

Abstract
The 4H+/4e− reduction of O2 to water, a key fuel-cell reaction also carried out in biology by oxidase enzymes, includes the critical O–O bond reductive cleavage step. Mechanistic investigations on active-site model compounds, which are synthesized by rational design to incorporate systematic variations, can focus on and resolve answers to fundamental questions, including protonation and/or H-bonding aspects, which accompany electron transfer. Here, we describe the nature and comparative reactivity of two low-spin heme–peroxo–Cu complexes, LS-4DCHIm, [(DCHIm)F8FeIII-(O22−)-CuII(DCHIm)4]+, and LS-3DCHIm, [(DCHIm)F8FeIII-(O22−)-CuII(DCHIm)3]+ (F8 = tetrakis(2,6-difluorophenyl)-porphyrinate; DCHIm = 1,5-dicyclohexylimidazole), toward different proton (4-nitrophenol and [DMF·H+](CF3SO3−)) (DMF = dimethylformamide) or electron (decamethylferrocene (Fc*)) sources. Spectroscopic reactivity studies show that differences in structure and electronic properties of LS-3DCHIm and LS-4DCHIm lead to significant differences in behavior. LS-3DCHIm is resistant to reduction, is unreactive toward weakly acidic 4-NO2–phenol, and stronger acids cleave the metal–O bonds, releasing H2O2. By contrast, LS-4DCHIm forms an adduct with 4-NO2–phenol, which includes an H-bond to the peroxo O-atom distal to Fe (resonance Raman (rR) spectroscopy and DFT). With addition of Fc* (2 equiv overall required), O–O reductive cleavage occurs, giving water, Fe(III), and Cu(II) products; however, a kinetic study reveals a one-electron rate-determining process, ket = 1.6 M−1 s−1 (−90 °C). The intermediacy of a high-valent [(DCHIm)F8FeIV = O] species is thus implied, and separate experiments show that one-electron reduction-protonation of [(DCHIm)F8FeIV=O] occurs faster (ket2 = 5.0 M−1 s−1), consistent with the overall postulated mechanism. The importance of the H-bonding interaction as a prerequisite for reductive cleavage is highlighted.
Graphical Abstract
INTRODUCTION
Because of ever-growing global environmental and economic concerns, researchers are pushing to find alternative and, ideally, renewable energy sources to fill the needs of the modern generation.1 In nature, energy is consumed to make chemical bonds and released when those bonds are broken. Importantly, this concept can be exhibited by the natural processes of photosynthesis and cellular respiration, which make up the life-sustaining oxygen cycle: O2 + 4e− + 4H+ ⇄ 2H2O.2,3 Here, there exists a critical interplay between acid– base and redox events4 including transformations of high-valent metal–oxo species.5,6 These processes have inspired chemists to design and study synthetic constructs that resemble activesite species and/or duplicate function, which can lead to new fundamental insights and the generation of practical systems. 7–12 Advancements involving Photosystem II-inspired model systems for the purposes of light harvesting and/or water oxidation to O2 quickly rise to the forefront of popular science due to their potential applications in solar energy conversion.13 However, to complete the biochemical oxygen cycle, the O–O bond is cleaved during cellular respiration to give back water, and the mechanistic similarities of how redox-active transition metal ions make or break the strong O–O bond in these two natural systems3 make Cytochrome c Oxidase (CcO) an extremely important enzyme. For this reason, considerable research efforts have been undertaken to understand the mechanistic details of dioxygen reduction, which occurs at the active site.
CcO’s heterobinuclear active site’s four-electron/four-proton reduction of O2 to water (Figure 1) is accompanied by membrane proton-translocation leading to ATP synthesis.14 While there is a consensus about many aspects of the CcO enzymatic mechanism, debate remains concerning the identity of certain intermediates and the details of the process triggering the O–O bond cleavage event, where the release of destructive, partially reduced oxygen species (i.e., H2O2, ·OH) is prevented.15 It has been proposed and widely accepted16,17 that a hydrogen-bonding network in the active site provides a channel from the cross-linked tyrosine (Tyr) residue to the putative bridging peroxo moiety (~5 Å away) and facilitates a proton-coupled electron transfer (PCET), which cleaves the O–O bond (Figure 1).16,18–22
Figure 1.

Net 4H+/4e− O2 reduction reaction by CcO (top), and the proposed mechanistic involvement of a hydrogen-bonding network in the active site to promote O–O cleavage (bottom).
(Bio)chemical systems require protons to promote O–O reductive cleavage; however, the specific nature of an acid moiety (e.g., identity, proximity to the O–O moiety, pKa, H-bonding capability, and number of protons) is crucial.23–25 To specifically facilitate O–O heterolytic cleavage in a heme-(hydroperoxo) intermediate, cytochrome P450 monooxygenases employ an acid–alcohol residue pair, located adjacent to the Fe–O–O distal O-atom to provide a key H-bond and proton source. If that H-bonding group is changed (e.g., by mutation), uncoupling occurs, and the outcome is Fe–O cleavage and H2O2 production.26–28 Nonheme iron and copper proteins are also found to have specific requirements (e.g., proximity, acid strength) to control proton-promoted O–O reductive cleavage chemistries.29–32
A few synthetic heme–Cu complexes can reduce O2 to water via a (su)peroxo intermediate,33–35 yet neither in these cases, nor in CcO, is the nature of H-bonding or protonation reactivity understood with respect to O–O cleavage. To provide the insights and fundamentals needed for this very important reaction, several different approaches can be taken. Studies on a diverse, ever-growing family of model complexes (both synthetic coordination complexes11,36–38 and modified protein systems12,22,23,39) have aided researchers in gaining a fundamental understanding about certain aspects of the chemical mechanism of O2 reduction.12,14 Our research group has for many years been systematically studying the O2 adducts of synthetic model systems in which we can define or control certain aspects and monitor the effects of varying coordination environments/geometries, redox properties, sterics of the Cu and/or iron centers, and nature of a given substrate on the outcomes of different reactions. The bridging heme–peroxo– copper formulation depicted in Figure 1 (center), while not (yet) observed in the enzyme, is considered by some11,16,40–42 as a likely intermediate, and several recent computational studies invoke the likelihood of a peroxo-level intermediate in CcO.43–45 A heme–peroxo–Cu complex is a convenient starting point from which to study how slight changes in structure can tune the functional O2-reduction capabilities of synthetic model complexes.46 Alternatively, it is also relevant to consider factors (chemical or structural) that lead to the affiliated nonredox reaction where protonation of O2-derived atom(s) results in M–O cleavage and release of H2O2, although this pathway is importantly avoided in the enzymatic mechanism.
