

Abstract
Alcohol (ethanol) at concentrations reached in blood following moderate to heavy drinking (30–80 mM) reduces cerebral artery diameter via inhibition of voltage- and calcium-gated potassium channels of large conductance (BK) in cerebral artery smooth muscle. These channels consist of channel-forming α and regulatory β1 subunits. A high-cholesterol diet protects against ethanol-induced constriction via accumulation of cholesterol within the vasculature. The molecular mechanisms of this protection remain unknown. In the present work, we demonstrate that in vitro cholesterol enrichment of rat middle cerebral arteries significantly increased cholesterol within arterial tissues and blunted constriction by 50 mM of ethanol. Ethanol-induced BK channel inhibition in inside-out patches excised from freshly isolated cerebral artery myocytes was also abolished by cholesterol enrichment. Enrichment of arteries with enantiomeric cholesterol (ent-cholesterol) also blunted BK channel inhibition and cerebral artery constriction in response to ethanol. The similar protection of cholesterol and ent-cholesterol against ethanol action indicates that this protection does not require protein site(s) that specifically sense natural cholesterol. Cholesterol-driven protection against ethanol-induced BK channel inhibition and vasoconstriction was replicated in myocytes and middle cerebral arteries of C57BL/6 mice. BK β1 subunits are known to regulate vascular diameter and its modification by ethanol. However, blunting of an ethanol effect by in vitro cholesterol enrichment was observed in arteries and myocyte membrane patches from BK β1 (KCNMB1) knockout mice. Thus, BK β1 subunits are not needed for cholesterol protection against ethanol effect on BK channel function and cerebral artery diameter.
Keywords: MaxiK channel, Cerebral artery, ent-Cholesterol, KCNMB1 knockout, Lipid-protein interaction
1. Introduction
Episodic, moderate to heavy ethyl alcohol intake (such as binge drinking) is the most common form of excessive alcohol use in the United State (http://www.cdc.gov/alcohol/fact-sheets/binge-drinking.htm; accessed 7.19.2016). Recent data report that nearly 25% of people 18 or older engaged into binge drinking within the past month (http://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics; accessed 7.19.2016). Episodic, moderate to heavy alcohol intake with blood alcohol levels reaching 35–80 mM results in constriction of cerebral arteries and represents a widely recognized risk factor for several cerebrovascular disorders [1,2]. Alcohol-induced cerebral artery constriction has been demonstrated in many species, including rats, mice, and humans [3–5]. Despite more than a decade of research, the molecular mechanisms that mediate and control ethanol-induced BK channel inhibition and the resulting cerebral artery constriction continue to be unraveled.
It has been demonstrated that alcohol-induced cerebral artery constriction is caused by ethanol inhibition of calcium/voltage-gated potassium channels of large conductance (BK) channels [4]. These channel complexes consist of channel-forming α (slo1) and accessory, regulatory (β1) subunits. The latter are particularly abundant in vascular and non-vascular smooth muscle but scarce in other tissues [6]. Located on the plasma membrane of vascular smooth muscle cells, BK channels generate outward potassium currents, tend to hyperpolarize the membrane, and diminish vascular contraction [6,7]. Thus, BK channel inhibition by ethanol results in arterial constriction.
Ethanol-induced BK channel inhibition is independent of alcohol metabolism and freely diffusible cytosolic signals. Rather, the cerebral artery myocyte BK channel complex (cbv1 + β1 subunits) and a cell-free membrane environment are sufficient [4–5]. Cholesterol is a common component of Western diet and is essential for mammalian cell function and membrane architecture. We identified dietary cholesterol as a critical regulator of the alcohol effect on cerebral artery diameter [8]. In particular, a high-cholesterol diet blunts ethanol-induced constriction of cerebral arteries via accumulation of cholesterol within the cerebral artery smooth muscle layer [8]. However, the molecular mechanism(s) underlying/mediating cholesterol antagonism of ethanol-induced cerebral artery constriction remains unknown and is the focus of our current study.
2. Material and methods
2.1. Ethical aspects of research
The care of animals and experimental protocols were reviewed and approved by the Animal Care and Use Committee of The University of Tennessee Health Science Center, which is an institution accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
2.2. Cerebral artery diameter measurement
Adult male Sprague-Dawley rats (≈250 g) and 8- to 12-weeks-old male KCNMB1 knockout (KO) and C57BL/6 (control) mice were decapitated using a guillotine and sharp scissors, respectively. Middle cerebral arteries were isolated from the rat or mouse brain on ice under a microscope (Nikon SMZ645; Nikon) and cut into 1- to 2-mm-long segments. Arteries were incubated in physiologic saline solution (PSS; composition below) containing methyl-beta-cyclodextrin/steroid (either cholesterol or ent-cholesterol) complex for 1 h immediately prior to endothelium removal and artery cannulation. Control arteries were also incubated in PSS for 1 h. After either cholesterol-enriching or control incubation, endothelium was removed by passing an air bubble into the vessel lumen for 90 s before vessel cannulation. A segment was cannulated at each end in a temperature-controlled, custom-made perfusion chamber. Using a Dynamax RP-1 peristaltic pump (Rainin Instruments, Inc., Oakland, CA), we continuously perfused the chamber at a rate of 3.75 mL/min with PSS (mM): 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.023 EDTA, 11 glucose, and 24 NaHCO3. PSS was equilibrated at pH 7.4 with a 21/5/74% mix of O2/CO2/N2 and maintained at 35–37 °C. Arteries were observed with a CCD camera (Sanyo VCB-3512T; Sanyo Electric Co., Moriguchi, Japan) attached to an inverted microscope (Nikon Eclipse TS100; Nikon). The artery external wall diameter was measured using the automatic edge-detection function of IonWizard software (IonOptics, Milton, MA) and digitized at 1 Hz. Steady-state changes in intravascular pressure were achieved by elevating an attached reservoir filled with PSS and were monitored using a pressure transducer (Living Systems Instruments, Burlington, VT). Arteries were first incubated at an intravascular pressure of 10 mm Hg for 10 min. Then, intravascular pressure was increased to 60 mm Hg and held steady throughout the experiment to develop and maintain arterial myogenic tone. Drugs were dissolved to make stock solutions, diluted in PSS to their final concentration, and applied to the artery via chamber perfusion. The effect of drug application on vessel diameter was evaluated at the time the latter reached a maximal, steady level. Absence of endothelium was confirmed by the absence of the vessel’s response to an endothelium-dependent (10 μM acetylcholine) vasodilator after artery pressurization [4,9].
