

Abstract
Objective
Cytosolic DNA sensors detect microbial DNA and promote type I interferon and pro-inflammatory cytokine production through the adaptor stimulator of interferon genes (STING) to resolve infection. Endogenous DNA also engages the STING pathway, contributing to autoimmune disease. We identified a novel role for STING in bone in arthritic DNase II/IFNaR double deficient (DKO) mice, and sought to define the bone phenotype in these mice and to address mechanism.
Methods
Bone parameters were evaluated in DKO, STING/DNaseII/IFNaR triple deficient and control mice by microcomputed tomography and histomorphometry. Cell culture techniques were employed to determine parameters of osteoclast and osteoblast differentiation and function. Nanostring and Affymetrix array analyses were performed to identify factors promoting ectopic bone formation.
Results
Despite the expression of pro-inflammatory cytokines that would be expected to induce bone loss in the skeleton in DKO mice, we demonstrate the paradoxical accumulation of bone in the long bones and spleen, sites of erythropoiesis and robust DNA accrual, as well as the induction of factors promoting osteoblast recruitment and function. STING deficiency significantly inhibits this bone accrual.
Conclusions
These data reveal a novel role for cytosolic DNA sensor pathways in bone in the setting of autoimmune disease. We demonstrate the requirement of an intact STING pathway for bone formation in this model, a finding that may have relevance to autoimmune diseases in which DNA plays a pathogenic role. Identification of pathways linking innate immunity and bone could reveal novel targets for the treatment of bone abnormalities in autoimmune diseases.
Introduction
Innate immune pattern recognition receptors, including cytosolic DNA sensors, detect nucleic acids from microbial organisms and orchestrate immune events and the production of cytokines to clear infection (1). Upon detection of DNA, cytosolic sensors including cGAS, IFI16/Ifi204 and others, signal through STING, leading to the nuclear translocation of IRF-3 and NF-κB, and production of type I interferons and pro-inflammatory cytokines, respectively (2).
Endogenous DNA derived from stressed or dying cells can activate these same cytosolic DNA sensing pathways, contributing to autoimmune disease (3–5). Inactivation of intracellular DNases in animal models leads to host DNA accrual, resulting in autoinflammatory and autoimmune disease. For example, deficiency in the endonuclease DNase III (TREX1) in mice results in systemic inflammation and myocarditis due to accrual of cytosolic DNA and STING-dependent production of type 1 interferons and pro-inflammatory cytokines by non-hematopoietic cells (6, 7). In humans, loss-of-function mutations in TREX1 are associated with Aicardi-Goutieres Syndrome, chilblain lupus, and SLE (8–13). Further evidence for a role for the STING pathway in autoimmunity is provided by the discovery of human gain of function mutations in Sting (Tmem173), leading to constitutive activation of STING in fibroblasts and endothelial cells, resulting in the clinical syndrome STING-associated vasculopathy with onset of infancy (SAVI). Patients with SAVI develop vasculopathy, skin lesions, pulmonary fibrosis, and arthritis (14).
Finally, endogenous DNA accrual from inactivation of DNase II, a lysosomal nuclease that degrades double-stranded DNA, is also associated with autoimmunity. In humans, SNPs in the promoter region of the DNase II gene that result in reduced DNase II activity are associated with the development of rheumatoid arthritis (15). In mice deficient in DNase II, DNA accumulates in phagolysosomes and secondarily in the cytosol of multiple cell types including macrophages and fibroblasts (16). Macrophages also engulf nuclei from apoptotic cells, leading to DNA accrual and the production of TNF, IL-1β, and IL-6, as well as type I interferons. Excessive type I IFN production leads to anemia-driven embryonic lethality, from which these mice are rescued when the gene for the type I interferon receptor (IFNaR) is also deleted. DNase II−/− IFNaR−/− double deficient (DKO) mice survive, but develop a distal, erosive inflammatory arthritis by 3 months of age, which is absent in DNaseII+/− IFNaR−/− (Het) controls (17). This arthritis is entirely abrogated in the setting of STING deficiency (4, 18), and is significantly attenuated by loss of the inflammasome-promoting cytosolic DNA sensor AIM2 (4, 5). DKO mice also develop clinical manifestations of SLE through pathways that are independent of STING but rely on Unc93B1, an adaptor protein required for endosomal TLR activity (4, 19).
In the setting of inflammatory arthritis, pro-inflammatory cytokines contribute to both articular and systemic bone loss due to enhanced osteoclast-mediated bone resorption and inhibition of osteoblast-mediated bone formation (20–23). In DKO mice, we therefore anticipated the development of osteoporosis due to the systemic production of TNF and IL-1β (17, 24), which should induce osteoclastogenesis and bone resorption (25–28). In addition, type I IFNs inhibit osteoclast differentiation, and IFNaR deficient mice demonstrate enhanced osteoclastogenesis and lose bone systemically (29). However, in the setting of DNA accrual in the DKO model of arthritis, we found an unexpected and dramatic enhancement of bone formation in the long bones and spleens, two sites of erythropoiesis and local DNA accumulation (17). Furthermore, STING deficiency abrogated bone accrual, revealing a potential role for this cytosolic DNA sensor pathway in bone. Collectively, these data may provide insights into bone disorders occurring in the context of autoimmunity.
Materials and Methods
Mouse Strains
C57BL/6 DNase II+/− embryos were provided by Dr. S. Nagata (Osaka Medical School) through the RIKEN Institute, and mice were crossed to IFNaR−/− C57BL/6 mice to produce DNase II−/− IFNaR−/− double knockout (DKO) and DNase II+/− IFNaR−/− heterozygous (Het) mice. DKO mice were intercrossed with STING-deficient mice to yield STKO mice (4). All animals were handled according to protocols approved by the IACUC at the University of Massachusetts Medical School.