For CcO, or for a given efficient molecular catalyst able to promote four-electron, four-proton O2-reduction to water (E°MeCN = 1.21 V vs Fc+/0),47 these particular nuances relevant to the promotion of the O–O cleavage process need to be delineated and understood within the framework of reduction/protonation chemistry. The breadth of these PCET studies involves probing different (or perhaps simultaneous) orderings of the events of (i) electron transfer (from Fe, Cu, or the Tyr phenol moiety), and (ii) proton transfer derived from different sources. Yet, it is surely only by investigation of chemical model constructs and/or computational inquiries, where one has the ability to break down such a complicated overall process into individual steps, so that deep fundamental insights can be obtained.48,49 The latter are in the future highly relevant to a broad range of fields, that is, (i) the understanding of mechanisms of action of other synthetic and/or metalloenzyme O2-activating systems, or (ii) the design of practical catalysts for O2-reduction or substrate oxidation/oxygenation by O2-derived metal–oxy complexes.
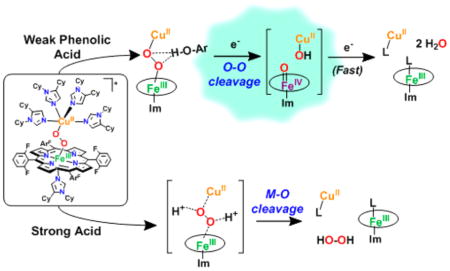
In this report, we investigate the fundamental interactions of model complexes with exogenous substrates as they relate to the enzymatic CcO mechanism, and insights into the structure–function relationship in the O–O versus M–O cleavage pathways are evaluated. Specifically, we detail how different acids and a reductant behave toward two similar model heme-(O22−)-Cu complexes, LS-3DCHIm and LS-4DCHIm (Figure 2). Such strikingly similar complexes show an interesting difference in their reactivities toward proton and electron sources. In the case of LS-4DCHIm, only when treated with relatively weak acid (4-NO2–phenol) and reductant (Fc*) is the coveted O–O cleavage reaction accomplished. Spectroscopic and kinetic evidence points toward a phenolic hydrogen-bond-associated intermediate, which triggers the electron transfer from Fc* necessary for O–O scission; a strong acid source instead leads to H2O2 release. Interestingly, the related complex, LS-3DCHIm (see Figure 2), shows no reactivity toward the same substrates, and with the support of DFT calculations, we believe this difference in reactivity can be attributed to steric factors and the degree to which the weakly acidic, phenolic substrate can access the bridging peroxo moiety to create a critical, activated H-bonded adduct. We are very interested in the chemistry of metal–oxy interactions with H+ and e− sources of varying strengths and the insights these reactions may provide into O–O activation and cleavage chemistry in general.50
Figure 2.

In situ generation of low-spin peroxo complexes, LS-3DCHIm and LS-4DCHIm, derived from our “naked” high-spin heme–peroxo–Cu complex, HS-3MeTHF,51 showing their reactivities toward strong acid (H+ = [DMF·H+](CF3SO3−)) and electron sources alone, that is, in the absence of proton sources.
RESULTS AND DISCUSSION
Reactivity with Fc* and/or Strong Acid
The peroxo complex, LS-4DCHIm, which has been previously characterized, 51 is stable at −90 °C in 2-methyltetrahydrofuran (MeTHF), even in the presence of excess (20 equiv) reductant, Fc* (E°MeTHF = −270 mV vs Ag+/0).52 However, when strong acid, [DMF·H+](CF3SO3 −) (pKa(CH3CN) = 6.1),53 is added to a solution of LS-4DCHIm, a reaction is immediately observed in the UV–vis spectrum. Rapid disappearance of the low energy absorption features associated with peroxo-to-Fe CT bands51 further suggests that the peroxo-bridged structure is no longer intact. In fact, chemical and spectroscopic interrogation of the product mixture reveals that H2O2 and mononuclear iron and copper species are produced (Figure 2). Employing the quantitative horseradish peroxidase spectrophotometric assay52,54 verifies that 95% of the expected H2O2 was released, meaning that both M–O bonds were cleaved via protonation (Figure 2) (see Experimental Section and Table S1). An EPR spectrum of the reaction product solution indicates full formation of a copper(II)-imidazolyl species (see the Supporting Information for further explanation), and a predominantly (>95%) low-spin heme-imidazolyl product (Figure 2, see also Figures S2–4), confirming that the dinuclear complex has come apart.
Thus, the occurrence of metal–oxygen bond cleavage is the result of simple acid–base chemistry. Reductive cleavage of an O–O bond requires protons and electrons, however, Fc* does not reduce the H2O2 present in the product mixture (Figure 2), nor does it reduce the iron(III) or Cu(II) species present; that is, the one-electron oxidized byproduct of a reduction event, the ferrocenium cation Fc*+, is not formed, as would be observed by an increase in absorption at 795 nm.
The analogous complex with three imidazolyl ligands on the copper ion, LS-3DCHIm, shows similar reactivity toward these relatively strong proton or electron sources; that is, there is no observable reaction with Fc*, and release of H2O2 (100%) occurs with addition of [DMF·H+](CF3SO3−) (Figure 2, Table S1). Because, with both of these heme–peroxo–copper complexes, the addition of strong acid in combination with reductant still leads to only acid–base chemistry, we reasoned that a certain degree of “finesse” was likely necessary with respect to peroxo–H+/e− interactions along the reaction pathway. Therefore, we have probed this idea further by employing milder acidic substrates (phenols) to find conditions that lead to O–O activation and reductive cleavage, but not H2O2 release. In support, a recent study involving electrochemical reduction of a synthetic Mn–peroxo species revealed that strong acid favored M–O cleavage (H2O2 evolution), whereas employment of a weak acid facilitated O–O reductive cleavage.55
Weak (Phenolic) Acid Association
The reactivity of interest here is revealed when the low-spin complex is tested in reactions with phenols. First, we observe no reaction (as monitored by UV–vis) when LS-4DCHIm is tested with phenols having low bond dissociation free energies (BDFE) and low acidities such as 4-MeO–phenol or 4-tBu–phenol, which are typically used as H-atom sources.56,57 With a slightly more acidic phenol, 4-CN–phenol (pKa(CH3CN) = 22.7),58 still, no reaction is observed. However, upon addition of excess (≥10 equiv) 4-NO2–phenol (pKa(CH3CN) = 20.758 and pKa(tetrahydrofuran, THF) = 1859)50,60 to a solution of LS-4DCHIm (Figure 3, black spectra), a new species is formed, [LS-4DCHIm(ArOH)] (Figure 3, blue spectra) (λmax = 414 (Soret), 540, and 840(b) nm). The fact that excess phenol is necessary to see full formation of this intermediate leads us to tentatively assign this as an equilibrium process (see kinetic analysis below). The blue-shifted Soret and upshifted ν4 oxidation state marker band from resonance Raman (rR) spectroscopy of the ArOH-adduct (Figure 3, bottom right, λex = 413 nm for both species) indicate partial oxidation of the FeIII center (more specifically, decreased donation from Fe (dπ) into porphyrin (π*) orbitals),61 in agreement with DFT calculations of H-bonding to the peroxo core (vide infra, see Supporting Information). We associate the weak intensity retained in the low-energy bands in the UV–vis spectrum with the binuclear complex remaining intact. The fact that this intermediate is EPR silent (<5% mononuclear CuII or FeIII ion species), Figure 3, corroborates that conclusion, due to the coupling of the CuII (S = 1/2) and LS-FeIII (S = 1/2) through the bridging peroxo moiety.