2.3. Rat cerebral artery myocyte isolation
Middle cerebral arteries were isolated from adult male Sprague-Dawley rats (≈250 g) as described in detail [9,10]. Immediately prior to the myocyte isolation procedure, arteries were incubated in PSS containing methyl-beta-cyclodextrin (MβCD):steroid complex (8:1 M ratio) for 1 h [9,11]. Control arteries were also incubated in PSS for 1 h. The myocyte isolation procedure rendered a cell suspension containing relaxed, individual myocytes (≥5 myocytes/field using a 20× objective) that could be identified under an Olympus IX-70 microscope (Olympus American Inc., Woodbury, NY). The cell suspension was stored in ice-cold dissociation media (DM) (mM): 0.16 CaCl2, 0.49 EDTA, 10 HEPES, 5 KCl, 0.5 KH2PO4, 2 MgCl2, 110 NaCl, 0.5 NaH2PO4, 10 NaHCO3, 0.02 phenol red, 10 taurine, 10 glucose, and supplemented with 0.06% soybean trypsin inhibitor and 0.06% BSA. Myocytes were used for electrophysiology up to 4 h after isolation.
2.4. Electrophysiology data acquisition and analysis
Currents were recorded from excised, inside-out (I/O) patches at single-channel resolution allowing for identification of individual channel openings. Both bath and electrode solutions contained (mM) 130 KCl, 5 EGTA, 1 MgCl2, and 15 HEPES, pH 7.35. In all experiments, the free Ca2+ in solution was adjusted to the desired value by adding CaCl2. In all cases, the nominal free Ca2+ was calculated with MaxChelator Sliders (C. Patton, Stanford University, Stanford, CA). Patch-clamp electrodes were pulled from glass capillaries (Drummond Scientific Co.). Immediately before recording, the tip of the electrode was fire-polished on a microforge (World Precision Instruments, Sarasota, FL) to give resistances of 8–10 MΩ when filled with extracellular solution (for composition, see above). The high pipette resistance corresponded to the small pipette tip sizes which allowed to record patches that only contained a few channels. An agar bridge with chloride as the main anion was used as the ground electrode. After excision from the cell, the membrane patch was exposed to a stream of bath solution containing either bath solution (control) or 50 mM ethanol. Solutions were applied onto the patch cytosolic side by using a pressurized, automated OctaFlow system (ALA Scientific Instruments, Farmingdale, NY) via a micropipette tip with an internal diameter of 100 μm. Experiments were performed at room temperature (20°– 22 °C). The ionic current was recorded using an EPC8 amplifier (HEKA, Lambrecht, Germany) at 1 kHz. Data were digitized at 5 kHz by using a Digidata 1320 A A/D converter and pCLAMP 8.0 (Molecular Devices, Sunnyvale, CA). The product of the number of channels in the patch (N) and channel open probability (Po) was used as an index of channel steady-state activity. NPo was obtained using a built-in option in Clampfit 9.2 (Molecular Devices) from 1-min, gap-free recordings under each condition or at each time-point.
2.5. Steroid and protein determination
Cerebral arteries from two to three animals were pulled together; the steroid-enriching procedure was performed as described in 2.2., 2.3, and 2.6. The procedure was repeated three times on different days with different sets of animals. Each time, arteries were homogenized in standard physiologic phosphate-buffered solution (PBS) supplemented with 1% Triton detergent. Free natural cholesterol, ent-cholesterol, and total protein levels were determined using the Amplex Red Cholesterol Assay kit (Molecular Probes, Inc.) and the Pierce BCA Protein Assay kit (Thermo Scientific), respectively, following manufacturers’ instructions.
2.6. Chemicals
Ent-cholesterol was synthesized as described elsewhere [12]. For cholesterol or ent-cholesterol enrichment, PSS contained 5 mM MβCD + 0.625 mM of either cholesterol or ent-cholesterol. Molar ratio of MβCD:steroid was set at 8:1 [9,11]. To ensure MβCD saturation with cholesterol, the solution was vortexed and sonicated for 30 min at room temperature, then shaken at 37 °C overnight and filtered on the morning of the experiment. Alcohol (ethyl alcohol, ethanol) was purchased from American Bioanalytical (Natick, MA); ethanol-containing solution was prepared fresh on the day of the experiment. Low density lipoprotein (LDL) was purchased from Kalen Biomedical (Montgomery Village, MD). For enrichment of arteries with cholesterol via LDL, arteries were incubated in 200 mg/dL LDL for 24 h at room temperature. All other chemicals were purchased from Sigma-Aldrich (Saint-Louis, MO).