Histopathologic Analyses
Hind limbs were fixed in 4% paraformaldehyde, decalcified in 15% EDTA, embedded in paraffin, and sectioned at 5μm. Sections were deparaffinized and stained with either H&E or TRAP (30). Spleens were fixed, embedded in methylmethacrylate, sectioned and then stained with H&E, TRAP, Von Kossa, or Goldner’s trichrome.
Microcomputed tomography (microCT)
Fixed bones were imaged using a Scanco Medical μCT 40 at 70kVp and 114μA. Analyses included trabecular bone within the entire femur from the proximal to distal growth plates, a 0.5 mm section in the central diaphysis of cortical bone, and trabecular bone within the rostral to caudal growth plates of the L3 lumbar vertebrae. Segmentation parameters included the values: 0.8 Gauss sigma, 1.0 Gauss support, and a threshold of 220–1000 Hounsfield units.
Static histomorphometric analysis
Femurs were fixed in 10% neutral buffered formalin and embedded in methylmethacrylate (22). The proximal femoral metaphysis was sectioned longitudinally (5μm), mounted to slides with non-fluorescent medium, and stained with McNeal’s trichrome (osteoid) and TRAP (osteoclasts). A region of interest approximately 4μm2 within the secondary spongiosa (~0.5mm distal to the growth plate) was defined, and osteoid area, bone area, bone surface, osteoblast surface and osteoclast surface were measured using a Nikon Optiphot 2 microscope interfaced to a semiautomatic analysis system (Bioquant OSTEO 7.20.10; Bioquant Image Analysis). Measurements were performed on two sections/sample (separated by ~25μm) and summed prior to normalization to obtain a single measure per sample. All parameters were measured and defined in accordance with ASBMR standards (31).
ELISAs
Serum levels of osteocalcin (Biomedical Technologies), TRAP-5b (Immunodiagnostic Systems), and CTX-1 (Immunodiagnostic Systems) were determined by ELISA.
Colony Forming Unit (CFU) Assays
Total bone marrow was flushed from femurs and RBCs were lysed. 2 × 10^6 cells/well were plated in medium containing α-MEM without ascorbic acid, 20% FBS, and pen/strep. After 2 days, cells were washed and exposed to osteoblast differentiation medium (50ug/ml ascorbic acid and 10mM β-glycerophosphate) for 14 days. Colony formation was assessed by staining for alkaline phosphatase (Sigma).
Calvarial Osteoblast Cultures
Primary osteoblasts were isolated from calvariae of C57BL/6 pups (Charles River). 8×10^4 cells/well were cultured in α-MEM supplemented with 10% FBS and treated with 50μg/ml ascorbic acid and 10mM β-glycerophosphate. On day 4, the cells were transfected with 1μg/μl of poly(dA:dT) using Lipofectamine 2000 (Invitrogen). RNA was isolated 5 days after transfection and subjected to quantitative polymerase chain reaction (qPCR).
qPCR
500ng of total RNA was amplified. Gene expression was normalized to the housekeeping gene hydroxymethylbilane synthase (HMBS). Primers were obtained from Qiagen. Data are expressed as the fold increase in gene expression compared to normalized lipofectamine controls, using the 2−ΔΔCT method.
Osteoclast Assays
Experiments were performed in α-MEM containing 10% FBS and pen/strep. Cells were flushed from the bone marrow and differentiated in 40ng/ml M-CSF (R&D) for 4 days. For differentiation, precursors were seeded at a density of 6,000 cells/well and differentiated in medium containing 20ng/ml M-CSF and 10ng/ml RANKL (R&D) for 5 days. Half of the medium was replaced with fresh medium/cytokines on day 3. On day 5, cells were fixed and stained for TRAP (Sigma). TRAP-stained osteoclasts with 3–10 nuclei were counted.
Precursors were also seeded at a density of 40,000 cells/well on hydroxyapatite-coated plates (Corning) and differentiated in α-MEM containing 40ng/ml of M-CSF and 20ng/ml of RANKL for 10 days. Half of the medium was replaced with fresh medium/cytokines every two days. Cells were removed with 10% bleach. Wells were rinsed and scanned on a flatbed scanner (Microtek 9800 XL). The percentage of resorbed area was determined using NIH ImageJ software.
Xrays
Organs were imaged for 1 second at 35kV using the Faxitron MX-20 machine.
Nanostring
Total RNA was isolated (RNeasy kit, Qiagen) and quantitated via a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). 100ng of RNA was hybridized and quantified with the NanoString nCounter analysis system (NanoString Technologies). Gene-expression data were normalized to internal positive and negative control sets and to three housekeeping genes: GAPDH, β-glucuronidase (GUSB), and hypoxanthine phosphoribosyltransferase 1 (HPRT1). All values were scaled by a log2(X-min(X)+1) function and a heatmap was generated using open-source R-based software at UMMS.
Gene Array
Total RNA was purified from spleens using the RNeasy kit (Qiagen), and cDNA was generated from 200ng of total RNA using the SensationPlus FFPE Amplification and WT Labeling Kit (Affymetrix). Samples were run on Affymetrix GeneChip Mouse Transcriptome Arrays 1.0 (MTA), and quality control was performed using Expression Console. Expression values were RMA normalized and detection p-values for each probe set determined with the detectable above background (DABG) algorithm. Biological replicate average, fold change, and ANOVA p-values were calculated between groups using the transcriptome analysis console (TAC, Affymetrix). Differential expression of mRNAs was identified as those significantly changing at least 1.5 fold with an ANOVA p-value <0.05. All values were scaled by a log2(X-min(X)+1) function and a heatmap was generated using open-source R-based software.