Figure 3.

UV–vis spectra of LS-4DCHIm reacting with 4-NO2– phenol to give the [LS-4DCHIm(ArOH)] adduct, and rR difference spectra showing the shifts that occur upon phenolic acid association.
Evidence that the O–O bond is still intact in [LS-4DCHIm(ArOH)] (Figure 3, top) is provided by experimental quantification of the H2O2 produced (85% yield) when this adduct is exposed to a strong acid, here [DMF·H+](CF3SO3−) (horseradish peroxidase assay; see Table S1). Resonance Raman data provide compelling support that the O–O bond is both intact and perturbed by phenol addition, revealing clear differences in the ν(Fe–O) and ν(O–O) stretching vibrations between the LS-4DCHIm species (λex = 413 nm) and the [LS-4DCHIm(ArOH)] adduct (λex = 825 nm for better selective enhancement) (Figure 3, bottom left). Specifically, the ν(Fe– O) increases slightly (by ~10 cm−1) from ~590 to 598 cm−1 (Δ18O2 = 29 cm−1), while the ν(O–O) decreases by >40 cm−1 from ~870 to 827 cm−1 (Δ18O2 = 48 cm−1) (Figure 3). It is valuable to note that for both LS-4DCHIm and [LS-4DCHIm(ArOH)], the Fe–O and O–O stretches are observed at both high and low energy excitation (413 nm and ~800–900 nm, Figure S5). This indicates that (1) the chromophore responsible for the low energy absorption bands also has a strong absorption at 413 nm (for the respective samples), (2) the O–O stretches observed for LS-4DCHIm and [LS-4DCHIm(ArOH)] are due to a heme-bound species (considering the significant resonance enhancement at 413 nm with low power (<2 mW)), and (3) the low energy bands around 850 nm can be reasonably assigned as peroxo-to-Fe CT bands (because both the ν(Fe–O) and the ν(O–O) are resonantly enhanced).
The phenomenon of H-bonding interactions inducing small (5–10 cm−1) shifts in the Fe–O stretching mode has been reported for dioxygen adducts of cytochrome P450 mutants (not including a copper ion), which contain potential H-bonding residues in various active site positions. Furthermore, it was concluded on the basis of experimental and theoretical studies that a shift to higher energy, as observed here, is indicative of an H-bonding interaction with the distal O-atom with respect to iron.62–64 The aforementioned spectral evidence leads us to propose the structure of the intermediate formed to be a 4-NO2–phenol associated heme–peroxo– copper adduct depicted in Figure 3 wherein the phenolic O–H associates via H-bonding with the Cu-bound O-atom (OCu).78 Considering that it is logically favorable to strengthen the Fe– O bond and weaken the O–O bond to effect O–O reductive cleavage, the weakly acidic phenol is therefore poised to participate in a full PCET reaction, resulting in homolytic O–O bond cleavage.
DFT calculations support the association of 4-NO2–phenol with LS-4DCHIm to form an adduct (Figure 4, also see the Experimental Section). This H-bonding interaction is indicated in the DFT calculated structure of [LS-4DCHIm(ArOH)] (Figure 4, right) by the dotted green line (rcalc(H–OCu) = 1.530 Å; rcalc(OPh–OCu) = 2.536 Å). Further calculations comparing possible binding modes (see Supporting Information for details) provide strong support for this assignment, predicting the OCu–phenol adduct to be ~2 kcal/mol lower in energy than the OFe–phenol adduct. This is expected given that LS-4DCHIm has a greater partial negative charge on OCu as compared to OFe, which would favor phenol association on OCu. Furthermore, phenol binding to OCu results in a shorter Fe–O bond and greater Fe–O bond order, consistent with the rR observed frequency change, whereas binding to OFe would give a longer Fe–O bond and lower Fe–O bond order (both cases result in a longer O–O bond and lower O–O bond order, consistent with the lower ν(O–O) from rR spectroscopy). We note that DFT-calculated frequencies showed substantial mixing of Fe–O, Cu–O, and O–O modes that was sensitive to small changes in core bond angles, therefore complicating interpretation of the individual two-atom centered modes. However, employing Badger’s rule to predict a change in the Fe–O stretching frequency on the basis of the change in bond length in Figure 4 yields a Δ(ν(Fe–O)) of +9 cm−1 for the OCu-bound structure relative to that in the peroxo complex, LS-4DCHIm, providing excellent agreement with the rR data shown in Figure 3 (as compared to Δ (ν(Fe–O)) = –29 cm−1 for the OFe-bound structure).65
Figure 4.

DFT-calculated structures for LS-4DCHIm and [LS-4DCHIm(ArOH)] (with porphyrin ArF groups and most H-atoms omitted for clarity). The table shows selected DFT-calculated properties for the H-bonding and H+-transfer structures discussed.
Calculations regarding the protonation states along the O–O cleavage pathway have suggested the possible formation of an Fe–OOH species following proton transfer from an active site Tyr residue.44 Therefore, we also computationally evaluated the possibility that the H+ has transferred from 4-NO2–PhOH in the adduct, leaving 4-NO2–PhO− strongly bound as an H-bond acceptor (although UV–vis data for 4-NO2–PhO− appearance and kinetic data (vide infra) suggest this is not the case). However, the optimized structure for the H+ transferred to the OCu reveals an O–O bond elongation (relative to LS 4DCHIm) that is far too large, corresponding to a ~195 cm−1 downshift in ν(O–O) as predicted by Badger’s rule65,66 (as compared to an ~92 cm−1 downshift when 4-NO2–PhOH is the H-bond donor). As it is also 2 kcal/mol higher in energy than the H-bonded adduct (Figure 4), a structure in which the H+ has transferred to yield a hydroperoxo species is unlikely.
It is interesting to briefly compare the effects of protonating the peroxo moiety (or to a lesser extent, H-bonding to it) in LS-4DCHIm with those observed in Cu2O2 and H2O2 chemistry, where protonation results in an increase, rather than a decrease, in the O–O stretching frequency (and DFT-predicted bond strength).67 This difference in the ν(O–O) behavior can be attributed to the fact that Fe(III) has a dπ hole, which receives additional (π-symmetry) donation from the O22− π* orbital in the peroxo structure. The interaction with the proton lowers the energy of the O2 2− orbitals, greatly decreasing this donation (thereby increasing π* occupation and weakening the O–O bond). Note that in the heme–Cu system, protonation on the OCu atom localizes charge on the OFe atom (originally in a pπ orbital) and strengthens the Fe–O σ bond (thus the net effect is a stronger Fe–O bond).