2.7. Data analysis
Final plotting, fitting, and statistical analysis of the data were conducted using Origin 8.5.1 (OriginLab, Northampton, MA) and InStat 3.0 (GraphPad, La Jolla, CA). Statistical analysis was conducted using either one-way analysis of variance and Bonferroni’s multiple comparison test or paired Student’s t-test, according to the experimental design. P < 0.05 was considered statistically significant. Data are expressed as the mean ± SEM, and n = number of patches/arteries. Each patch was obtained from a different myocyte, and each pressurized artery measurement was obtained from a separate animal.
3. Results
3.1. Protective effect of dietary cholesterol against alcohol-induced cerebral artery constriction is mimicked by in vitro enrichment of cerebral artery with cholesterol
We recently showed that a protracted (18–23 weeks) high-cholesterol diet blunts alcohol-induced cerebral arteriole constriction in vivo and cerebral artery constriction in vitro [8]. This protection is mediated by the build-up of cholesterol in arterial tissue. In the present work, we mimicked the in vivo cholesterol accumulation observed during a high-cholesterol diet by “acute” enrichment of cerebral arteries with cholesterol in vitro. After being dissected from rat brains, middle cerebral arteries were placed in the media with MβCD/cholesterol complex that is a proven tool for tissue cholesterol enrichment in vitro [11]. Indeed, brief 15-min immersion of cerebral arteries into cholesterol-enriching solution resulted in significant (~1.5-fold) increase in tissue cholesterol level (Fig. 1A).
Fig. 1.
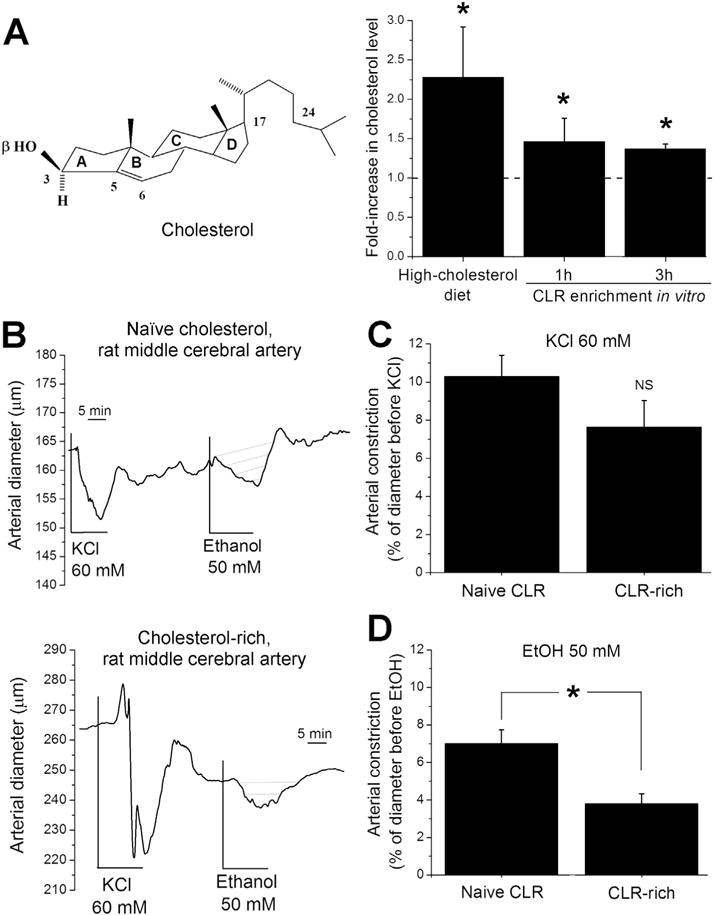
In vitro cholesterol enrichment of rat middle cerebral arteries blunted ethanol-induced constriction. (A) Averaged data showing similar fold-increase in artery cholesterol level observed in cerebral arteries from rats on a high-cholesterol diet (18–23 weeks of diet, n = 5) when compared to in vitro cholesterol enrichment (1 h and 3 h following the in vitro cholesterol-enriching treatment, n = 3). *Different from naïve cholesterol (P < 0.05). A dashed line indicates no change in cholesterol level. The insert shows the chemical structure of cholesterol. (B) Original traces showing changes in middle cerebral artery diameter in response to 60 mM KCl, 50 mM ethanol, and Ca2+-free solution in arteries with naïve cholesterol levels vs. cholesterol-enriched vessels. (C) Averaged constriction by 60 mM KCl (n ≥ 10 in each group). (D) Averaged constriction by 50 mM ethanol (n ≥ 6 in each group). *Different from naïve cholesterol (P < 0.05).
This increase is not statistically different (P > 0.05) from the cholesterol increase in rat cerebral artery tissue following 18–23 weeks on a high-cholesterol diet and remains unchanged for at least 3 h after placing cerebral arteries in physiologic saline without cholesterol-enriching treatment (Fig. 1A). Cholesterol-enriched and corresponding control arteries were pressurized in vitro as described [10]. After myogenic tone development at 60 mm Hg, arteries were probed with 60 mM KCl to evaluate their contractile response to depolarization. KCl-induced cerebral artery constriction was not affected by a high-cholesterol diet: KCl caused an 8–12% decrease in arterial diameter in the control (naïve cholesterol, n = 44) and cholesterol-enriched (n = 10) groups (Fig. 1B–C). At the end of each experiment, the artery was perfused with Ca2+-free PSS to confirm the presence of myogenic tone (Fig. 1B).
