

Abstract
Thrombosis within small-diameter vascular grafts limits the development of bioartificial, engineered vascular conduits, especially those derived from extracellular matrix (ECM). Here we describe an easy-to-implement strategy to chemically modify vascular ECM by covalently linking a collagen binding peptide (CBP) to heparin to form a heparin derivative (CBP–heparin) that selectively binds a subset of collagens. Modification of ECM with CBP–heparin leads to increased deposition of functional heparin (by ~7.2-fold measured by glycosaminoglycan composition) and a corresponding reduction in platelet binding (>70%) and whole blood clotting (>80%) onto the ECM. Furthermore, addition of CBP–heparin to the ECM stabilizes long-term endothelial cell attachment to the lumen of ECM-derived vascular conduits, potentially through recruitment of heparin-binding growth factors that ultimately improve the durability of endothelialization in vitro. Overall, our findings provide a simple yet effective method to increase deposition of functional heparin on the surface of ECM-based vascular grafts and thereby minimize thrombogenicity of decellularized tissue, overcoming a significant challenge in tissue engineering of bioartificial vessels and vascularized organs.
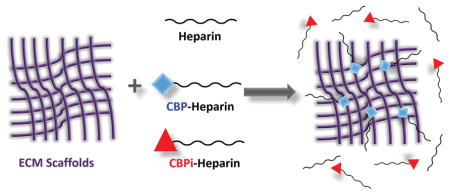
Graphical abstract
1. INTRODUCTION
Vascular thrombosis continues to be a major problem and contributor to patient morbidly in clinical vascular surgery and limits progress toward development of small caliber synthetic and biosynthetic blood vessels. Thrombosis, precipitating vessel occlusion, is the result of platelet activation when the subendothelial basement membrane containing collagens and other extracellular matrix (ECM) proteins are exposed to circulating blood at the site of anastomosis during vascular surgery.1 Thrombosis is also a major obstacle in organ and tissue engineering, where exposed ECM may be a nidus for clot formation. Despite reconstitution of decellularized tissues and organs with endothelial cells, naturally occurring three-dimensional biologic scafiolds derived from heart,2 liver,3 kidney,4 and lung5 often times require systemic anticoagulation and lead to low patency of limited duration (hours to days) after implantation into recipient animal models. Systemic administration of prophylactic, anticoagulant (e.g., heparin), or antiplatelet agents (e.g., aspirin, clopidogrel) to prevent thrombosis increases the risk of undesired bleeding, which may result in debilitating or life-threatening hemorrhage.6
Strategies to locally deliver and immobilize heparin to sites of injury at the subendothelial matrix often involves chemical cross-linking7 that changes the ultrastructure and mechanical properties of native ECM, resulting in macrophage activation, inflammation, and vascular calcification.8 To deliver heparin without altering the compliance of biosynthetic vascular grafts to ultimately reduce thrombogenicity, we synthesized a bifunctional macromolecule linking the anticoagulant heparin with a ten-amino-acid peptide sequence that specifically recognizes and binds to a subset of the collagen family of ECM macromolecules. Collagen-binding peptide, or CBP (amino acid sequence CQDSETRTFY, or Cys-Gln-Asp-Ser-Glu-Thr-Arg-Thr-Phe-Tyr), is a fragment encoding a collagen-binding domain found in fibronectin that was originally identified in 1984.9,10 We used this fragment as a targeting sequence to selectively confer the properties of heparin to decellularized tissue (ECM), where collagens and other structural macromolecules are ubiquitously present. In this study, we determine the binding specificity of CBP within ECM to demonstrate that it binds selectively to collagen, and specifically discriminates between its subtypes, to reduce the thrombogenicity of ECM compared to unmodified heparin. Moreover, we demonstrate that targeted modification of the ECM with CBP–heparin recruits heparin-binding growth factors to the matrix and improves longevity of endothelial cells grown on heparin-modified ECM used to reconstitute vascular grafts, which may provide an added benefit to improve the long-term thromboresistance of the vascular graft by maintaining an endothelial lining that protects the subendothelial matrix from circulating blood.
2. EXPERIMENTAL SECTION
2.1. Materials
CBP (CQDSETRTFY) and its inactive, non-binding form CBPi (CDEFQRSTTY) with and without biotinylation were custom synthesized by ABI Scientific, Inc. (Sterling, VA). Heparin sodium (average molecular weight 15 kDa) was purchased from Celsus Glycoscience, Inc. (Cincinnati, OH). Collagen types I (rat), II (chicken), III (human), and IV (mouse), laminin (mouse), 2-(N-morpholino) ethanesulfonic acid (MES), 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), and resazurin were obtained from Sigma-Aldrich (St. Louis, MO). Streptavidin (Alexa Fluor 594 conjugate), phalloidin (Alexa Fluor 488 conjugate), fibronectin from human plasma, and Matrigel were obtained from Thermo Fisher Scientific Inc. (Waltham, MA).
2.2. Peptide and ECM Binding Specificity
Collagens I, II, III, and IV, fibronectin, laminin, and Matrigel were coated onto 96-well black plates at 10 μg/cm2. Biotinylated CBP or CBPi were added to coated plates at 0, 1, 10, 100, and 1000 μM overnight at 4 °C, after which the plates were washed three times with PBS. Fluorescent streptavidin-594 (4 μg/mL) was added to each well and incubated at room temperature for 2 h, then thoroughly washed with PBS. The plates were read with a fluorescence microplate reader for fluorescence intensity (Ex 591 nm, Em 614 nm). All data were normalized to the background fluorescence intensity of Streptavidin-594 alone directed at each coated protein.