Statistical analysis
Statistical significance was analyzed with the unpaired, two-tailed Student’s t test or ANOVA for multiple comparisons. Data are presented as the mean ± SEM. Statistical significance is represented by the following notation in the figures: p<0.05=*, p<0.01=**, and p<0.001=***.
Results
Deficiency of DNase II promotes trabecular bone formation
As reported previously, DNase II/IFNaR double deficient (DKO) mice develop a distal polyarthritis accompanied by bone erosion, resulting from the local expression of pro-inflammatory cytokines (17). We confirmed the presence of synovitis, pannus formation and osteoclast-mediated articular erosion (17) in the distal joints of DKO mice and the absence of these findings in Het littermate controls (Figure 1A). Thus, in the joint microenvironment, typical osteoclast-mediated bone loss occurs. However, despite local and systemic production of TNF, IL-1β and IL-6 that promote osteoclastogenesis, there is a surprising and significant accrual of trabecular bone in the tibiae adjacent to inflamed ankle joints, compared to Het controls (Figure 1B). This trabecular bone formation occurs despite the presence of an increased number of TRAP-expressing osteoclasts (Figure 1B) compared to control mice, and is preceded by a marrow infiltrate that we have previously shown to be dependent on the expression of both the STING and endosomal TLR pathways (32).
Figure 1. Trabecular bone accrual occurs over time in long bones of DKO mice.

A) Midfoot, 10 month-old mice (female, n=4 mice/genotype): Representative H&E (upper panels) and TRAP (lower panels) stained images of inflammation and articular bone erosions (arrows). Magnification 4x and 20x, respectively. B) Tibiae, 10 month-old mice (female, n=6 mice/genotype). Representative images of H&E and TRAP stained sections. Arrows indicate trabecular bone. Magnification 4x. C) Micro-CT images of femurs at 2, 6, 10, and 16 months (female, n=3–7 mice/genotype/age). Left images: cortical and trabecular bone. Right images: 3D reconstruction of trabecular bone. D) Quantitation of micro-CT data from 10 month-old mice (female, n=6–7 mice/genotype) for trabecular bone volume per total volume (BV/TV), trabecular bone surface, and trabecular connectivity (con.) density. Data represent 3 individual experiments. Values are the mean ± SEM compared to Het: **=p<0.01, ***=p<0.001.
To quantitate bone accrual in the long bones, we analyzed the femurs of female DKO mice by microCT. As expected, Het control mice lose trabecular bone over time due to aging, as well as to the absence of type I interferon signaling, which promote osteoclastogenesis and bone loss (29). In contrast, arthritic DKO mice (that are also deficient in type I interferon signaling) paradoxically demonstrate a dramatic accrual of trabecular bone that appears by 5–6 months and progresses over time, such that by 16 months the marrow cavity is largely replaced by bone (Figure 1C). MicroCT analyses of femurs from female mice confirmed a significant increase in trabecular bone volume/total volume (BV/TV), trabecular surface, and trabecular connectivity density in 10 month-old DKO mice compared to controls (Figure 1D). Further analysis revealed a trend toward a decrease in cortical bone volume/total volume and a significant decrease in cortical thickness in DKO compared to Het mice (Figure S1), demonstrating a loss of cortical bone over time in female DKO mice compared to Het controls. This loss may result from the accrual of trabecular bone, which provides mechanical support and reduces load on cortical bone, or alternatively may be the result of a differential effect on bone cells in trabecular versus cortical bone. The trabecular bone phenotype was more dramatic in female mice. However, microCT analyses of femurs from male DKO mice revealed a similar phenotype, with a trend toward increased trabecular bone surface and trabecular connectivity density as well as a significant loss of cortical bone compared to controls (Figure S2). Thus, despite the expression of pro-inflammatory cytokines, the effects of aging on bone, and the lack of type I interferon signaling, all of which would be predicted to result in bone loss, mice with DNase II deficiency develop accrual of trabecular bone in the long bones over time.
Bone accrual is accompanied by an increase in osteoblast number and function
To determine mechanism, parameters of osteoblast and osteoclast number and function were examined. Static histomorphometric measurements of trabecular bone in femurs from 10-month old female mice showed a marked increase in osteoblasts (osteoblast surface, Ob.S) and production of osteoid (osteoid area, O.Ar.) in DKO mice compared with controls (Figure 2A). The ratios of Ob.S/bone surface (BS) and O.Ar./B.Ar were not significantly increased, demonstrating that there are appropriate numbers of osteoblasts for the amount of bone produced (Figure 2A). A significant increase in bone surface covered by osteoclasts (osteoclast surface, OC.S) was also observed in DKO mice compared to controls, demonstrating a concomitant increase in osteoclast number (Figure 2B). Again, similar ratios of OC.S/BS exist between Het and DKO mice, suggesting that the number of osteoclasts lining bone is the expected number for the bone present. In this model, the rapid deposition of woven bone has precluded attempts at dynamic measurement of bone formation rates.
Figure 2. Osteoblast and osteoclast parameters in DKO mice.

A and B) Static histomorphometry, femurs, 10 months (female, n=4–5 mice/genotype). Abbreviations: osteoblast surface (Ob. S), bone surface (BS), osteoid area (O. Ar.), bone area (B.Ar.), and osteoclast surface (OC. S). C) Representative images from osteoblast colony forming unit (CFU) assays, 2.5 month-old mice (male, n=5 mice/genotype). Colonies are stained for alkaline phosphatase (pink). D and E) Serum bone turnover markers: Osteocalcin, C-terminal peptides of type I collagen (CTX-1), and Trap5b. 10 month-old mice (female, n=6–12 mice/genotype). F) Number of Trap+ cells in osteoclast differentiation cultures. 2.5 month-old mice (male, n=3 mice/genotype). G) Percentage of hydroxyapatite area resorbed by osteoclasts. 2.5 month-old mice (female and male, n=6–7 mice/genotype). Values are the mean ± SEM compared to Het; *=p<0.05, **=p<0.01, ***=p<0.001.