Applying 50 mM ethanol to pressurized middle cerebral arteries from control rats resulted in ≈8% decrease in arterial diameter (Fig. 1B, D). This effect disappeared upon washout with ethanol-free PSS (Fig. 1B). In contrast, applying 50 mM ethanol to pressurized middle cerebral arteries that were cholesterol-enriched via MβCD (see Methods) resulted in only a 3–4% decrease in arterial diameter (Fig. 1B, D). This constriction is significantly smaller (P < 0.05) than that found in the control group (Fig. 1D). To ensure that blunting of ethanol constriction was not due to the exposure of arteries to MβCD itself, we performed artery cholesterol enrichment using LDL. Cholesterol-enrichment with LDL resulted in a statistically significant decrease in ethanol-induced constriction (Fig. S1). Thus, the protective effect of a high-cholesterol diet against the alcohol effect [8] can be mimicked by in vitro enrichment of isolated cerebral arteries with cholesterol. The similar blunting of ethanol-induced vasoconstriction by a high-cholesterol diet [8] and cholesterol in vitro enrichment of isolated arteries (Fig. 1, Fig. S1) is in agreement with the idea that a high-cholesterol diet protects against the ethanol effect on cerebral arteries by accumulating cholesterol in artery tissue and does not require circulating factors or complex neuronal integrity. Finally, the fact that two different methods to enrich arteries in vitro with cholesterol rendered a similar result, strongly suggests that cholesterol blunting of alcohol-induced cerebrovascular constriction is independent of the cholesterol-carrier used but linked to cholesterol itself.
3.2. Cholesterol blunting of an alcohol effect is observed at the level of BK channel – a major target of alcohol in cerebral arteries
Alcohol-induced cerebral artery constriction is mediated by alcohol-induced inhibition of BK channels in vascular smooth muscle [4–5]. To determine whether cholesterol blunts alcohol-induced BK channel inhibition, we used a conventional patch-clamp approach to record alcohol responses of BK channels in freshly isolated myocytes from control vs. cholesterol-enriched middle cerebral arteries of rat. Arteries were treated with either PSS or PSS containing MβCD/cholesterol complex for 15 min immediately prior to myocyte isolation. Recordings were performed in I/O patches to evaluate the ethanol effect on BK function in the absence of cell metabolism or channel modulators. Transmembrane voltage was set at −40 mM and Ca2+i = 30 μM to mimic membrane voltage in vascular myocytes and calcium level in the vicinity of the BK channel during myocyte contraction [13,14]. Immediately after excision, each I/O patch was perfused with control (bath) solution for 1 min to obtain basal BK function before applying ethanol; 50 mM ethanol perfusion lasted 10 min with continuous monitoring of NPo (Fig. 2A, left column, B). As previously reported [4,9], 50 mM ethanol significantly reduced BK channel activity by ≈20% (Fig. 2). Ethanol-induced BK channel inhibition could be clearly observed during the 3–7 min of ethanol application. Remarkably, this time course reflects maximal in vivo constriction of cerebral arterioles by ethanol [8]. Pre-incubation of cerebral arteries with MβCD/cholesterol complex totally blunted ethanol-induced BK channel inhibition (Fig. 2A right column, B). Considering that cholesterol antagonism of ethanol’s effect was found in cell-free myocyte membranes, cholesterol protection against ethanol-induced BK channel inhibition and resulting vasoconstriction are independent of freely diffusible cytosolic signals and cell metabolism of cholesterol or ethanol.
Fig. 2.

Cholesterol enrichment abolished ethanol-induced BK channel inhibition in membrane patches excised from middle cerebral artery myocytes of rat. (A) BK recordings from an I/O patch excised from an arterial myocyte with naïve cholesterol content (left column) or cholesterol-enriched (right column). Channel activity is shown before and during exposure to either ethanol-free (control, bath) or 50 mM ethanol solution. Here and in all other figures, channel openings are shown as downward deflections, arrows indicate baseline (all channels are closed). Vm = −40 mV, free Ca2+i = 30 μM. (B) Averaged changes in BK channel activity over time in myocyte membrane patches from arteries with naïve (n = 4) vs. enriched cholesterol levels (n = 4). (C) Averaged fold-changes in BK channel NPo. Each bar represents an average of data from no less than four myocytes. The dotted line underscores pre-application level of activity *Statistically significant difference (P < 0.05).
3.3. Cholesterol protection against alcohol-induced BK channel inhibition and resulting cerebral artery constriction is unlikely to be mediated by a protein site that is specific for natural cholesterol
Our data from cell-free membrane patches clearly point at the BK channel protein or its immediate membrane environment as the target of cholesterol action that results in blunting the ethanol effect. To determine whether cholesterol blunting of the alcohol effect is mediated by a cholesterol-specific protein site(s), we tested alcohol’s effect on BK channel-containing myocyte membrane patches from rat middle cerebral arteries enriched with cholesterol enantiomer “ent-cholesterol” (Fig. 3A). Ent-cholesterol is a “mirror image” of natural cholesterol, with both compounds sharing major physicochemical properties. Because cholesterol chirality has a minor effect on protein-free phospholipid media [15], differential effects of cholesterol vs. ent-cholesterol have been widely used as a criterion to establish that cholesterol modification of membrane protein function is the result of cholesterol recognition by a specific protein site(s) [16–19]. Ent-cholesterol enrichment of rat middle cerebral arteries was performed and verified following the protocol used for enrichment with natural cholesterol (Fig. S2; see Material and Methods). At experimental conditions identical to those described in Section 3.2, 50 mM ethanol failed to inhibit BK channels in myocyte membrane patches from arteries subjected to ent-cholesterol enrichment (Fig. 3). Thus, both cholesterol and ent-cholesterol were able to abolish ethanol-induced BK channel inhibition (Fig. 2C vs. Fig. 3C).