2.3. Peptide–Heparin Conjugation and Characterization
2.3.1. Synthesis of Heparin–Peptide Conjugates
Heparin was conjugated with CBP or CBPi via carbodiimide chemistry (Figure 1A). Heparin sodium (1 mM) was dissolved in MES buffer (pH 6.5) and activated in the presence of EDC (120 mM) and NHS (60 mM) for 4 h at room temperature. CBP or CBPi was then added to the activated heparin at 1 mM (molar ratio of heparin/peptide 1:1) and allowed to react at 4 °C overnight. The product was dialyzed against Milli-Q water with MWCO 3500 membrane to remove unbound peptides and small molecules, and lyophilized to dry.
Figure 1.

Schematic representation of (A) heparin conjugated with the collagen binding or nonbinding peptide (CBP or CBPi), and (B) ECM-modified by CBP–heparin, but not CBPi–heparin or unfractionated heparin alone.
2.3.2. Concentration of Heparin and Peptide in Conjugates
To characterize the heparin to peptide mass and molar ratio after the reaction, the lyophilized product was dissolved in water at 1 mg/mL. The concentration of heparinized macromolecule in the solution was quantified using a dimethylmethylene blue (DMMB) based glyco-saminoglycan (GAG) assay as previously described.11 Serial dilutions of heparin sodium were used to generate standard curve. The concentration of peptide was quantified using Ellman’s reagent assay12 to detect cysteine in CBP or CBPi. Serial dilutions of L-cysteine were used to generate a standard curve. The mass and molar concentrations of heparin and peptide were then calculated separately and compared to obtain the mass and molar ratios.
2.3.3. Anti-Factor Xa Activity of Heparin–Peptide Conjugates
The anticoagulant activity of heparin after conjugation with the CBP or CBPi peptide was quantified using a chromogenic anti-Factor Xa assay (Chromogenix, West Chester, OH). CBP–heparin or CBPi–heparin was diluted in PBS to 10 μg/mL and added to antithrombin to form the CBP/CBPi–heparin–antithrombin complex to neutralize factor Xa, which is proportional to the amount of active heparin in the conjugated macromolecule. The remaining activated factor Xa hydrolyzes the chromogenic substrate S-2222, which was read on a colorimetric detector at 405 nm. Heparin sodium (anti-Factor Xa activity 200 U/mg) was diluted to 0.1–0.7 U/mL in PBS as a reference standard. The anti-Factor Xa activity of CBP–heparin and CBPi–heparin is represented as U/mg.
2.4. Heparin Immobilization onto Rat Aorta ECM
All animal experiments and procedures were approved by the Animal Care and Use Committee of Northwestern University and animal care was performed in accordance with the NIH Guide for Care and Use of Laboratory Animals. Male Sprague–Dawley rats weighing 200–250 g (Charles River Laboratories, Chicago, IL) were used as donors to recover the abdominal segment of the aorta. Rat aorta ECM was isolated via decellularization as described13 using 1% Triton X for 48 h and 1.5% SDS for 48 h at room temperature, followed by 100 U/mL DNase I solution at 37 °C for 4 h. Aorta ECM was subsequently incubated with heparin sodium, CBP-conjugated heparin or CBPi-conjugated heparin (1 mg/mL) overnight at 4 °C, followed by three washes with PBS to remove excessive and unbound heparin (Figure 1B). ECM without heparin incubation served as negative, untreated controls.
2.5. Heparinized ECM Characterization
2.5.1. Quantification of Heparin Deposited onto ECM
The amount of heparin immobilized onto aorta ECM was quantified using a GAG assay.14 ECM samples (PBS, heparin, CBPi–heparin, or CBP–heparin-treated) were weighed and digested with Proteinase K, and the digested material was added to DMMB solution for colorimetric measurement. Retention of heparin on ECM after CBP–heparin treatment was assessed by incubating heparinized ECM in endothelial growth medium (EGM-2, Lonza) at 37 °C and measuring the amount of heparin remaining on ECM weekly using a GAG assay. ECM samples without heparin modification served as a baseline control.
2.5.2. Surface Morphology Characterization
Scanning electron microscopy (SEM) was used to evaluate the surface topography of modified ECM. Treated ECM (PBS, heparin, CBPi–heparin, or CBP–heparin) were fixed with 2.5% glutaraldehyde for 2 h, followed by incubation with 0.1 M sodium cacodylate buffer overnight at 4 °C. The samples were then dehydrated in a series of ethanol solutions, followed by critical point drying and gold sputter coating at the Northwestern University Biological Imaging Facility (BIF, Evanston, IL). The luminal surface of each graft was imaged with a Hitachi S4800–II cFEG SEM at the Electron Probe Instrumentation Center (EPIC) at Northwestern University (Evanston, IL).
2.5.3. Platelet Adhesion
The thrombogenic properties of modified ECM were evaluated by a platelet adhesion assay described previously.13 ECM grafts were incubated in diluted rat platelet rich plasma (2–5 ×108 platelet/mL) at 37 °C for 1 h, rinsed with warm PBS, and lysed with 2% Triton-X. Lactate dehydrogenase (LDH) released into the medium was measured by an LDH assay following the manufacturer’s protocol (Roche Molecular Diagnostics, Pleasanton, CA). LDH measured from lysates of serially diluted platelets standardized the assay to quantify the concentration of deposited platelet on ECM.