Reductive O–O Cleavage
The above results show that the H-bonding interaction weakens the O–O bond and “activates” the [LS-4DCHIm(ArOH)] adduct peroxo moiety to be able to accept an electron from a reducing agent (Fc*), leading to a PCET-type, O–O cleavage reaction.
The products of the reaction, obtained in high yield, are shown in the diagram at the top of Figure 5, as deduced from our spectroscopic analysis. The heme-containing product exhibits UV–vis features at λmax = 414 (Soret) and 542 nm (Figure 5, green spectrum), characterized as a low-spin, six-coordinate ferric-heme. Two equivalents of decamethylferrocenium ion (Fc*+) are formed on the basis of the new absorptions at λmax = 785, 805 nm (quantified as ΔAbs(785 nm –870 nm); ε = 580 M−1 cm−1),68 signifying that a twoelectron reduction of [LS-4DCHIm(ArOH)] has occurred. Additionally, an increase in absorbance corresponding to the 4-NO2–PhO− anion appears as a shoulder of the heme Soret band, indicating that proton(s) have also been transferred in significant quantities (see Figure S18).
Figure 5.

Scheme, UV–vis, and EPR spectra showing the 2-electron reduction of the H-bonded adduct, [LS-4DCHIm(ArOH)], by Fc*. This step includes O–O cleavage and therefore decoupling of the Fe(III) and Cu(II) metal centers.
Further supporting our conclusions about the nature and identity of species in the product mixture is the latter’s EPR spectrum showing the emergence of nearly 100% of the CuII ion expected, and mostly low-spin (~95%) FeIII species, which match an EPR spectrum of an authentic mixture of these products (see Figure 5, bottom). Quantification of the iron(III) and copper(II) products was carried out by comparing the reaction product spectrum with intensity calibration curves obtained by independently generating these species (Figure S11).
A negligible amount of H2O2 (<5%, Table S1) is released in this step, and, furthermore, rR data collected at 413 nm for the reaction products show no isotope-sensitive stretching vibrations, and indicate the final heme product is the low-spin bis-imidazole F8Fe(III) species (Figure S12). While these observations do not definitively prove that water has formed, the consumption of two (stoichiometrically required) reductive equivalents, confirmation of oxidized metal centers in the final product mixture, and lack of H2O2 released do provide compelling evidence that the O–O bond has been broken. Recall that both protons and electrons are required for O–O cleavage, as addition of up to 20 equiv of Fc* alone to a solution of LS-4DCHIm causes no spectral changes (Figure 2).
Kinetic Measurements and Mechanistic Insights
For the processes occurring in Figures 3 and 5, pseudo-first-order kinetic experiments were carried out to determine reaction orders for LS-4DCHIm, 4-NO2–phenol, and Fc* (see Supporting Information for details). While the reaction rate was found to be independent of [LS-4DCHIm] and linear with respect to [Fc*], saturation behavior was observed with increasing concentrations of 4-NO2–phenol (Figure 6A,B). This observation is consistent with a reversible equilibrium or formation of an intermediate prior to the rate-determining step.52,69,70 In this case, it can be attributed to the association of the 4-NO2–phenol with the peroxo core. Notably, the UV–vis changes that occur upon addition of 4-NO2–phenol to LS-4DCHIm are not reversed following addition of strong base; the entire phenolic moiety inserts into the structure of LS-4DCHIm (vide supra). After measuring the rate as a function of Fc*+ appearance (ΔAbs(785–870 nm)) over time (also see Figures S14–16) and applying the kinetic model in Figure 6C, we calculate an association constant, KH+ = 604 ± 21 M−1, as well as a rate constant for the second-order electron transfer step, ket1 = 1.6 ± 0.1 M−1 s−1.
Figure 6.

Kinetic data for the weak acid/electron reactions with LS-4DCHIm. Dependence of kobs on [Fc*] (a) and on [4-NO2–phenol] (b). Kinetic model used for fitting the data (c).
All kinetic experiments were carried out by initial addition of the reductant to the LS-4DCHIm complex, which caused no spectral changes, followed by addition of 4-NO2–phenol. In all cases, immediate formation of the intermediate species, [LS-4DCHIm(ArOH)], was observed, leading to the formation of the reaction products. This experimental observation is consistent with the proposed mechanism, in which construction of an H-bonding network around the Fe–peroxo–Cu complex precedes and facilitates the electron transfer. The linear dependence of kobs on the concentration of Fc* indicates that during the rds, only one molecule of reductant is involved, which we believe results in O–O cleavage and transient formation of a FeIV=O, and the second electron transfer occurs immediately following that formation. No H2O2 is released when [DMF·H+](CF3SO3−) is added to the product mixture (Table S1), indicating that all peroxide has been reductively cleaved and protonated to give water (Figure 5). This result, along with the Cmpd II experimental results (vide infra, Figure 7) and nature of the final products formed (Fe(III), Cu(II), and 2 equiv of Fc*+), support this claim that a transient ferryl must have formed in the initial reduction (1-electron) and protonation.
Figure 7.

Scheme (top) and UV–vis spectra showing (A) formation and 1e− reduction reactivity of FeIV=O in the presence of 4-NO2–phenol. Initial rates (B) and UV–vis spectra (C) for formation of 1 equiv of Fc*+ in the reduction of Cmpd II (blue) and 2 equiv of Fc*+ for [LS-4DCHIm(ArOH)] (green).
Deuterated 4-NO2–phenol(OD) was employed for kinetic isotope effect analysis (see Supporting Information). The resultant KIE values of 1.6 for the phenol-association step (KArOD = 374 ± 28 M−1) and 1.9 for the rate-determining step (ket(D+) = 0.89 ± 0.1 M−1 s−1) suggest that the phenolic O–H/D bond is broken, and the proton is transferred in the ratedetermining step. This conclusion is consistent with the DFT interrogation of [LS-4DCHIm(ArOH)] (vide supra) and the overall proposed mechanism. While smaller than expected for a phenolic HAT reaction, these KIE values are in the range for PCET reactions involving phenol O–H(D) bond activation by metal–oxygen species.71,72
Mechanistic Insights from Independent FeIV=O Studies
In CcO, it is proposed that delivery of a H+ and e− induces O–O cleavage of an FeIII–peroxo–CuII moiety, resulting in an FeIV=O (Cmpd II-type) species, which is subsequently reduced and protonated to give a low-spin FeIII– OH product.73 To probe the possibility that such a ferryl species forms during reduction of [LS-4DCHIm(ArOH)], as implied by the kinetic results just described, initial rates were measured for the protonation/reduction of an authentic solution of F8FeIV=O74 by addition of 4-NO2–phenol and Fc*.