Fig. 3.
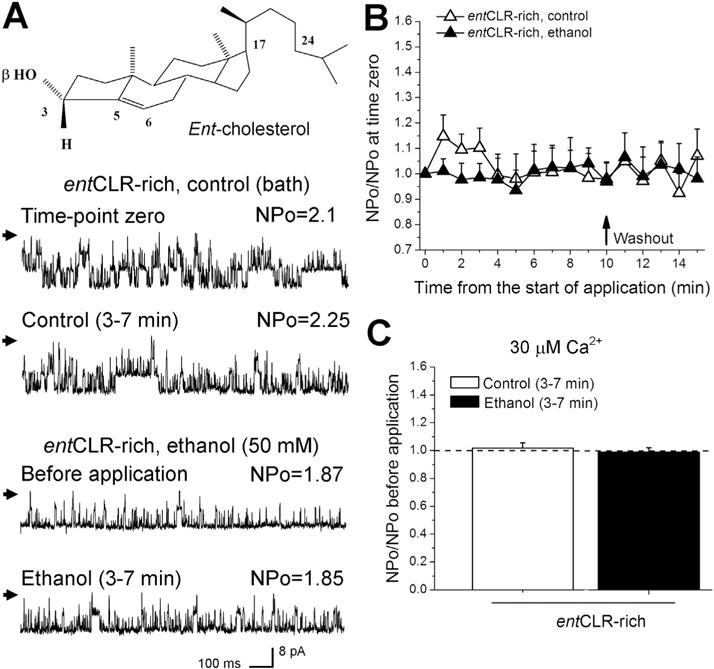
Ent-cholesterol enrichment abolished ethanol-induced BK channel inhibition in rat cerebral artery myocytes. (A) BK recordings from an I/O patch excised from rat myocyte of ent-cholesterol-enriched middle cerebral artery. Channel activity is shown before and during exposure to either ethanol-free (control, bath) or 50 mM ethanol solution. The insert depicts the chemical structure of ent-cholesterol. (B) Averaged changes in BK channel activity over time in myocyte membrane patches from ent-cholesterol-enriched middle cerebral arteries (n = 3). (C) Averaged fold changes in BK channel NPo. Each bar represents an average of data from three myocytes. A dotted line underscores pre-application level of activity.
In pressurized cerebral arteries, KCl-induced constriction was abolished by ent-cholesterol enrichment (Fig. 4A–B). Moreover, ent-cholesterol-enriched arteries were also resistant to ethanol-induced constriction (Fig. 4A, C). To ensure that blunting of ethanol-induced constriction was due to the presence of ent-cholesterol and was not a mere reflection of the overall reduction in artery’s ability to constrict in response to depolarization, we normalized ethanol-induced constriction to constriction by KCl within each experimental group. The ratio of ethanol-induced constriction over constriction evoked by 60 mM KCl remained unchanged between cholesterol-and ent-cholesterol-enriched vessels, and was significantly less than the ethanol-induced constriction in arteries with naïve cholesterol (Fig. 4D). The similar ability of cholesterol and ent-cholesterol to antagonize ethanol-induced BK channel inhibition and resulting cerebral artery constriction argues against the idea that cholesterol control over an alcohol effect requires cholesterol recognition by a protein site that specifically recognizes natural cholesterol. At the very least, this site does not discriminate between cholesterol and ent-cholesterol. Alternatively, cholesterol/ent-cholesterol control over an alcohol affect may be mediated by nonspecific, lipid-mediated mechanisms such as sterol-driven changes in membrane physical properties (Discussion).
Fig. 4.
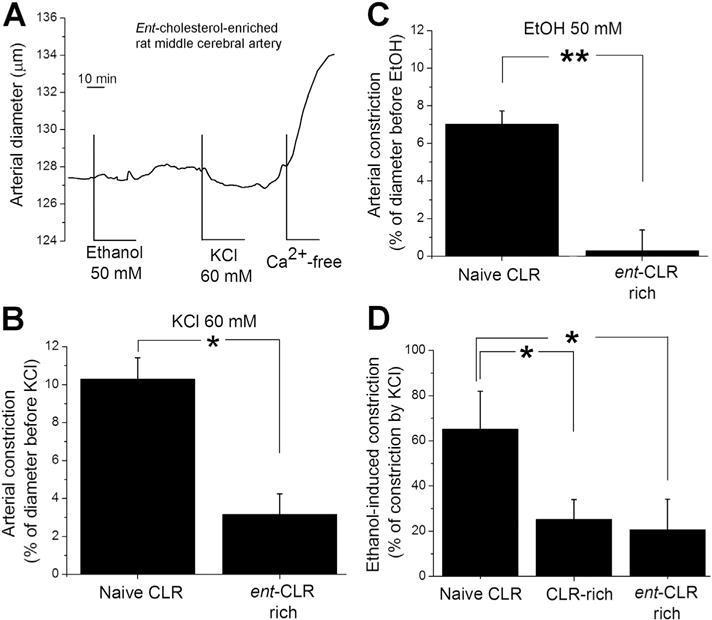
Ent-cholesterol blunted alcohol-induced constriction of rat cerebral artery. (A) Original trace showing changes in middle cerebral artery diameter in response to 60 mM KCl, 50 mM ethanol, and Ca2+-free solution in ent-cholesterol-enriched vessel. (B) Averaged constriction by 60 mM KCl (n ≥ 6). (C) Averaged constriction by 50 mM ethanol (n ≥ 6). (D) Averaged constriction by ethanol as percentage of corresponding constriction by KCl. In B–D, * different from naïve cholesterol (P < 0.05).