To determine whether this bone phenotype is a result of increased osteoblast number, colony forming unit (CFU) assays were performed. Cells from DKO bone marrow demonstrated an increased number of colonies compared with controls, consistent with an increased number of mesenchymal precursors in the marrow (Figure 2C). Serum markers of bone turnover including osteocalcin (bone formation), CTX-1 (bone resorption) and Trap5b (osteoclast number) in 10 month-old female mice demonstrated that serum levels of osteocalcin trended higher in DKO mice compared to controls (Figure 2D), consistent with an increase in osteoblast activity. Levels of CTX-1 and Trap5b were significantly elevated compared to controls (Figure 2E), indicating increased osteoclast activity and number. To determine whether there is an intrinsic alteration in either differentiation or function of osteoclasts that could be contributing to trabecular bone accrual, osteoclast differentiation and resorption assays were performed. These studies showed a trend toward increased osteoclast differentiation in DKO compared to controls that was not significant (Figure 2F) and no significant difference in resorption on hydroxyapatite-coated plates (Figure 2G). Overall, these data suggest that the trabecular bone accrual in DKO mice results from an increase in osteoblast number, with an accompanying increase in osteoclasts. The balance between osteoblast and osteoclast activity over time thus appears to skew in favor of bone formation, and likely does not result from a decrease in osteoclast number or function, although the contribution of abnormal osteoclast-mediated bone resorption in vivo cannot be completely excluded.
Ectopic Bone Forms in DKO spleens with aging
Imaging of 10 month-old female mice was performed to evaluate the entire skeleton. Unexpectedly, X-ray images revealed multiple calcified nodules in the spleens of DKO mice. Splenic enlargement occurs in DKO mice compared to controls, as previously reported (17) and calcified nodules in the spleen measured up to 3mm (Figures 3A, B). Histologic staining demonstrated woven bone within splenic white pulp (Figure 3C), with osteoblasts (Figure 3D) and osteoclasts (Figure 3E) lining the surface of the bone. Additional stains showed that the bone is mineralized (Figure 3F), and robust osteoid production by osteoblasts is present at bone surfaces (Figure 3G). Analysis of spleens from younger mice revealed that ectopic bone formation begins at approximately 9 months of age in female mice and accrues over time (data not shown). Bone was not identified in lymph nodes, liver, kidney, heart, intestine or brain (Figure 3H), revealing that ectopic bone formation is unique to the spleen. While many mouse models of autoimmunity do show splenic enlargement, the presence of ectopic bone is extremely rare (33), and suggests that osteoblasts are generated from mesenchymal precursor cells recruited to the spleen or present locally within the spleens of DKO mice.
Figure 3. Ectopic bone forms in DKO spleens.
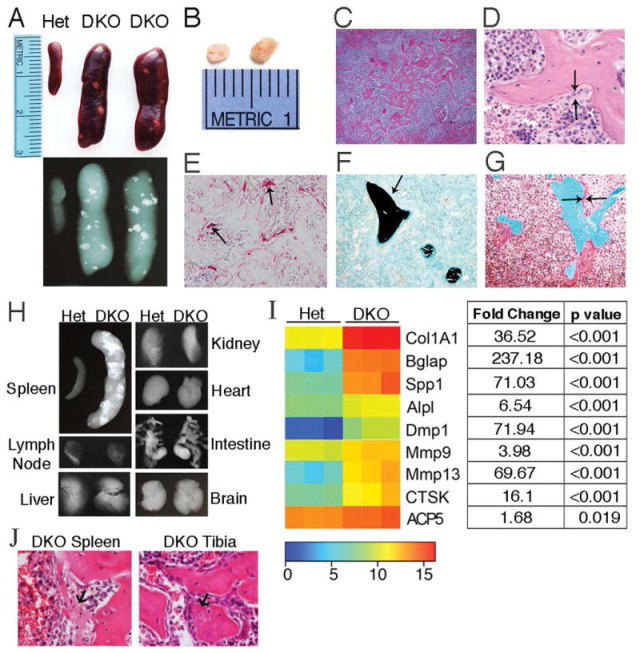
A) Representative images and X-rays of spleens from 10 month-old mice (female, n=6 mice/genotype). B) Islands of bone removed from 10 month-old female DKO spleens. C–G) Representative images of histologic stains performed on DKO spleen sections. 10 month-old mice (female, n=3 mice). C and D) H&E stained sections showing bone formation in white pulp (Magnification 4x & 20x, respectively). Arrows identify osteoblasts lining the surface of bone. E) TRAP stain, osteoclasts (arrows). F) Von Kossa/Fast Green stain shows mineralized bone (black, arrow). G) Goldner’s trichrome stain demonstrates osteoid production (dark pink, arrows). H) Representative X-rays of organs, 22 month-old mice (female, n=3 mice/genotype). I) Heatmap: Nanostring mRNA profiling of key bone remodeling genes, 10 month-old spleens (female, n=3 mice/genotype). Mean intensities of gene expression were transformed by a log2 function. Table lists fold change in DKO vs. Het gene expression and the corresponding p values. J) Representative H&E images of bone formation in DKO spleen and tibia. Arrows indicate fibrous tissue representing early bone matrix that may become mineralized.