Indeed, the same low-spin heme product was observed (Figure 7), and both the proton and electron sources were necessary to observe a reaction. However, the rate of reduction (1e−) of the Cmpd II-type complex is >3 times faster than the reduction of [LS-4DCHIm(ArOH)] (2e− overall, vide supra); therefore, this explains the finding that we could not spectroscopically (by UV–vis) observe the FeIV=O intermediate during the LS-4DCHIm reaction because excess reductant is present (note that in CcO and other model complexes,75 the stoichiometric supply of electrons can be controlled, allowing observation of a metastable FeIV=O species). This result demonstrates the mechanistic similarity between O–O cleavage in LS-4DCHIm and CcO, highlighting the relevance of this work among enzyme model complexes.
Impact of Structure on Reactivity
In addition to LS-4DCHIm, our lab has recently characterized a similar heme– peroxo–copper complex, LS-3DCHIm, in which the copper ion is ligated by only three monodentate DCHIm donors.51 LS-3DCHIm is only stable below −115 °C in MeTHF and is unchanged following addition of 4-NO2–phenol and/or Fc* (monitored by UV–vis and rR), illustrating the different chemical properties of the two low-spin imidazolyl complexes. It is important to note that LS-4DCHIm can be cooled below –115 °C and maintain its reactivity (at slower rates). However, no reaction is observed when an excess of a sterically hindered, albeit slightly more acidic, phenol, 2,6-di-tert-butyl-4-nitrophenol (pKa(CH3CN) = 19.1),57 is added to a solution of LS-4DCHIm, even in the presence of Fc*. In contrast, recall that reaction with strong acid, [DMF·H+](CF3SO3−), gives identical products for both LS-3DCHIm and LS-4DCHIm (UV–vis, EPR, H2O2 quantification, vide supra). To gain insight into this difference in reactivity, we compared the DFT-optimized structures for these two complexes (Figure 8). DFT predicts the lowest energy structure for LS-3DCHIm to have a distorted square planar geometry (τ = 0.4, where 0 = square planar, 1 = tetrahedral) with an Fe–Cu distance of 4.10 Å, and for LS-4DCHIm to have a square pyramidal geometry (τ = 0.1, where 0 = square pyramidal, 1 = trigonal bipyramidal)76 in which the additional DCHIm ligand occupies the axial position at the copper center, and the Fe–Cu distance is 4.47 Å. While these calculated structures do not give insight into the markedly high O–O stretch observed in rR experiments, it is clear that an additional monodentate ligand induces a structural rearrangement, which influences the accessibility of the peroxo core. In particular, the core geometries of LS-3DCHIm and LS-4DCHIm, depicted in Figure 8 (also Figure S9 for more selected bond lengths and angles), indicate a more accessible peroxo moiety in LS-4DCHIm. These results together suggest that steric factors inhibiting H-bond association greatly contribute to the difference in reactivity between the two complexes.
Figure 8.

DFT-optimized structures of LS-4DCHIm and LS-3DCHIm (porphyrin ArF groups and H-atoms omitted for clarity) depicting the relationship between structure and reactivity toward different acids.
CONCLUSIONS
The structural nature of the CcO-inspired heme–peroxo– copper complexes, and their reactivity in protonation-reduction chemistry, which has been described here, further emphasizes the realization concerning how acutely nature has constructed its systems to achieve the chemical transformations necessary for sustaining aerobic life. Rather than rigorous mimicking of proteins, we have taken the approach of designing synthetically simplified complexes in which we can control or manipulate certain aspects in our pursuit of a fundamental understanding of the factors involved in O2-reduction processes. This has required the use of cryogenic conditions (e.g. −90 °C) for generation of heme–Cu O2-adducts, those which are highly reactive in biological systems and currently inaccessible for spectroscopic study.77 In doing so, we have shown that both the structure of the bridging peroxo-complex and the nature of the proton source play a role in determining whether the protonation/reduction mechanism proceeds via acid/base chemistry (metal–O cleavage, release of H2O2) or substrate-mediated redox chemistry (O–O bond reductive cleavage). The use of monodentate DCHIm ligands allows for the formation of heme–(O22−)–Cu complexes possessing comparable yet unique structures, LS-3DCHIm and LS-4DCHIm, with intriguingly different reactivities.
The two complexes discussed differ in that a phenolic H-bond associated adduct can form with LS-4DCHIm, where we have shown that the weak acid, 4-NO2–phenol, associates with the Cu-bound Operoxo-atom. These observations contrast to the behavior of LS-3DCHIm, where a potentially more compressed peroxo core does not allow for formation of such an adduct. If a strong acid is employed, [DMF·H+](CF3SO3−), steric hindrance is no longer a contributing factor, as a (solvated) proton can easily reach either peroxo core to induce metal–O cleavage via acid–base chemistry, and release hydrogen peroxide. Therefore, we have observed that in a fashion similar to the generally proposed CcO enzymatic mechanism, reductive O–O cleavage is effected after first establishing a hydrogen-bonding network close to the peroxo, which then facilitates the proton-coupled electron transfer (in this case, from 4-NO2– phenol and Fc*, and in CcO, from the cross-linked Tyr residue) (Scheme 1). In the enzyme, this efficient manner of transferring the H+/e− to the peroxo prevents the production of partially reduced, detrimental, reactive oxygen species.15,79,80 Only a few previous CcO model systems have achieved O–O cleavage; however, they have employed rigid structural frameworks or “picket-fence porphyrins”. Now, we have shown that a new, potentially more flexible adduct, when paired with the appropriate reagents, can closer mimic the enzyme’s proposed stepwise transfer of H+/e− to an Fe–Cu bridging peroxo moiety. In this way, we continue to uncover the seemingly small, yet critical, chemical and/or structural aspects of transition metal–O2 assemblies, which greatly impact O–O cleavage chemistry.
Scheme 1.

Overall Proposed Mechanism for the Reaction of LS-4DCHIm with 4-NO2–Phenol and Fc*
EXPERIMENTAL SECTION
General
All reagents and solvents used were of commercially available quality and used without further purification except as noted. Inhibitor-free 2-MeTHF was distilled over Na/benzophenone under Ar and deoxygenated with Ar before use. [CuI(CH3CN)4(BArF)] and F8FeII were synthesized as previously described.81,82 All UV–vis measurements were carried out using a Hewlett-Packard 8453 diode Figure 8. DFT-optimized structures of LS-4DCHIm and LS-3DCHIm (porphyrin ArF groups and H-atoms omitted for clarity) depicting the relationship between structure and reactivity toward different acids. array spectrophotometer with a quartz Schlenk cuvette cell to monitor changes in Q-band and with a 0.2 cm path length cuvette to monitor changes in Soret band. The spectrometer was equipped with HP Chemstation software and a Unisoku thermostated cell holder for lowtemperature experiments. Resonance RAMAN samples were excited at a variety of wavelengths, using either a Coherent I90C-K Kr+ ion laser, a Coherent 25/7 Sabre Ar+ ion laser, or a Lighthouse Photonics Sprout-G pumped SolsTiS-PX Ti:Saph laser, while the sample was immersed in a liquid nitrogen cooled (77 K) EPR finger dewar (Wilmad). Power was ~2 mW at the sample for 413 nm excitation and 150–200 mW for lower energy excitation. Data were recorded while rotating the sample to minimize photodecomposition. The spectra were recorded using a Spex 1877 CP triple monochromator with either a 600, 1200, or 2400 grooves/mm holographic spectrograph grating, and detected by an Andor Newton CCD cooled to −80 °C (for high-energy excitation) or an Andor IDus CCD cooled to −80 °C (for the low-energy excitation). Spectra were calibrated on the energy axis to toluene. Excitation profiles were intensity calibrated to the solvent (MeTHF) by peak fitting in the program Origin.