2.5.4. Clot Formation of Recalcified Whole Blood
A recalcified whole blood clotting assay was performed to assess hemocompatibility of ECM grafts treated with or without the addition of heparin, alone or conjugated to CBP/CBPi.15 Briefly, 0.01 M CaCl2 was added to anticoagulated porcine blood and incubated with ECM grafts for 1 h at room temperature. Each sample was weighed before and after incubation with recalcified blood, and the mass of the blood clotted on each sample was calculated and normalized to the weight of the ECM. Each sample containing clotted blood was subsequently embedded in paraffin, sectioned and stained with H&E for microscopic analysis.
2.5.5. Growth Factor Binding to Modified ECM
ECM samples treated with PBS alone, heparin alone, CBPi–heparin, or CBP–heparin were incubated in EGM-2 (containing 2 ng/mL recombinant human vascular endothelial growth factor, VEGF) for 3 days and washed with PBS to measure the degree of VEGF recruited to heparin-modified ECM. ECM grafts before and after EGM-2 incubation were weighed and proteins were extracted as previously described using a urea buffer.13 The amount of VEGF was measured using an ELISA for human and rat VEGF (Thermo Fisher, Waltham, MA).
2.6. Endothelial Cell Adhesion and Retention on ECM
2.6.1. Addition of Endothelial Cells to Modified ECM
Human umbilical vein endothelial cells (HUVECs, Lonza) at passages 3–8 were cultured in EGM-2 and used for cell seeding. The following groups of ECM vascular grafts (1 cm in length each, n = 3 per group) were prepared: (1) ECM control alone without heparin or peptide; (2) ECM incubated with free, unconjugated heparin sodium (1 mg/ mL); (3) ECM incubated with CBPi conjugated to heparin (1 mg/ mL); and (4) ECM incubated with CBP conjugated to heparin (1 mg/ mL). HUVECs were seeded twice onto the lumen of each graft (10000 cells/cm2 at each inoculation) with a 180° rotation of the graft at 30 min after initial seeding. Recellularized grafts were maintained in culture under static condition with medium changed every 2 days.
2.6.2. Cell Number and Viability Measurement
Resazurin was used to assess cell number and viability of HUVECs on ECM grafts over time.16 Resazurin sodium (44 μM in EGM-2) was added to cell-laden, ECM-modified grafts and incubated for 2 h and the fluorescence intensity (Ex 560 nm, Em 590 nm) was read. The number of cells was calculated based on the fluorescence reading of serially diluted HUVEC suspensions incubated in resazurin solution to develop a standard curve. Prior to performing each measurement, cell seeded grafts were moved to a new low-attachment, multiwell plate to eliminate off target readings from cells that may have migrated off the graft.
2.6.3. Cell Staining and Fluorescence Microscopy
At 4 weeks after cell seeding, each cell-seeded graft was fixed with paraformaldehyde and stained with Phalloidin-488. Fluorescence microscopy (Nikon TE2000U, Japan) was performed on the flattened luminal surface of the graft.
2.7. Statistical Analysis
All statistical data were expressed as mean ± standard deviation. Data were analyzed using one-way ANOVA with a Tukey–Kramer post-test on SigmaStat (San Jose, CA). For all comparisons, p < 0.05 was considered statistically significant.
3. RESULTS AND DISCUSSION
3.1. CBP Selectively Binds to ECM with High Specificity to Collagen IV
To test binding specificity of CBP to ECM proteins, biotinylated CBP (biotin-CQDSETRT-FY) or its inactive, nonbinding sequence CBPi (biotin-CDEFQRSTTY) was incubated with surfaces coated with a series of ECM proteins at varying peptide concentrations. Bound peptides were subsequently probed with fluorescently labeled streptavidin. CBP, but not CBPi, bound specifically to collagen IV coated surfaces with high relative affinity, and to collagen I coated surfaces to a much lesser extent. CBP bound to collagen IV in a dose-dependent manner, with a 3.85 ± 1.17-fold increase in surface fluorescence intensity at 100 μM CBP, and a 12.09 ± 1.59-fold increase in surface fluorescence intensity at 1000 μM CBP (Figure 2A), compared to background fluorescence on collagen IV coated surfaces (1.00 ± 0.07 at 0 μM CBP). CBPi, on the other hand, only resulted in a 1.22 ± 0.16-fold increase on the surface at 100 μM CBPi, and a 1.47 ± 0.09-fold increase at 1000 μM CBPi (Figure 2B), compared to background fluorescence. On collagen I coated surfaces, CBP bound to collagen I with a 3.75 ± 1.11-fold increase at 1000 μM CBP compared to the background fluorescence (1.00 ± 0.03 at 0 μM CBP), whereas CBPi at 1000 μM did not result in any increase in surface concentration. Neither CBP nor CBPi binds to collagen II, collagen III, or fibronectin. Both CBP and CBPi at 1000 μM showed minimal, through significant, levels of binding to laminin and Matrigel, suggesting a low degree of nonspecific adhesion to these ECM components.
Figure 2.

Biotinylated CBP (A) or CBPi (B) binding specificity to surfaces coated with ECM proteins, collagen I (COL I), collagen II (COL II), collagen III (COL III), collagen IV (COL IV), Matrigel (MGel), fibronectin (FN), and laminin (LN). *p < 0.05 (n = 4).