3.4. Blunting of ethanol-induced BK channel inhibition and resulting cerebral artery constriction by cholesterol enrichment does not require the presence of the BK β1 subunit
Disregarding the primary mechanism (membrane protein vs. membrane lipid-mediated) of cholesterol control over an alcohol effect on BK channel function, the consequences of cholesterol enrichment are ultimately translated to the BK channel complex, which is the effector of ethanol action on cerebral artery diameter. In smooth muscle (including vasculature), the BK channel complex is composed of channel-forming α and smooth muscle-specific accessory β1 subunits [6,20]. The latter have been shown to regulate cerebral vascular diameter [20] and are necessary for ethanol to constrict these vessels [5]. Therefore, we set out to determine whether cholesterol-driven protection against ethanol-induced BK channel inhibition requires the presence of β1 protein. Thus, I/O recordings under physiological conditions of voltage (Vm = −40 mV) and Ca2+i = 30 μM were conducted on myocytes freshly isolated from middle cerebral arteries of wt C57BL/6 vs. KCNMB1 KO mice with either naïve cholesterol or subjected to cholesterol enrichment.
As demonstrated with BK from rat cerebral artery myocytes (Fig. 2), BK NPo from wt mice was drastically and reversibly reduced by 50 mM ethanol (Fig. 5). Ethanol-induced channel inhibition in wt mouse myocytes was not present in myocyte membrane patches from cholesterol-enriched arteries (Fig. 5).
Fig. 5.
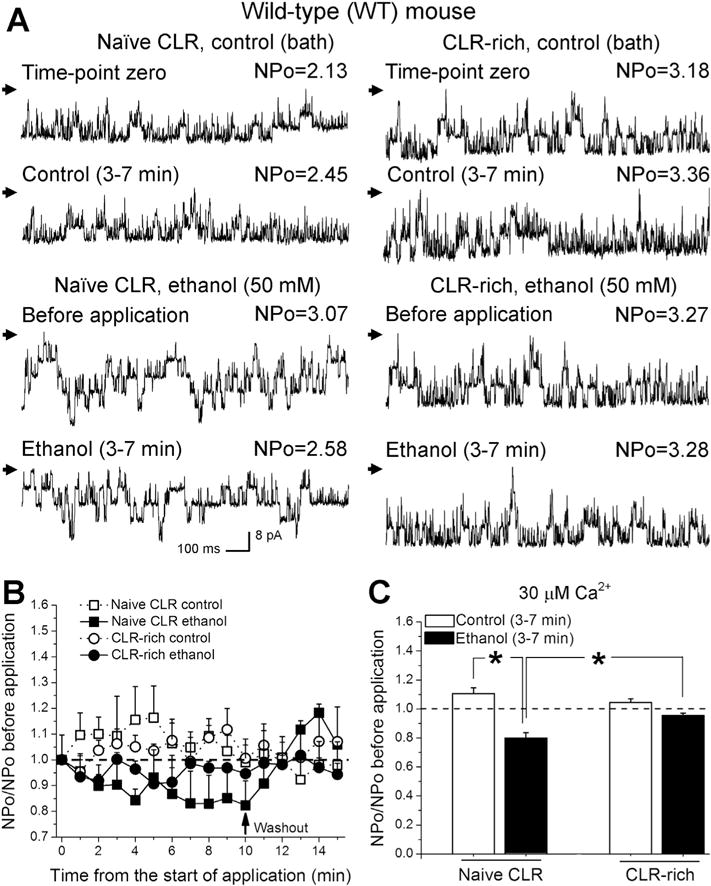
Cholesterol enrichment abolished ethanol-induced BK channel inhibition in membrane patches excised from middle cerebral artery myocytes of C57BL/6 mouse. (A) BK recordings from an I/O patch excised from an arterial myocyte with naïve cholesterol content (left column) or cholesterol-enriched (right column). Channel activity is shown before and during exposure to either ethanol-free (control, bath) or 50 mM ethanol solution. (B) Averaged changes in BK channel activity over time in myocyte membrane patches from arteries with naïve (n = 4) vs. enriched (n = 5) cholesterol levels. (C) Averaged fold changes in BK channel NPo. Each bar represents an average of data from four myocytes. A dotted line underscores pre-application level of activity. *Statistically significant difference (P < 0.05).
Ethanol-induced inhibition of BK activity was barely detectable in myocytes from KCNMB1 KO mice at 30 μM calcium at the intracellular side of the membrane patch (Fig. 6). The lack of ethanol-induced inhibition at this calcium level in the absence of the BK β1 subunit is consistent with previous reports [5]. Remarkably, cholesterol-enrichment further blunted ethanol action and turned it into BK channel activation (Fig. 6B, C). To ensure that cholesterol blunting of ethanol-induced BK channel inhibition is present when ethanol-induced inhibition is comparable with inhibition observed in myocytes from wt mice, we performed experiments at higher levels of calcium that would enable ethanol-induced BK channel inhibition in the absence of BK β1 subunits [5]. As expected, at a higher calcium level (300 μM), ethanol-induced BK channel inhibition was readily observed in membrane patches of cerebral artery myocytes from KCNMB1 KO mice (Fig. 7). Cholesterol-enrichment totally blunted this effect (Fig. 7). Thus, our data clearly demonstrated that antagonism of ethanol’s effect on BK channel activity does not require the presence of BK β1 subunits.
Fig. 6.