To further explore mechanism, we utilized a customized Nanostring code set containing 150 genes involved in innate immune and bone pathways. Analysis of gene expression in spleens revealed significant upregulation of genes associated with bone resorption and formation in DKO mice compared to controls, and confirmed the spleen as a site of active bone formation (Figure 3I). The Col1A1 gene that produces a component of type 1 collagen, a major constituent of bone, was highly upregulated in DKO mice compared to controls as were the Bglap and Spp1 genes encoding osteocalcin and osteopontin, respectively, both noncollagenous proteins found in bone. Moreover, genes encoding alkaline phosphatase, as well as DMP1, a critical factor for bone mineralization produced by osteocytes and other cell types, were also upregulated in DKO spleens compared to controls. Upregulation of the matrix metalloproteinase (MMP) genes MMP-9 and MMP-13 reflect remodeling of bone extracellular matrix. Additionally, the CTSK and ACP5 genes, encoding cathepsin K and Trap5b, were also significantly upregulated in DKO compared to Het spleens, demonstrating enhanced osteoclast numbers.
Histologic analysis strongly suggests that the process of bone formation in DKO mice is similar in the long bones and spleen (Figure 3J), with lack of evidence for endochondral ossification at either site. Rather, the production of organic bone matrix, with subsequent mineralization of that matrix, is noted at both sites (Figure 3J). In DKO mice, the long bones and spleen are sites of erythropoiesis (17, 24). To determine whether bone accrual was present in bones in which erythropoiesis typically does not occur in mice, such as the vertebrae (34), vertebral bodies were subjected to microCT analysis, revealing a significant decrease in BV/TV, trabecular surface, and trabecular connectivity density in DKO mice compared to controls (Figure S3). These data suggest that local factors that promote the differentiation of mesenchymal precursor cells to osteoblasts may be released at sites of erythropoiesis (long bones and spleen) (17, 35).
DNase II deficiency promotes enhanced osteoblast differentiation and function in the spleen
The finding of bone in the spleen of DKO mice provides additional evidence that bone accrual in this model is driven by the excessive differentiation and activity of osteoblasts, leading to the production and mineralization of bone matrix. This could result from an increase in mesenchymal osteoblast precursors, or from enhanced osteoblast differentiation due to DNA accumulation within the cytosol of mesenchymal precursor cells themselves. To test the effect of DNA on differentiation, calvarial osteoblasts were transfected with the dsDNA mimetic poly(dA:dT) and differentiation was determined by expression of alkaline phosphatase. Interestingly, transfection with DNA upregulated the expression of p204 in calvarial osteoblasts, a cytosolic DNA sensor previously shown to also act as a transcriptional coactivator for bone formation (36–38). However, alkaline phosphatase levels were significantly reduced in cells transfected with dsDNA (Figure 4), suggesting that accumulation of dsDNA in osteoblast precursors inhibits, rather than promotes, differentiation. These results support an alternative hypothesis that there is an increase in osteoblast precursor cells and/or that extrinsic factors expressed by other cell types drive the differentiation of mesenchymal precursors to the osteoblast lineage in this model.
Figure 4. Transfection of DNA in osteoblasts induces p204, but inhibits differentiation.
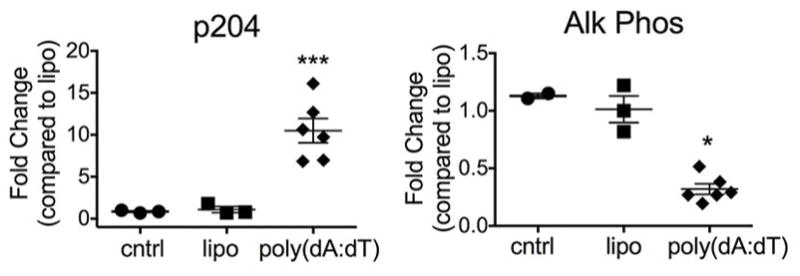
Fold change in p204 and alkaline phosphatase gene expression in calvarial osteoblasts stimulated with the DNA ligand poly(dA:dT), lipofectamine, or media control (cntrl).
To identify factors promoting osteoblast differentiation and bone formation, gene expression in 10 month-old DKO spleens was examined by array analysis, revealing upregulated expression of numerous genes regulating osteoblast differentiation and bone formation (Figure 5). Among the most highly regulated osteoblast-related genes in whole spleen were two genes in the transforming growth factor beta (TGFβ) family, Tgfbi and Tgfbr1. TGF-β signaling promotes the recruitment of osteoblast progenitors and contributes to the early differentiation of osteoblasts (39). Moreover, the gene encoding the BMP-signaling transducer Smad1, which regulates expression of the osteoblast-specific transcription factors Runx2 and Osterix (39), as well as the Runx2 gene itself, were upregulated in DKO mice compared to controls. The Bmp1 gene that encodes a type I collagen C-propeptidase required for mature collagen maturation in bone was also upregulated. We did not detect upregulation of other BMP receptor ligands known to promote osteoblast differentiation, including BMP2, 4, 6 or 7. In addition, we found an upregulation of the Gdf3 gene, a member of the TGF-β superfamily and an inhibitor of BMP signaling, which would be expected to inhibit bone formation and may represent a compensatory mechanism to inhibit local osteoblast differentiation.
Figure 5. STING-dependent induction of factors regulating osteoblast differentiation in DKO mice.

Heatmap: Mean intensities of expression in gene arrays, spleen, 10 month-old mice (n=3–4 mice/genotype). Signals were transformed by a log2 function. Table lists fold change in DKO vs. Het and STKO vs. DKO gene expression and the corresponding p values.