UV–Vis
The complexes LS-3DCHIm and LS-4DCHIm were generated as previously described at 0.1 mM concentrations at –125 or –90 °C in a 1 cm path length, rubber septum-capped, quartz Schlenk cuvette.51 50 μL of a solution containing the desired number of equivalents of acid (10 for 4-NO2–phenol, 2 for [DMF·H+](CF3SO3−)) and/or reductant (Fc*, 10 equiv) was added via gastight syringe and mixed by bubbling Ar. To obtain conditions optimal for observing Soret peaks, the same concentration (~0.1 mM) was used in a 2 mm path length Schlenk cuvette. For kinetic measurements, spectra were recorded every 1.0 s to obtain “initial rate” data. Reactions were monitored until stable by UV–vis (i.e., no more spectral changes are observed); however, we do note that in reactions with excess Fc*, slow reduction of the metal centers may eventually occur.
EPR
0.6 mL solutions of the complexes LS-3DCHIm and LS-4DCHIm were generated anaerobically as previously described51 at 2.0 mM concentrations at −125 °C (pentane/liqiod N2 bath) and −90 °C (acetone/liquic N2 bath), respectively, in a 5 mm, rubber septumcapped, EPR tube. The desired substrates (acids and/or reductant) were added in the desired amounts, mixed by bubbling Ar, and frozen prior to recording spectra. Spectra were taken with an ER 073 magnet equipped with a Bruker ER041 X-Band microwave bridge and a Bruker EMX 081 power supply: microwave frequency = 9.41 GHz, microwave power = 0.201 mW, attenuation = 30 db, modulation amplitude = 10 G, modulation frequency = 100 kHz, temperature = 10 K.
rRaman
The complexes LS-3DCHIm and LS-4DCHIm were generated in the same fashion as EPR samples, above, at 1.0 mM concentrations at −125 °C (pentane/liquid N2 bath) and −90 °C (acetone/liquid N2 bath), respectively, in a 9-in., 5 mm, rubber septum-capped, NMR tube using either 16O2 or 18O2.51 The desired substrates (acids and/or reductant) were added in the desired amounts and mixed by bubbling Ar. The tubes were then frozen and flamesealed. Spectra were obtained with spinning tubes at 77 K with various excitation wavelengths noted for each case.
H2O2 Quantification by Horseradish Peroxidase (HRP) Test
The spectrophotometric quantification of hydrogen peroxide was achieved by recording the intensity of the diammonium 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate (AzBTS-(NH4)2) peaks (different wavelengths, monitored at 418 nm to minimize error, Figure S1) oxidized by horseradish peroxidase (HRP), which was adapted from published procedures.52 In a typical experiment, 3 mL of the desired heme–peroxo–copper complex [0.135 mM] was generated at −90/−125 °C. If required, two sample solutions of 100 μL containing Fc* [2 mM] and 100 μL containing 4-NO2–phenol [2 mM] were added, and the reaction was allowed to proceed until completion (no further spectral changes were observed). The reaction crude was subjected to the H2O2 analysis as described below, both before and after quenching by addition of a 100 μL solution containing 2.5 equiv of [DMF· H+](CF3SO3−) [0.338 mM]. Hydrogen peroxide (H2O2) was detected using the procedure described using the following stock solutions: 300 mM sodium phosphate buffer, pH 7.0 (solution A), 1 mg/mL AzBTS- (NH4)2 (solution B), 4 mg of HRP (type II salt free (Sigma)), and 6.5 mg of sodium azide in 50 mL of water (solution C). Quantification of hydrogen peroxide was achieved by adding 100 μL of the cold MeTHF sample solution to a cuvette containing 1.3 mL of water, 500 μL of solution A, 100 μL of solution B, and 50 μL of solution C (all chilled). After being mixed for 15 s, the samples were allowed to incubate for ~2 min until full formation of the 418 nm band was achieved (Table S1).
DFT Calculations
Density functional theory (DFT) calculations were performed with the Gaussian 09, version D.01 software package. All calculations were done using the BP86 functional within the spin-unrestricted formalism and broken-symmetry singlet surface, employing a split basis set as follows: 6-311g* for Fe, Cu, and peroxo O-atoms; 6-31g* for all metal-bound N atoms; and 6–31g for all remaining atoms. Tight SCF convergence and an ultrafine integration grid were used. All cyclohexyl substituents on DCHIm ligands were truncated as isopropyl groups to lower the computational cost, yet still capture some of the inherent steric effects. (A structure for LS-4DCHIm was optimized using the complete DCHIm ligands, which yielded very similar core bond distances and vibrational frequencies, indicating that the truncated model adequately represents the full ligand system for correlating to our experimental data.) Free energies were calculated at 183 K. For each binding orientation (phenol H-bonding to OFe or OCu), several positions of the phenol were attempted in which the O–H was directed toward the particular peroxo O-atom. Unconstrained optimization from these starting orientations yielded one minimum for each of the H-bonded structures (OFe-bound and OCu-bound). Structures for which the H+ was transferred to the respective peroxo O-atoms were obtained by constraining the OPh–H distance at the same distance as the OCu–H in the H-bonding structure (thus the distance from the H+ to the H-bond acceptor remains the same) and optimizing the remaining atoms.
Supplementary Material
Acknowledgments
This research was supported by the U.S. NIH (GM60353 to K.D.K. and DK31450 to E.I.S.). I.G.-B. thanks the European Commission for a Marie Curie IOF Fellowship.
Footnotes
The authors declare no competing financial interest.
ORCID
Kenneth D. Karlin: 0000-0002-5675-7040
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11322.