The binding between collagens (types I and IV) and CBP, is predicted to be mediated by intermolecular forces between discrete amino acids within collagen and CBP17 that contribute to the binding specificity observed above. The molecular composition of ECM is largely heterogeneous, including fibrous proteins such as collagens, soluble proteins such as fibronectin and laminin, and proteoglycans such as heparan sulfate, keratan sulfate, and chondroitin sulfate.18 The detailed composition of ECM proteins also varies from tissue to tissue, however, ECM from most tissue types contains collagen I (interstitial matrix) and collagen IV (basement membrane).18 Each collagen subtype is retained in decellularized tissues, specifically vascular (Supporting Information, Figure S1A) and renal tissues (Supporting Information, Figure S1C). To evaluate binding to collagens within these tissues, biotinylated CBP was administered at 100 μM (Supporting Information, Figure S1B,D) and colocalized with each subtype but was more prominent around collagen IV, in agreement with binding studies to individual ECM proteins described above. Taken together, this delivery strategy using CBP as a localizing moiety specifically targets collagens (type IV in particular) and may be used to selectively carry bioactive molecules to the vasculature where collagen IV defines the subendothelial basement membrane that forms the vascular network, including the macro- and micro- vasculatures within all tissues and organs. Though the use of ECM-binding peptides have been characterized elsewhere, such as a domain within placenta growth factor-2 (PlGF-2123–144)19 that strongly and promiscuously binds various ECM proteins (including fibronectin, vitronectin, fibrinogen, collagen I, etc.), this strategy using CBP imparts a high degree of specificity for a subset of ECM and collagen matrix components.
3.2. CBP–Heparin Delivers Functional Heparin to the ECM
Heparin sodium (average molecular weight 15000 Da) was conjugated to CBP or CBPi using the same carbodiimide chemistry used to link CBP to biotin. To accomplish this, CBP (or CBPi) and heparin sodium were reacted together at a 1:1 peptide to heparin molar ratio, resulting in a conjugated product consisting of 1.60 ± 0.22 mol of CBP per mol of heparin or 1.77 ± 0.05 mol of CBPi per mol of heparin (p = 0.7929, CBP–heparin vs CBPi–heparin). These results confirm that the compositions of each CBP- and CBPi–heparin variants were similar. The peptide–heparin conjugates contain >80% heparin by mass (82.9% in CBP–heparin vs 81.6% in CBPi–heparin) and >10% peptide (11.1% in CBP–heparin vs 12.0% in CBPi–heparin; Figure 3A). The bioactivity of the resulting heparin conjugate, measured by anti-Factor Xa activity, did not result in any significant difference between the two variants (36.4 ± 5.7 U/mg in CBP–heparin vs 31.6 ± 3.1 U/mg in CBPi–heparin, p = 0.4056; Figure 3B). However, there is a significant decrease in anti-Factor Xa activity compared to unconjugated heparin (200 U/mg, p < 0.01).
Figure 3.

(A) Mass ratio and (B) antifactor Xa bioactivity of peptide–heparin conjugates (CBP vs CBPi). No significant difference (p > 0.05) was found between CBP–heparin and CBPi–heparin (n = 3).
Vascular ECM from rodent aortas were modified with CBP–heparin (1 mg/mL) and led to a 7.2-fold increase in deposition of glycosaminoglycans (e.g., heparin) within ECM (15.25 ± 4.29 μg GAG/mg ECM), beyond the baseline, untreated level (2.12 ± 0.18 μg GAG/mg ECM, PBS-treated, p = 0.0056 compared to CBP–heparin-treated). Moreover, conjugation of heparin to CBP led to a 5.4-fold increase in glycosaminoglycan content beyond ECM treated with unconjugated heparin sodium alone (2.81 ± 0.19 μg GAG/mg ECM, heparin-sodium-treated, p = 0.0062 compared to CBP–heparin-treated; Figure 4A). CBPi–heparin (1 mg/mL) treated aorta ECM showed no statistically significant increase in the level of glycosaminoglycans beyond baseline (3.28 ± 0.30 μg GAG/mg ECM, CBPi–heparin-treated). An increase in the molar ratio of CBP to heparin in the preparation of CPB–heparin beyond 1:1 (peptide/heparin) also did not lead to improved heparin binding to ECM (Supporting Information, Figure S2A); therefore, a molar ratio of 1:1 was used for all further studies.
Figure 4.

Heparin concentration measured in ECM (A) immediately after treatment with PBS, heparin sodium, CBPi–heparin or CBP–heparin, and (B) up to 4 weeks after ECM modification. The dash line indicates baseline level of GAGs measured in ECM at t = 0 (n = 3).
The durability of the ECM modification by CBP–heparin was investigated by evaluating the retention of surface-bound heparin over time on ECM treated with CBP–heparin during incubation in complete EGM-2 growth medium at 37 °C (Figure 4B). Within the first week, heparin concentration decreased by 68% from 15.25 ± 4.29 μg GAG/mg ECM to 4.77 ± 1.57 μg GAG/mg ECM, but still remained significantly above the baseline, untreated level (2.12 ± 0.18 μg GAG/mg ECM, p < 0.05). By the end of the second week, heparin concentration decreased to 2.46 ± 1.55 μg GAG/mg ECM, similar to the baseline concentration (2.12 ± 0.18 μg GAG/mg ECM, p = 0.8759), and remained unchanged thereafter. This window of durability is still considered acceptable because the peak time for acute thrombosis after implantation is at early time points after restoration of blood flow.1 The risk of thrombosis decreases as endothelial cells migrate and proliferate to cover discontinuous, exposed areas at the collagen-blood interface. To evaluate the surface morphology of ECM linked to CBP–heparin, we imaged the luminal surfaces of ECM modified with each heparin derivative or heparin alone (Supporting Information, Figure S3). However, we found no qualitative differences between ECM treated with PBS (Supporting Information, Figure S3A), heparin sodium alone (Supporting Information, Figure S3B), CBPi–heparin (Supporting Information, Figure S3C), or CBP–heparin (Supporting Information, Figure S3D).