Regulation of several other genes supports the interpretation of splenic tissue as a site at which mesenchymal precursor cells are differentiating to osteoblasts. The Vdr gene, encoding the vitamin D receptor, is upregulated in DKO compared to Het mice. This receptor induces expression of factors that enhance osteoblast differentiation and inhibit apoptosis of osteoblasts. Recently, transgenic overexpression of Vdr in mature osteoblasts was shown to increase trabecular bone volume (40). The Serpinf1 gene, encoding pigment epithelium-derived factor (PEDF), was also upregulated in DKO spleens. PEDF modulates human and murine mesenchymal stem cell (MSC) differentiation by promoting osteogenesis and inhibiting adipogenesis (41). In addition, the Serpinh1 gene, encoding heat shock protein 47 (HSP47), is upregulated in DKO spleens compared to controls. Hsp47 is localized in the endoplasmic reticulum and acts as a molecular chaperone for the maturation of collagen (42). Finally, expression of the Pth1r gene encoding the PTHR was upregulated, which would promote osteoblast differentiation in the presence of PTH (43).
The STING pathway is required for bone formation
We then examined the role of the STING pathway in bone formation in DKO mice. In this model, cytosolic sensors detect host DNA and signal through STING to induce Type I IFN, TNF, IL-6, and IL1β. We and others have previously shown that arthritis in DKO mice is abrogated by deletion of STING in STING−/−DNase II−/−IFNaR−/− triple knock out (STKO) and in STING−/−DNase II−/− mice (4, 18). In order to determine whether the osteogenic factors identified in DKO mice were upregulated in a STING-dependent fashion, we performed gene array analysis in STING deficient DKO mouse (STKO) spleens. Expression of pro-osteogenic genes including Tgfbi, Tgfbr1, Smad1, Runx2, Vdr, Serpinf1, and Pth1r is not induced in the setting of STING deficiency (Figure 5).
Importantly, in vivo, STING deficiency in DKO mice ameliorates not only arthritis, but also bone accrual in both the long bones (Figure 6A) and spleens (Figure 6B). Trabecular BV/TV and trabecular bone surface are dramatically decreased in STKO compared to DKO mice. Moreover, the expression of genes associated with osteoblast activity (Col1a1, Spp1, Col1a2, Bglap, Omd, Alpl, Ibsp, Phex, Dmp1), matrix bone remodeling (MMP13), and osteoclasts (CTSK, ACP5) are significantly decreased in STKO compared to DKO spleens (Figure 6C), demonstrating that local bone formation in the setting of DNA accrual is dependent upon an intact STING pathway, and revealing a novel mechanism for aberrant bone formation.
Figure 6. Bone accrual in DKO mice requires the STING pathway.

A) Sagittal and transverse micro-CT images of femurs and quantitation of micro-CT data for trabecular bone volume/total volume (BV/TV) and trabecular bone surface. The Het and DKO values are the same as those shown in Figure 1D. 10 month-old mice (female, n=4–7 mice/genotype). Values are the mean ± SEM compared to Het; *=p<0.05, **=p<0.01, ***=p<0.001. B) Representative images and X-rays of spleens. All analyses performed on 10 month-old mice (female, n=4–6 mice/genotype). C) Heatmap: Mean intensities of expression in gene arrays, spleen, 10 month-old mice (n=3–4 mice/genotype). Signals were transformed by a log2 function. Table lists fold change in DKO vs. Het and STKO vs. DKO gene expression and the corresponding p values.
Discussion
In this study we demonstrate a novel pathway regulating bone and show that accrual of undegraded DNA and the resulting inflammatory response promotes the activation of cytosolic nucleic acid sensors, leading to trabecular bone accrual in the long bones and spleen. This occurs despite the presence of arthritis and osteoclast-mediated articular bone erosion, and the production of pro-inflammatory cytokines that promote bone loss. Bone formation results from an increase in osteoblast number and function and the bone phenotype manifests late, beginning at approximately 5–6 months of age in long bones, while bone formation in the spleen appears by 9–10 months. This late manifestation is likely due to the requirement for accrual of DNA over time, as well as to the time required for the enhancement of bone formation over bone resorption to ultimately favor bone accrual. DKO mice develop splenomegaly associated with excessive numbers of Ter119+ cells by an early age. Thus, both the long bones and spleen are sites of erythropoiesis and the chronic accrual of extruded, undegraded nuclei from reticulocytes. The inability of DKO mice to degrade DNA eventually leads to entrance of DNA into cytosolic compartments and activation of cytosolic DNA sensors that converge on STING. In addition, the ensuing inflammation and cell death most likely release ligands that can engage endosomal TLRs, which have been implicated in both autoantibody formation and splenomegaly in these mice (4, 19, 32). These pathways may contribute to bone formation in this model as well.
Affymetrix data from spleen samples demonstrated a major drive towards osteogenesis and identified a number of upregulated osteogenic factors at this site of extramedullary erythropoiesis. Among the most highly regulated were genes in the TGF-β family. Although the role of TGF-β signaling in bone is complex, TGF-β isoforms and their receptors (including type I receptor (TGFβRI)) are known to expand the pool of mesenchymal osteoblast progenitor cells and promote early differentiation and commitment to the osteoblast lineage (44). Moreover, mice with tissue-specific removal of TGFβRI show reduced trabecular bone in the long bones and decreased proliferation and differentiation of osteoblasts (45). Additionally, the genes encoding the osteogenic BMP-signaling transducer Smad1 and the osteoblast-specific transcription factor Runx2 were upregulated in DKO mice compared to controls.
Bone formation in the long bones and spleen appears to occur by similar mechanisms, given the gene expression profiles and histologic features at these sites. However, bone formation in the spleen requires the presence of a population of mesenchymal osteoblast precursor cells that are either recruited to splenic tissue, or are resident within this site. One possible mesenchymal precursor with osteogenic potential is the pericyte. Microvascular pericytes have long been known to serve as a reservoir for multiple cellular lineages in joints, including osteoblasts (46). It has been shown that vascular pericytes implanted into athymic mice reproducibly form cartilage and bone. Furthermore, these cells can secrete components of bone matrix associated with initiation of mineralization and nucleation of hydroxyapatite (46). Bone forms in DKO spleens in a process similar to what is seen in implanted vascular pericyte cultures. Pericytes may arise from a CD34-expressing progenitor cell within vessel walls (47), and it is of interest that bone in the spleen forms in regions of white pulp, areas rich in vasculature.