•H2O2 quantification details, kinetic and KIE data, strong acid reactivity product analysis, control experimental details, and supplementary DFT calculations (PDF)
•X-ray crystallographic data for [(DCHIm)2CuI](BArF) (CIF)
•X-ray crystallographic data for [(DCHIm)4CuII](ClO4)2· Et2O (CIF)
References
- 1.Cook TR, Dogutan DK, Reece SY, Surendranath Y, Teets TS, Nocera DG. Chem Rev. 2010;110:6474–6502. doi: 10.1021/cr100246c. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 3.Hoganson CW, Pressler MA, Proshlyakov DA, Babcock GT. Biochim Biophys Acta, Bioenerg. 1998;1365:170–174. doi: 10.1016/s0005-2728(98)00057-7. [DOI] [PubMed] [Google Scholar]
- 4.Warren JJ, Mayer JM. Biochemistry. 2015;54:1863–1878. doi: 10.1021/acs.biochem.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ray K, Felix F, Wang B, Nam W. J Am Chem Soc. 2014;136:13942–13958. doi: 10.1021/ja507807v. [DOI] [PubMed] [Google Scholar]
- 6.Engelmann X, Monte-Pérez I, Ray K. Angew Chem, Int Ed. 2016;55:7632–7649. doi: 10.1002/anie.201600507. [DOI] [PubMed] [Google Scholar]
- 7.Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 8.Kim E, Chufán EE, Kamaraj K, Karlin KD. Chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 9.Cracknell JA, Vincent KA, Armstrong FA. Chem Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 10.Chufán EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moénne-Loccoz P, Karlin KD. Inorg Chem. 2007;46:6382–6394. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- 11.Kieber-Emmons MT, Li Y, Halime Z, Karlin KD, Solomon EI. Inorg Chem. 2011;50:11777–11786. doi: 10.1021/ic2018727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y, Lombardi A. Chem Soc Rev. 2016;45:5020–5054. doi: 10.1039/c5cs00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cady CW, Crabtree RH, Brudvig GW. Coord Chem Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshikawa S, Shimada A. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 15.Muramoto K, Ohta K, Shinzawa-Itoh K, Kanda K, Taniguchi M, Nabekura H, Yamashita E, Tsukihara T, Yoshikawa S. Proc Natl Acad Sci U S A. 2010;107:7740–7745. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blomberg MRA, Siegbahn PEM, Wikstrom M. Inorg Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 17.Yoshikawa S, Muramoto K, Shinzawa-Itoh K, Mochizuki M. Biochim Biophys Acta, Bioenerg. 2012;1817:579–589. doi: 10.1016/j.bbabio.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Gorbikova EA, Belevich I, Wikstrom M, Verkhovsky MI. Proc Natl Acad Sci U S A. 2008;105:10733–10737. doi: 10.1073/pnas.0802512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshikawa S, Muramoto K, Shinzawa-Itoh K. Biochim Biophys Acta, Bioenerg. 2011;1807:1279–1286. doi: 10.1016/j.bbabio.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Kaila VRI, Verkhovsky MI, Wikstrom M. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 21.Siegbahn PEM, Blomberg MRA. Chem Rev. 2010;110:7040–7061. doi: 10.1021/cr100070p. [DOI] [PubMed] [Google Scholar]
- 22.Petrik ID, Davydov R, Ross M, Zhao X, Hoffman BM, Lu Y. J Am Chem Soc. 2016;138:1134–1137. doi: 10.1021/jacs.5b12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Lv X, Li J, Zhou Q, Cui C, Hosseinzadeh P, Mukherjee A, Nilges MJ, Wang J, Lu Y. J Am Chem Soc. 2015;137:4594–4597. doi: 10.1021/ja5109936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soper JD, Kryatov SV, Rybak-Akimova EV, Nocera DG. J Am Chem Soc. 2007;129:5069–5075. doi: 10.1021/ja0683032. [DOI] [PubMed] [Google Scholar]
- 25.Barry BA. J Photochem Photobiol, B. 2011;104:60–71. doi: 10.1016/j.jphotobiol.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinis SA, Atkins WM, Stayton PS, Sligar SG. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- 27.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 28.Shook RL, Borovik AS. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaguchi S, Nagatomo S, Kitagawa T, Funahashi Y, Ozawa T, Jitsukawa K, Masuda H. Inorg Chem. 2003;42:6968–6970. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 30.Augustine AJ, Quintanar L, Stoj CS, Kosman DJ, Solomon EI. J Am Chem Soc. 2007;129:13118–13126. doi: 10.1021/ja073905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solomon EI, Augustine AJ, Yoon J. Dalt Trans. 2008;9226:3921. doi: 10.1039/b800799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collman JP, Devaraj NK, Decréau RA, Yang Y, Yan YL, Ebina W, Eberspacher TA, Chidsey CED. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halime Z, Kotani H, Li Y, Fukuzumi S, Karlin KD. Proc Natl Acad Sci U S A. 2011;108:13990–13994. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh S, Xie X, Dey A, Sun Y, Scholes CP, Solomon EI. Proc Natl Acad Sci U S A. 2009;106:4969–4974. doi: 10.1073/pnas.0900995106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collman JP, Sunderland CJ, Berg KE, Vance MA, Solomon EI. J Am Chem Soc. 2003;125:6648–6649. doi: 10.1021/ja034382v. [DOI] [PubMed] [Google Scholar]
- 37.Kim E, Kamaraj K, Galliker B, Rubie ND, Moénne-Loccoz P, Kaderli S, Zuberbuhler AD, Karlin KD. Inorg Chem. 2005;44:1238–1247. doi: 10.1021/ic048907b. [DOI] [PubMed] [Google Scholar]
- 38.Chufán EE, Puiu SC, Karlin KD. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Yu Y, Hu C, Zhang W, Lu Y, Wang J. Angew Chem, Int Ed. 2012;51:4312–4316. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. J Am Chem Soc. 2000;122:12848–12858. [Google Scholar]
- 41.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma V, Wikström M, Kaila VRI. Biochim Biophys Acta, Bioenerg. 2011;1807:813–818. doi: 10.1016/j.bbabio.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Du WGH, Noodleman L. Inorg Chem. 2013;52:14072–14088. doi: 10.1021/ic401858s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du WGH, Götz AW, Yang L, Walker RC, Noodleman L. Phys Chem Chem Phys. 2016;18:21162–21171. doi: 10.1039/c6cp00349d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blomberg MRA. Biochemistry. 2016;55:489–500. doi: 10.1021/acs.biochem.5b01205. [DOI] [PubMed] [Google Scholar]
- 46.Chatterjee S, Sengupta K, Hematian S, Karlin KD, Dey A. J Am Chem Soc. 2015;137:12897–12905. doi: 10.1021/jacs.5b06513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pegis ML, Roberts JAS, Wasylenko DJ, Mader EA, Appel AM, Mayer JM. Inorg Chem. 2015;54:11883–11888. doi: 10.1021/acs.inorgchem.5b02136. [DOI] [PubMed] [Google Scholar]
- 48.Bhagi-Damodaran A, Petrik I, Lu Y. Isr J Chem. 2016;61801:773–790. doi: 10.1002/ijch.201600033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hematian S, Garcia-Bosch I, Karlin KD. Acc Chem Res. 2015;48:2462–2474. doi: 10.1021/acs.accounts.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.We report pKa and E0 values in MeCN at room temperature (although our chemistry is carried out in MeTHF under cryogenic conditions), as a general guide, because the RT and/or nonaqueous solvent values are documented in the literature, internally consistent, and serve the purpose of providing a relative scale of these parameters in organic solvent.