3.3. CBP–Heparin Reduces Thrombogenicity of the ECM
Platelets bind to ECM and, in particular, collagens, via surface receptors such as von Willebrand factor (VWF) and glycoprotein Ib (GPIb),20 which triggers platelet activation and clot formation. We next investigated the biological effect of heparin linked to ECM via CBP by assessing the thrombogenicity of modified ECM. After incubation with platelet-rich plasma, ECM treated with CBP–heparin at 1 mg/ mL exhibited a 71.5 ± 5.0% reduction (p < 0.01, CBP–heparin vs PBS) in the number of adherent platelets compared to PBS-treated ECM (control; Figure 5A). ECM treated with heparin sodium alone at 1 mg/mL and CBPi–heparin at 1 mg/mL exhibited minimal antiplatelet activity, with only a 5.7 ± 6.2% (p > 0.05, compared to PBS) or 12.3 ± 18.0% (p > 0.05, compared to PBS) reduction in the number of platelets adherent to ECM, respectively. Our early investigation into the influence of the molar ratio of CBP to heparin on the biological function of the conjugated molecule did not demonstrate a correlation as altering the molar ratio 1:1 to 10:1 did not result in a further reduction in the number of platelets adherent to the ECM (Supporting Information, Figure S2B). Therefore, a molar ratio of 1:1 (peptide/heparin) was determined to be the optimal reaction conditions to synthesize the conjugate and optimize the biological function.
Figure 5.

(A) Platelet adhesion to arterial ECM treated with PBS, heparin sodium, CBPi–heparin, or CBP–heparin (n = 3). The number of platelets binding to ECM treated with each condition was normalized to the number of platelets adherent to ECM treated with PBS alone as a reference. (B) Degree of clot formation by recalcified whole blood on arterial ECM with each separate modification (n = 3). H&E images show the degree of blood clot formation on ECM treated with (C) PBS, (D) heparin sodium, (E) CBPi–heparin, or (F) CBP–heparin. Percent clot formation was determined by comparing mass of the blood clot formed to the mass of the ECM graft before addition of whole blood. Scale bar = 200 μm.
To investigate the effect of CBP–heparin on clot formation, we incubated treated ECM with recalcified anticoagulant blood, which clots within 30 min in the absence of ECM. ECM linked to heparin via the CBP–heparin conjugate resulted in a significant decrease in the mass of the resulting clot, relative to the mass of ECM, at the same time 30 min point (108.1 ± 73.5%, [mass of clot/mass of ECM ×100%]) compared to ECM treated with PBS (573.7 ± 258.8%, p = 0.0134), heparin alone (612.1 ± 126.0%, p < 0.001), or CBPi–heparin (520.2 ± 212.8%, p = 0.0105; Figure 5B). Thick blood clots were observed inside the lumen and along the exterior surface of decellularized vascular ECM grafts treated with PBS alone (Figure 5C), heparin alone (Figure 5D), or CBPi–heparin (Figure 5E). However, minimal formation of thrombotic clots were observed within CBP–heparin-treated grafts, supporting the quantitative mass measurements of the retained clots noted above (Figure 5F).
The addition of heparin to ECM to reduce its thrombogenicity has been investigated by other groups using various technologies, including covalent cross-linking,21 layer-by-layer deposition,22 and polymer–ECM hybridization.13 We previously published several strategies to immobilize heparin to vascular ECM via synthetic polymer–ECM composites to reduce ECM thrombogenicity.13,14 Specifically, poly(1,8-octa-methylene citrate) (POC) or its derivative poly(1,8-octa-methylene citrate)-co-cysteine (POC-Cys) was hybridized onto vascular ECM to form a polymer–ECM composite, which allowed for heparin immobilization via either carbodiimide chemistry13 or maleimide–thiol “click” chemistry.14 However, neither POC nor POC-Cys is soluble in water, and the hybridization process involves complete ECM dehydration and subsequent rehydration. This hybridization strategy may not be optimal for tissues with a highly organized microvascular network, such as vital organs, and may damage the ECM ultrastructure during dehydration and rehydration. In contrast, the method presented here utilizing CBP as an intermediate linker to immobilize heparin can be carried out entirely in a hydrophilic environment with an equivalent antithrombotic effect as our previous polymer–ECM composite strategies.
3.4. Immobilization of Heparin to ECM via CBP Stabilizes Endothelial Cell Attachment
An intact, continuous endothelium is critical to maintain proper function of the vascular system, including maintaining the selective barrier for solute filtration within the kidney, preventing thrombosis within the vasculature, and controlling inflammation and trafficking of leukocytes from the circulation and into extravascular tissues.23 Endothelial cells are often seeded onto the lumen of vascular networks to provide these functions in various tissue engineering applications such as vascular grafts or vascularized thick scaffolds using decellularized tissues as matrices.24,2,25 The long-term stability of endothelial cells is critical to maintaining vascular integrity, in general, including the functional durability of bioengineered tissues.