These findings may also be relevant to several human diseases in which abnormal bone formation occurs in soft tissues. Heterotopic ossification (HO) is a debilitating condition associated with formation of lamellar bone in extra-skeletal sites. The etiology of acquired HO is unknown, although heterotopic lesions occur following soft tissue trauma and viral infections (48–50). These triggers precede ectopic bone formation and strongly implicate a role for inflammatory innate immune pathways in the pathogenesis of HO. It is possible that viral DNA or DNA released from damaged cells may overwhelm the activity of DNase II, triggering the activation of innate immune DNA sensors. Further investigation into these mechanisms should provide novel pathways for the prevention and treatment of bone abnormalities in autoimmune disease and in heterotopic bone formation.
Supplementary Material
Transverse micro-CT images of femurs and quantitation of cortical bone volume/total volume (BV/TV) and cortical thickness for 10 month-old mice (female, n=6–7 mice/genotype). Values are the mean ± SEM compared to Het: **=p<0.01.
Representative micro-CT images of trabecular and cortical bone in femurs, and quantitation of trabecular bone surface, trabecular connectivity density, cortical bone volume/total volume (BV/TV), and cortical thickness (10.5–11.5 month-old mice, male, n=4–6 mice/genotype). Values are the mean ± SEM compared to Het: **=p<0.01.
Micro-CT images of trabecular vertebral bone and quantitation of trabecular bone volume/total volume (BV/TV), trabecular bone surface, and trabecular connectivity density. Values are the mean ± SEM compared to Het; *=p<0.05, **=p<0.01, ***=p<0.001.
Acknowledgments
Grant Support
This work was supported by grants from the National Institutes of Health: AR067394 (E.M.G.), T32 AI 095213 (R.B.), AI093752 and AI083713 (K.A.F.), AR050256 (A.M.R.), and by grants from the Musculoskeletal Center of Excellence Grants Program, University of Massachusetts (E.M.G.) and the Alliance for Lupus Research (S.S.).
We would like to thank Catherine Manning and Kelly Army for assistance with animal husbandry, Stacey Russell for micro-CT assistance, and Vicki Rosen PhD for helpful discussions.
References
- 1.Sharma S, Fitzgerald KA. Innate immune sensing of DNA. PLoS pathogens. 2011;7:e1001310. doi: 10.1371/journal.ppat.1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat N, Fitzgerald KA. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. European journal of immunology. 2014;44:634–40. doi: 10.1002/eji.201344127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stetson DB. Endogenous retroelements and autoimmune disease. Current opinion in immunology. 2012;24:692–7. doi: 10.1016/j.coi.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. Journal of immunology. 2015;194:873–7. doi: 10.4049/jimmunol.1402573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakobs C, Perner S, Hornung V. AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PloS one. 2015;10:e0131702. doi: 10.1371/journal.pone.0131702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–98. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–31. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nature genetics. 2006;38:917–20. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 9.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nature genetics. 2007;39:1065–7. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 10.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Human molecular genetics. 2009;18:R130–6. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis and rheumatism. 2010;62:1469–77. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 12.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes and immunity. 2011;12:270–9. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nature reviews Immunology. 2015;15:429–40. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. The New England journal of medicine. 2014;371:507–18. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura-Kataoka K, Yasuda T, Fujihara J, Toga T, Ono R, Otsuka Y, et al. Genetic and expression analysis of SNPs in the human deoxyribonuclease II: SNPs in the promoter region reduce its in vivo activity through decreased promoter activity. Electrophoresis. 2012;33:2852–8. doi: 10.1002/elps.201200260. [DOI] [PubMed] [Google Scholar]
- 16.Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell reports. 2014;9:180–92. doi: 10.1016/j.celrep.2014.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 18.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19386–91. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pawaria S, Moody K, Busto P, Nundel K, Choi CH, Ghayur T, et al. Cutting Edge: DNase II deficiency prevents activation of autoreactive B cells by double-stranded DNA endogenous ligands. Journal of immunology. 2015;194:1403–7. doi: 10.4049/jimmunol.1402893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. The American journal of pathology. 2001;159:1689–99. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nature reviews Rheumatology. 2012;8:656–64. doi: 10.1038/nrrheum.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh NC, Reinwald S, Manning CA, Condon KW, Iwata K, Burr DB, et al. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2009;24:1572–85. doi: 10.1359/jbmr.090320. [DOI] [PubMed] [Google Scholar]
- 23.Baum R, Gravallese EM. Impact of inflammation on the osteoblast in rheumatic diseases. Current osteoporosis reports. 2014;12:9–16. doi: 10.1007/s11914-013-0183-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19432–7. doi: 10.1073/pnas.1010603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh NC, Crotti TN, Goldring SR, Gravallese EM. Rheumatic diseases: the effects of inflammation on bone. Immunological reviews. 2005;208:228–51. doi: 10.1111/j.0105-2896.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 26.Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, et al. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Experimental cell research. 1999;247:84–93. doi: 10.1006/excr.1998.4320. [DOI] [PubMed] [Google Scholar]