- 51.Garcia-Bosch I, Adam SM, Schaefer AW, Sharma SK, Peterson RL, Solomon EI, Karlin KD. J Am Chem Soc. 2015;137:1032–1035. doi: 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peterson RL, Ginsbach JW, Cowley RE, Qayyum MF, Himes RA, Siegler MA, Moore CD, Hedman B, Hodgson KO, Fukuzumi S, Solomon EI, Karlin KD. J Am Chem Soc. 2013;135:16454–16467. doi: 10.1021/ja4065377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carver CT, Matson BD, Mayer JM. J Am Chem Soc. 2012;134:5444–5447. doi: 10.1021/ja211987f. [DOI] [PubMed] [Google Scholar]
- 54.Tosha T, Kagawa N, Arase M, Waterman MR, Kitagawa T. J Biol Chem. 2008;283:3708–3717. doi: 10.1074/jbc.M707338200. [DOI] [PubMed] [Google Scholar]
- 55.Ching HYV, Anxolabéhère-Mallart E, Colmer HE, Costentin C, Dorlet P, Jackson TA, Policar C, Robert M. Chem Sci. 2014;5:2304. [Google Scholar]
- 56.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicoleti CR, Marini VG, Zimmermann LM, Machado VG. J Braz Chem Soc. 2012;23:1488–1500. [Google Scholar]
- 58.Klamt A, Eckert F, Leito I, Kaljurand I, Ku A, Diedenhofen M. J Comput Chem. 2009;30:799–810. doi: 10.1002/jcc.21103. [DOI] [PubMed] [Google Scholar]
- 59.Nam E, Alokolaro PE, Swartz RD, Gleaves MC, Pikul J, Kovacs JA. Inorg Chem. 2011;50:1592–1602. doi: 10.1021/ic101776m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.For reference and comparison: unsubstituted PhOH, pKa(H2O) = 9.95, pKa(MeCN) = 22.7, pKa(THF) = 21, and for p-NO2–ArOH, pKa(H2O) = 7.18, pKa(MeCN) = 20.7, pKa(THF) = 18 (from refs 58, 59). Also, see ref 12: Vazdar K, Kunetskiy R, Saame J, Kaupmees K, Leito I, Jahn U. Angew Chem, Int Ed. 2014;53:1435–1438. doi: 10.1002/anie.201307212.
- 61.Kitagawa T, Abe M, Kyogoku Y. Chem Phys Lett. 1977;48:55–58. [Google Scholar]
- 62.Gregory M, Mak PJ, Sligar SG, Kincaid JR. Angew Chem, Int Ed. 2013;52:5342–5345. doi: 10.1002/anie.201300760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spiro TG, Soldatova AV, Balakrishnan G. Coord Chem Rev. 2013;257:511–527. doi: 10.1016/j.ccr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR. J Phys Chem A. 2008;112:13172–13179. doi: 10.1021/jp8017875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Green MT. J Am Chem Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 66.Cramer CJ, Tolman WB. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Root DE, Mahroof-Tahir M, Karlin KD, Solomon EI. Inorg Chem. 1998;37:4838–4848. doi: 10.1021/ic980606c. [DOI] [PubMed] [Google Scholar]
- 68.Kakuda S, Peterson RL, Ohkubo K, Karlin KD, Fukuzumi S. J Am Chem Soc. 2013;135:6513–6522. doi: 10.1021/ja3125977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wijeratne GB, Corzine B, Day VW, Jackson TA. Inorg Chem. 2014;53:7622–7634. doi: 10.1021/ic500943k. [DOI] [PubMed] [Google Scholar]
- 70.Hatcher LQ, Karlin KD. JBIC, J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 71.Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J Am Chem Soc. 2003;125:11027–11033. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
- 72.Kundu S, Miceli E, Farquhar ER, Ray K. Dalt Trans. 2013;43:2–5. doi: 10.1039/c3dt52644e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han S, Ching Y, Rousseau DL. Nature. 1990;348:89–90. doi: 10.1038/348089a0. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Bosch I, Sharma SK, Karlin KD. J Am Chem Soc. 2013;135:16248–16251. doi: 10.1021/ja405739m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collman JP, Decréau RA. Chem Commun. 2008:5065. doi: 10.1039/b808070b. [DOI] [PubMed] [Google Scholar]
- 76.Marlin DS, Olmstead MM, Mascharak PK. Inorg Chem. 2001;40:7003–7008. doi: 10.1021/ic010523n. [DOI] [PubMed] [Google Scholar]
- 77.(a) The low temperatures and conditions employed here, somewhat extreme, are however not at all a detriment to the goal of obtaining fundamental insights via modeling. With the constructs employed, and in fact via the experimental approach of cyrogenic conditions, we are able to limit the number of factors that vary, and directly interrogate a system where the addition of phenol or acid, or Cu-ligation, are under our control and understood; these limited variations result in dramatic influences upon reactivity (i.e., resulting in O–O reductive cleavage vs breaking metal–O bonds and releasing H2O2). The simplification of a complex system, to address limited numbers of key factors pertinent to reactivity (in this case), is basic to modeling in all of science: (i) “…the synthetic modeling approach, whereby inorganic complexes that replicate aspects of the metalloenzyme active site are characterized and their reactivity is examined…”;77b (ii) “…synthetic analogues that approach or achieve one or more significant properties of a protein active site…”;77c and (iii) “…artificial model systems composed of only a few components are being used to further our understanding of…”.77dTolman WB. Inorg Chem. 2013;52:7307–7310. doi: 10.1021/ic4013813.Holm RH, Solomon EI. Chem Rev. 2004;104:347–348. doi: 10.1021/cr0206364.Marsden HR, Tomatsu I, Kros A. Chem Soc Rev. 2011;40:1572–1585. doi: 10.1039/c0cs00115e. (note: on studies of model systems for membrane fusion)
- 78.The previously noted observation that 4-CN-phenol does not interact with LS-4DCHIm requires further investigation; it is not sterically inhibited and the small difference in acidity compared to 4-NO2-phenol is not expected to translate in a major way to H-bonding ability. See Abraham MH, Grellier PL, Prior DV, Duce PP, Morris JJ, Taylor PJ. J Chem Soc, Perkin Trans 2. 1989:699–711.
- 79.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 80.Konstantinov AA. FEBS Lett. 2012;586:630–639. doi: 10.1016/j.febslet.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 81.Liang HC, Kim E, Incarvito CD, Rheingold AL, Karlin KD. Inorg Chem. 2002;41:2209–2212. doi: 10.1021/ic010816g. [DOI] [PubMed] [Google Scholar]
- 82.Ghiladi RA, Kretzer RM, Guzei I, Rheingold AL, Neuhold YM, Hatwell KR, Zuberbuhler AD, Karlin KD. Inorg Chem. 2001;40:5754–5767. doi: 10.1021/ic0105866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.