We found that endothelial cells seeded effectively onto the lumen of ECM-based vascular grafts with good biocompatibility (i.e., cell adhesion and viability) and developed a continuous endothelial cell layer during the first day, regardless of the ECM modification strategy used (Figure 6A). However, as cell-laden grafts were maintained in culture for up to 4 weeks, the number of adherent endothelial cells bound to ECM decreased significantly for ECM modified with PBS, heparin alone, or CBPi–heparin (Figure 6A,B). Only ECM modified with CBP–heparin supported and maintained the level of adherent endothelial cells over the 4-week time course study (Figure 6A,B). ECM grafts treated with CBP or CBPi peptides alone, both without conjugation to heparin, likewise did not preserve endothelial cell adhesion at 4 weeks (Supporting Information, Figure S4). Taken together, these data suggest a precise, and important role for specifically linking heparin to ECM via the CBP–heparin conjugate, which results in both improved anticoagulation and long-term endothelial cell attachment and maintenance of an endothelial layer, important biological functions that are specifically mediated by the heparin moiety within the conjugate.
Figure 6.

(A) Phalloidin staining of endothelial cells seeded on the lumen of arterial ECM treated with PBS, heparin sodium, CBPi–heparin, or CBP–heparin at days 1 and 28 (scale bar = 100 μm). (B) Quantitative analysis of adherent cell number on ECM at day 28, with the dash line indicating the initial cell seeding density at day 1 (n = 3). (C) Quantitative analysis of VEGF concentration on ECM after incubating in endothelial cell complete medium (EGM-2, n = 3).
To investigate this observation further, we hypothesized that improved long-term attachment of endothelial cells to the ECM by CBP–heparin may be due to recruitment early on of soluble heparin-binding growth factors in the EGM-2 media, including vascular endothelial growth factor (VEGF),26,27 basic fibroblast growth factor (bFGF)28 and heparin-binding EGF-like growth factor (HB-EGF).29 A quantitative analysis to measure VEGF within ECM grafts after incubation in EGM-2 for 3 days revealed that VEGF concentrations increased for all groups from the baseline level (26.8 ± 8.4 pg/mg ECM, before incubation in EGM-2). However, the increase was significantly more pronounced for ECM pretreated with CBP–heparin (132.0 ± 37.3 pg/mg ECM), compared to ECM treated with PBS alone (54.4 ± 6.1 pg/mg ECM, p < 0.05), ECM prepared with unconjugated heparin (46.5 ± 12.1 pg/mg ECM, p < 0.05), and ECM prepared with CBPi–heparin (58.0 ± 23.3 pg/ mg ECM, p < 0.05; Figure 6C). ECM-associated growth factors, such as VEGF, promote endothelial adhesion, migration, and survival,30 which may explain the long term adhesion and maintenance of endothelial cells growing on CBP–heparin-treated ECM at 4 weeks. The dual effect of the CBP–heparin to both promote the long-term retention of endothelial cells on the lumen and improve thromboresistance at early time points will together reduce the risk of vascular occlusion and stenosis within the engineered tissue vasculature and improve graft function.
3.5. Limitations and Prospects
This strategy to non-covalently immobilize heparin onto ECM using CBP as an intermediate linker to bring heparin to the ECM also provides a platform for the immobilization of other bioactive molecules using a variety of ECM binding peptides. In this study, we chose to use CBP (CQDSETRTFY) as a proof-of-principle to develop targetable ECM modifiers to deliver bioactive agents (i.e., heparin) to vascular ECM. CBP has been utilized by a number of groups for its specific binding to collagens. For example, Panitch et al. used CBP as an anchor to immobilize various bioactive molecules for modulating collagen fibrillogenesis,31,32 inhibiting MMP-mediated collagen degradation,33 and inhibition of platelet adhesion and activation on collagen during balloon angioplasty.34 Sistiabudi et al. also employed CBP to modify the retinal Bruch membrane, to restore the retinal pigment epithelial (RPE) layer.35–37
In contrast, our study demonstrates the utility of heparin immobilization to prevent clot formation due to the removal of endothelial cells during decellularization and incomplete revascularization of the endothelial layer during recellularization. However, one limitation using CBP to immobilize heparin onto the ECM, as demonstrated in our study, is its limited long-term durability when bound to ECM (Figure 4B). This limitation could potentially be overcome by selecting or identifying other peptide sequences with higher affinity to ECM proteins or using technologies to uncover novel sequences.38 Another limitation is reduced heparin anticoagulant activity after peptide conjugation, as demonstrated by decreased anti-Factor Xa activity after conjugation. Using chemistry other than the EDC/NHS carbodiimide chemistry to conjugate peptide with heparin may help preserve heparin’s bioactivity. Nevertheless, it is important to point out that although CBP–heparin has lower anti-Factor Xa activity in solution compared to heparin alone, once bound to ECM the effective antithrombotic activity of the conjugated heparin is significantly enhanced beyond ECM treated with unconjugated heparin alone due to its higher affinity for the matrix (Figure 5).