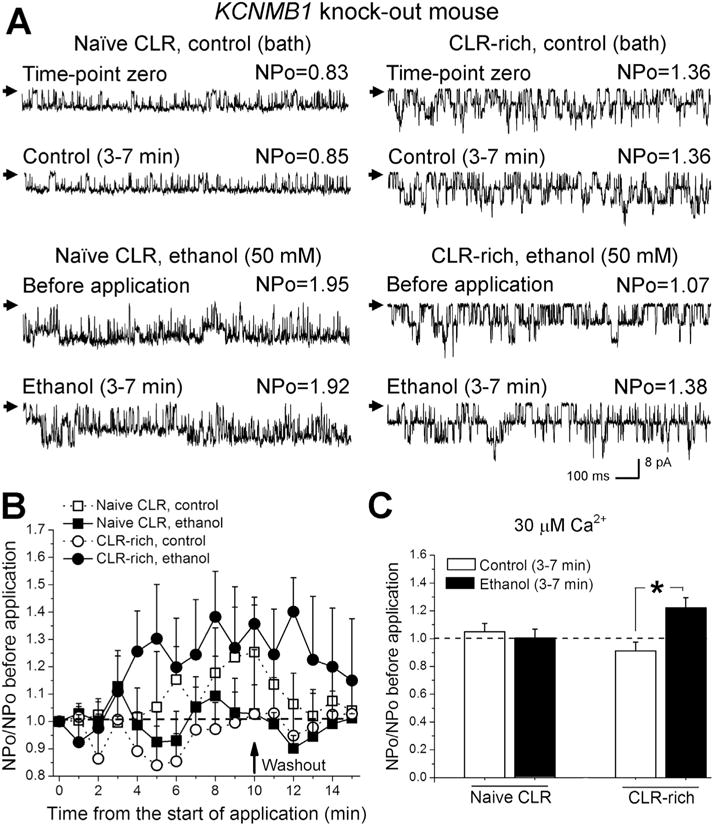
Cholesterol enrichment resulted in ethanol-induced BK channel activation in membrane patches excised from middle cerebral artery myocytes of KCNMB1 KO mice. (A) BK recordings from an I/O patch excised from an arterial myocyte with naïve cholesterol content (left column) or cholesterol-enriched (right column). Channel activity is shown before and during exposure to either ethanol-free (control, bath) or 50 mM ethanol solution. (B) Averaged changes in BK channel activity over time in myocytes membrane patches from arteries with naïve (n = 6) vs. enriched (n = 5) cholesterol levels. (C) Averaged fold changes in BK channel NPo at 30 μM Ca2+ at the intracellular side of membrane patch. Each bar represents an average of data from no less than five myocytes. A dotted line underscores pre-application level of activity. *Different from control (bath) application (P < 0.05).
Fig. 7.
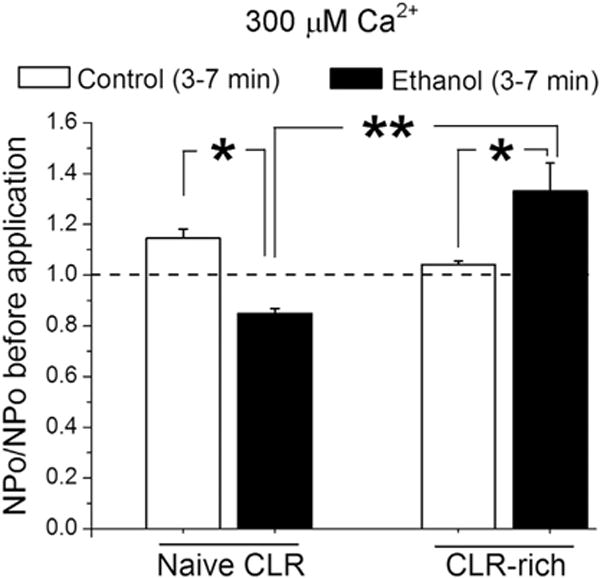
Cholesterol enrichment antagonizes ethanol-induced BK channel inhibition in membrane patches excised from middle cerebral artery myocyte of KCNMB1 KO mice at a high calcium levels. Averaged fold changes in BK channel NPo at 300 μM Ca2+ at the intracellular side of membrane patch. Each bar represents an average of data from four myocytes. A dotted line underscores pre-application level of activity. *Different from control (bath) application (P < 0.05). **Different from ethanol effect at naïve cholesterol level (P < 0.01).
3.5. Cholesterol antagonism of ethanol action in the absence of BK β1 subunits has a profound effect on organ function
To validate the patch-clamp results at the level of organ function, we used de-endothelialized, pressurized cerebral arteries from wt C57BL6 vs. KCNMB1 KO mice. These arteries retained their contractile machinery as was previously determined by probing C57BL6 and KCNMB1 KO cerebral arteries with vasoconstrictor phenylephrine [5]. In the present work, we probed artery diameter with 50 mM ethanol when the cholesterol level in the arterial wall was kept unmodified (naïve) or after cholesterol-enriching treatment. In all cases, the presence of myogenic tone was verified as previously described for rat cerebral arteries. As found with rat arteries (Fig. 1), 50 mM ethanol caused a significant reduction in wt mouse artery diameter (up to 14%) from pre-ethanol levels (Fig. 8A–B). Ethanol-induced constriction, however, was blunted after wt mouse arteries were pre-exposed to cholesterol-enriching treatment (Fig. 8A,B). This result underscores the similarity between cholesterol’s effect on alcohol responses in rat vs. mouse cerebral arteries.
Fig. 8.