- 27.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. The Journal of biological chemistry. 2000;275:4858–64. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- 28.Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. The Journal of clinical investigation. 2005;115:282–90. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–9. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 30.Matzelle MM, Gallant MA, Condon KW, Walsh NC, Manning CA, Stein GS, et al. Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis and rheumatism. 2012;64:1540–50. doi: 10.1002/art.33504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 32.Baum R, Nundel K, Pawaria S, Sharma S, Busto P, Fitzgerald KA, et al. Synergy between Hematopoetic and Radioresistant Stromal Cells is Required for Autoimmune Manifestations of DNase II−/− IFNaR−/− Mice. Journal of Immunology. 2016;3:1348–54. doi: 10.4049/jimmunol.1502130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macias MP, Fitzpatrick LA, Brenneise I, McGarry MP, Lee JJ, Lee NA. Expression of IL-5 alters bone metabolism and induces ossification of the spleen in transgenic mice. The Journal of clinical investigation. 2001;107:949–59. doi: 10.1172/JCI11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macki M, Bydon M, Papademetriou K, Gokaslan Z, Bydon A. Presacral extramedullary hematopoiesis: an alternative hypothesis. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 2013;20:1664–8. doi: 10.1016/j.jocn.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Nagata S. Autoimmune diseases caused by defects in clearing dead cells and nuclei expelled from erythroid precursors. Immunological reviews. 2007;220:237–50. doi: 10.1111/j.1600-065X.2007.00571.x. [DOI] [PubMed] [Google Scholar]
- 36.Liu CJ, Chang E, Yu J, Carlson CS, Prazak L, Yu XP, et al. The interferon-inducible p204 protein acts as a transcriptional coactivator of Cbfa1 and enhances osteoblast differentiation. The Journal of biological chemistry. 2005;280:2788–96. doi: 10.1074/jbc.M412604200. [DOI] [PubMed] [Google Scholar]
- 37.Luan Y, Yu XP, Xu K, Ding B, Yu J, Huang Y, et al. The retinoblastoma protein is an essential mediator of osteogenesis that links the p204 protein to the Cbfa1 transcription factor thereby increasing its activity. The Journal of biological chemistry. 2007;282:16860–70. doi: 10.1074/jbc.M610943200. [DOI] [PubMed] [Google Scholar]
- 38.Luan Y, Yu XP, Yang N, Frenkel S, Chen L, Liu CJ. p204 protein overcomes the inhibition of core binding factor alpha-1-mediated osteogenic differentiation by Id helix-loop-helix proteins. Molecular biology of the cell. 2008;19:2113–26. doi: 10.1091/mbc.E07-10-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. International journal of biological sciences. 2012;8:272–88. doi: 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Triliana R, Lam NN, Sawyer RK, Atkins GJ, Morris HA, Anderson PH. Skeletal characterization of an osteoblast-specific vitamin D receptor transgenic (ObVDR-B6) mouse model. The Journal of steroid biochemistry and molecular biology. 2015 doi: 10.1016/j.jsbmb.2015.08.009. http://dx.doi.org/10.1016/j.jsbmb.2015.08.009. [DOI] [PubMed]
- 41.Gattu AK, Swenson ES, Iwakiri Y, Samuel VT, Troiano N, Berry R, et al. Determination of mesenchymal stem cell fate by pigment epithelium-derived factor (PEDF) results in increased adiposity and reduced bone mineral content. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:4384–94. doi: 10.1096/fj.13-232900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagata K. HSP47 as a collagen-specific molecular chaperone: function and expression in normal mouse development. Seminars in cell & developmental biology. 2003;14:275–82. doi: 10.1016/j.semcdb.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 43.Yu B, Zhao X, Yang C, Crane J, Xian L, Lu W, et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27:2001–14. doi: 10.1002/jbmr.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. The Journal of clinical investigation. 2014;124:466–72. doi: 10.1172/JCI70050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsunobu T, Torigoe K, Ishikawa M, de Vega S, Kulkarni AB, Iwamoto Y, et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Developmental biology. 2009;332:325–38. doi: 10.1016/j.ydbio.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doherty MJ, Ashton BA, Walsh S, Beresford JN, Grant ME, Canfield AE. Vascular pericytes express osteogenic potential in vitro and in vivo. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1998;13:828–38. doi: 10.1359/jbmr.1998.13.5.828. [DOI] [PubMed] [Google Scholar]
- 47.Modder UI, Khosla S. Skeletal stem/osteoprogenitor cells: current concepts, alternate hypotheses, and relationship to the bone remodeling compartment. Journal of cellular biochemistry. 2008;103:393–400. doi: 10.1002/jcb.21423. [DOI] [PubMed] [Google Scholar]
- 48.Ranganathan K, Loder S, Agarwal S, Wong VW, Forsberg J, Davis TA, et al. Heterotopic Ossification: Basic-Science Principles and Clinical Correlates. The Journal of bone and joint surgery American volume. 2015;97:1101–11. doi: 10.2106/JBJS.N.01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clinical orthopaedics and related research. 2004:275–9. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan FS, Pignolo RJ, Shore EM. Granting immunity to FOP and catching heterotopic ossification in the Act. Seminars in cell & developmental biology. 2016;49:30–6. doi: 10.1016/j.semcdb.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transverse micro-CT images of femurs and quantitation of cortical bone volume/total volume (BV/TV) and cortical thickness for 10 month-old mice (female, n=6–7 mice/genotype). Values are the mean ± SEM compared to Het: **=p<0.01.
Representative micro-CT images of trabecular and cortical bone in femurs, and quantitation of trabecular bone surface, trabecular connectivity density, cortical bone volume/total volume (BV/TV), and cortical thickness (10.5–11.5 month-old mice, male, n=4–6 mice/genotype). Values are the mean ± SEM compared to Het: **=p<0.01.
Micro-CT images of trabecular vertebral bone and quantitation of trabecular bone volume/total volume (BV/TV), trabecular bone surface, and trabecular connectivity density. Values are the mean ± SEM compared to Het; *=p<0.05, **=p<0.01, ***=p<0.001.