4. CONCLUSIONS
We developed a strategy to synthesize CBP–heparin that selectively bindings to ECM proteins collagen IV and, to a lesser extent, collagen I. Moreover, we show that vascular ECM modified with CBP–heparin exhibits reduced thrombogenicity, improved long-term adhesion of endothelial cells, and increased binding of the growth factor VEGF. Taken together, we describe an easy to implement approach to help overcome the challenge of thrombosis in various tissue engineering applications, where decellularized tissues are used as biologic scaold materials. This technology opens up new opportunities to develop patient-specific vascular grafts and vascular network in engineered organs using decellularized tissue ECM as scaffolds.
Supplementary Material
Acknowledgments
This work is supported by the McCormick Foundation via Northwestern Memorial Hospital, the National Institute of Health (5R01EB017129 and K08DK101757), the Dixon Translational Research Grants Innovation Award from the Northwestern Memorial Foundation, American Heart Association (AHA) Midwest Affiliate Postdoctoral Fellowship (14POST20160091), and the Chicago Biomedical Consortium (CBC) Postdoctoral Award (PDR-008). We thank the generous support of Dr. Frank A. Krumlovsky. The authors would also like to thank the Northwestern University Microsurgical Core for performing animal surgeries, and Chongwen Duan for performing SEM imaging.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-mac.6b01330.
Figure S1: Immunofluorescence staining of collagen I (green) and collagen IV(red) in decellularized rat aorta ECM (A) and renal ECM (C). Streptavidin-594 (red) staining of biotinylated CBP on decellularized rat aorta ECM (B) and renal ECM (D). Scale bar = 100 μm. Figure S2: Heparin quantification (A) and platelet adhesion (B) on ECM treated with conjugates of varying CBP to heparin molar ratio during optimization of CBP–heparin conjugates. *indicates p < 0.05. Figure S3: SEM images of the lumen surface of ECM treated with PBS (A), heparin sodium (B), CBPi–heparin (C), or CBP–heparin (D). Figure S4: Phalloidin staining of endothelial cells seeded on ECM treated with CBP or CBPi (both without heparin) at day 1 and day 28 (n = 3). Minimal to no cells remain on ECM at day 28 for both CBP and CBPi without heparin. Scale bar = 100 μm (PDF).
References
- 1.Wang Y, Gallant RC, Ni H. Extracellular matrix proteins in the regulation of thrombus formation. Curr Opin Hematol. 2016;23(3):280–7. doi: 10.1097/MOH.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 2.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat Med. 2008;14(2):213–21. doi: 10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- 3.Soto-Gutierrez A, Zhang L, Medberry C, Fukumitsu K, Faulk D, Jiang H, Reing J, Gramignoli R, Komori J, Ross M, Nagaya M, Lagasse E, Stolz D, Strom SC, Fox IJ, Badylak SF. A whole-organ regenerative medicine approach for liver replacement. Tissue Eng, Part C. 2011;17(6):677–86. doi: 10.1089/ten.tec.2010.0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caralt M, Uzarski JS, Iacob S, Obergfell KP, Berg N, Bijonowski BM, Kiefer KM, Ward HH, Wandinger-Ness A, Miller WM, Zhang ZJ, Abecassis MM, Wertheim JA. Optimization and critical evaluation of decellularization strategies to develop renal extracellular matrix scaffolds as biological templates for organ engineering and transplantation. Am J Transplant. 2015;15(1):64–75. doi: 10.1111/ajt.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva I, Ikonomou L, Kotton D, Vacanti JP. Regeneration and orthotopic transplantation of a bioartificial lung. Nat Med. 2010;16(8):927–33. doi: 10.1038/nm.2193. [DOI] [PubMed] [Google Scholar]
- 6.Momtahan N, Sukavaneshvar S, Roeder BL, Cook AD. Strategies and processes to decellularize and recellularize hearts to generate functional organs and reduce the risk of thrombosis. Tissue Eng, Part B. 2015;21(1):115–32. doi: 10.1089/ten.TEB.2014.0192. [DOI] [PubMed] [Google Scholar]
- 7.Murugesan S, Xie J, Linhardt RJ. Immobilization of heparin: approaches and applications. Curr Top Med Chem. 2008;8(2):80–100. doi: 10.2174/156802608783378891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt CE, Baier JM. Acellular vascular tissues: natural biomaterials for tissue repair and tissue engineering. Biomaterials. 2000;21(22):2215–31. doi: 10.1016/s0142-9612(00)00148-4. [DOI] [PubMed] [Google Scholar]
- 9.Yamada KM, Kennedy DW. Dualistic Nature of Adhesive Protein Function - Fibronectin and Its Biologically-Active Peptide-Fragments Can Autoinhibit Fibronectin Function. J Cell Biol. 1984;99(1):29–36. doi: 10.1083/jcb.99.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boucaut JC, Darribere T, Poole TJ, Aoyama H, Yamada KM, Thiery JP. Biologically-Active Synthetic Peptides as Probes of Embryonic-Development - a Competitive Peptide Inhibitor of Fibronectin Function Inhibits Gastrulation in Amphibian Embryos and Neural Crest Cell-Migration in Avian Embryos. J Cell Biol. 1984;99(5):1822–1830. doi: 10.1083/jcb.99.5.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farndale RW, Sayers CA, Barrett AJ. A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect Tissue Res. 1982;9(4):247–8. doi: 10.3109/03008208209160269. [DOI] [PubMed] [Google Scholar]
