2014.01.035. [DOI] [PubMed] [Google Scholar]; a Li X, Zuo Y, Tang G, Wang Y, Zhou Y, Wang X, Guo T, Xia M, Ding N, Pan Z. J Med Chem. 2014;57:5112. doi: 10.1021/jm4017762. [DOI] [PubMed] [Google Scholar]
- 15.a Long JZ, Nomura DK, Cravatt BF. Chem Biol. 2009;16:744. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Crow JA, Bittles V, Borazjani A, Potter PM, Ross MK. Biochem Pharmacol. 2012;84:1215. doi: 10.1016/j.bcp.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.