- 12.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7(2):88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 13.Jiang B, Akgun B, Lam RC, Ameer GA, Wertheim JA. A polymer-extracellular matrix composite with improved thromboresistance and recellularization properties. Acta Biomater. 2015;18:50–8. doi: 10.1016/j.actbio.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang B, Suen R, Wang JJ, Zhang ZJ, Wertheim JA, Ameer GA. Mechanocompatible Polymer-Extracellular-Matrix Composites for Vascular Tissue Engineering. Adv Healthcare Mater. 2016;5(13):1594–605. doi: 10.1002/adhm.201501003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshi RA, Van Lith R, Jen MC, Allen JB, Lapidos KA, Ameer G. The blood and vascular cell compatibility of heparin-modified ePTFE vascular grafts. Biomaterials. 2013;34(1):30–41. doi: 10.1016/j.biomaterials.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267(17):5421–6. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 17.Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24(6):389–99. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200(4):423–8. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 19.Martino MM, Briquez PS, Guc E, Tortelli F, Kilarski WW, Metzger S, Rice JJ, Kuhn GA, Muller R, Swartz MA, Hubbell JA. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science. 2014;343(6173):885–8. doi: 10.1126/science.1247663. [DOI] [PubMed] [Google Scholar]
- 20.Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102(2):449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 21.Conklin BS, Richter ER, Kreutziger KL, Zhong DS, Chen C. Development and evaluation of a novel decellularized vascular xenograft. Med Eng Phys. 2002;24(3):173–83. doi: 10.1016/s1350-4533(02)00010-3. [DOI] [PubMed] [Google Scholar]
- 22.Bruinsma BG, Kim Y, Berendsen TA, Ozer S, Yarmush ML, Uygun BE. Layer-by-layer heparinization of decellularized liver matrices to reduce thrombogenicity of tissue engineered grafts. J Clin Transl Res. 2015;1(1):04. doi: 10.18053/jctres.201501.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang B, Perrin L, Kats D, Meade T, Ameer G. Enabling non-invasive assessment of an engineered endothelium on ePTFE vascular grafts without increasing oxidative stress. Biomaterials. 2015;69:110–20. doi: 10.1016/j.biomaterials.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lichtenberg A, Tudorache I, Cebotari S, Ringes-Lichtenberg S, Sturz G, Hoeffler K, Hurscheler C, Brandes G, Hilfiker A, Haverich A. In vitro re-endothelialization of detergent decellularized heart valves under simulated physiological dynamic conditions. Biomaterials. 2006;27(23):4221–9. doi: 10.1016/j.biomaterials.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 25.Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A, Soker S. The Use of Whole Organ Decellularization for the Generation of a Vascularized Liver Organoid. Hepatology. 2011;53(2):604–617. doi: 10.1002/hep.24067. [DOI] [PubMed] [Google Scholar]
- 26.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular Endothelial Growth-Factor Is a Secreted Angiogenic Mitogen. Science. 1989;246(4935):1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 27.Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161(2):851–8. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- 28.Sakiyama-Elbert SE, Hubbell JA. Development of fibrin derivatives for controlled release of heparin-binding growth factors. J Controlled Release. 2000;65(3):389–402. doi: 10.1016/s0168-3659(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 29.Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A Heparin-Binding Growth-Factor Secreted by Macrophage-Like Cells That Is Related to Egf. Science. 1991;251(4996):936–939. doi: 10.1126/science.1840698. [DOI] [PubMed] [Google Scholar]
- 30.Hutchings H, Ortega N, Plouet J. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 2003;17(11):1520–2. doi: 10.1096/fj.02-0691fje. [DOI] [PubMed] [Google Scholar]
- 31.Paderi JE, Panitch A. Design of a synthetic collagen-binding peptidoglycan that modulates collagen fibrillogenesis. Biomacromolecules. 2008;9(9):2562–6. doi: 10.1021/bm8006852. [DOI] [PubMed] [Google Scholar]
- 32.Paderi JE, Sistiabudi R, Ivanisevic A, Panitch A. Collagen-binding peptidoglycans: a biomimetic approach to modulate collagen fibrillogenesis for tissue engineering applications. Tissue Eng, Part A. 2009;15(10):2991–9. doi: 10.1089/ten.tea.2009.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart K, Paderi J, Snyder PW, Freeman L, Panitch A. Collagen-binding peptidoglycans inhibit MMP mediated collagen degradation and reduce dermal scarring. PLoS One. 2011;6(7):e22139. doi: 10.1371/journal.pone.0022139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paderi JE, Stuart K, Sturek M, Park K, Panitch A. The inhibition of platelet adhesion and activation on collagen during balloon angioplasty by collagen-binding peptidoglycans. Biomaterials. 2011;32(10):2516–23. doi: 10.1016/j.biomaterials.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Sistiabudi R, Ivanisevic A. Collagen-binding peptide interaction with retinal tissue surfaces. Langmuir. 2008;24(5):1591–4. doi: 10.1021/la703561d. [DOI] [PubMed] [Google Scholar]
- 36.Sistiabudi R, Paderi J, Panitch A, Ivanisevic A. Modification of native collagen with cell-adhesive peptide to promote RPE cell attachment on Bruch’s membrane. Biotechnol Bioeng. 2009;102(6):1723–9. doi: 10.1002/bit.22215. [DOI] [PubMed] [Google Scholar]
- 37.Sistiabudi R, Ivanisevic A. Dip-Pen Nanolithography of Bioactive Peptides on Collagen-Terminated Retinal Membrane. Adv Mater. 2008;20(19):3678–3681. [Google Scholar]
- 38.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380(6572):364–6. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.