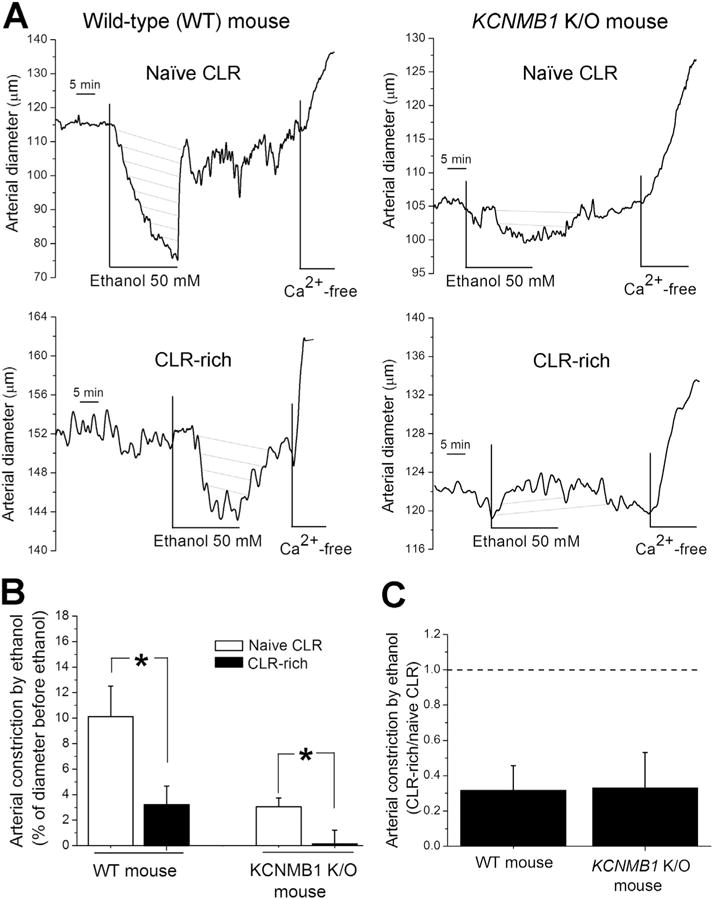
Blunting of ethanol-induced cerebral artery constriction by cholesterol enrichment does not require the presence of the BK β1 subunit as it is observed in KCNMB1 KO mouse artery. (A) Diameter traces of pressurized, de-endothelialized arteries from wt C57BL/6 (left column of traces) vs. KCNMB1 KO mouse (right column) show antagonism of ethanol-induced constriction in cholesterol-enriched vessels. (B) Averaged data showing statistically significant decrease in ethanol-induced middle cerebral artery constriction by cholesterol enrichment in arteries from both wt (C57BL/6) (n = 7) and KCNMB1 KO mice (n = 4). *Different from ethanol-induced constriction at naïve cholesterol level (P < 0.01). (C) Averaged data highlighting similar blunting of ethanol-induced constriction in cholesterol-enriched arteries in wt vs. KCNMB1 KO mouse middle cerebral arteries.
In contrast to wt mice and consistent with previous reports [5], de-endothelialized arteries from KCNMB1 KO mice barely constricted in the presence of 50 mM ethanol: ethanol-induced constriction did not exceed 3–4% (Fig. 8A–B). However, cholesterol-enriched KCNMB1 KO mouse arteries did not constrict in response to 50 mM ethanol at all (Fig. 8B). Moreover, in some cases mild dilation in response to ethanol was actually observed (Fig. 8A right column). On average, blunting of ethanol-induced constriction by cholesterol enrichment was similar between wt and KCNMB1 KO mouse arteries (Fig. 8C). The ability of cholesterol to antagonize ethanol’s effect on cerebral artery diameter in the absence of the BK β1 subunit highlights the fundamental role of β1 subunit-free BK channel complex as the effector of cholesterol-ethanol interactions leading to cholesterol antagonism of alcohol-induced cerebrovascular constriction.
4. Discussion
Cholesterol is a major constituent of mammalian cell plasma membrane and a critical modulator of ethanol’s effect on cerebral artery diameter. We recently showed that a high-cholesterol diet protects against ethanol-induced BK channel inhibition and resulting cerebral artery constriction via accumulation of cholesterol in the vasculature in vivo [8]. Our current work further expands these findings by showing that cholesterol enrichment in vitro fully mimics the effect of a high-cholesterol diet on ethanol-induced cerebral artery constriction (Fig. 1). However, the effect of cholesterol on ethanol-induced vasoconstriction is not monotonic: we have previously reported that cholesterol depletion from rat and mouse cerebral arteries blunts ethanol-induced BK channel inhibition and the resulting cerebral artery constriction [9]. Thus, there seems to be an optimal level of cholesterol to fully enable ethanol inhibition of BK channel function and reduction of cerebral artery diameter.
Modulation of ion channel pharmacology by increased cholesterol levels represents a widespread phenomenon. For example, an increase in cholesterol level significantly inhibits phenobarbital-induced block of voltage-gated Na+ channels [21]. A rise in membrane cholesterol increases the efficacy of nonsteroidal activators of GABA-A receptors, such as propofol, flunitrazepam, and pentobarbitone, while decreasing the action of steroid activators, such as pregnanolone and alphaxalone [22]. However, the mechanisms and channel subunits that enable cholesterol control over the pharmacological modulation of ion channel remain largely unknown. For the first time, our current work sheds light on possible distinct mechanisms that underlie cholesterol control over ethanol’s effect on BK channel function and cerebral artery diameter.
One of the critical findings of our work is that cholesterol and ent-cholesterol possess a similar ability to blunt ethanol-induced BK channel inhibition and the resulting constriction of cerebral arteries (Figs. 1C, 2C, 3C, 4D). Earlier work on artificial lipid bilayers demonstrated the differential ability of these sterols to modulate alcohol’s action on BK channel function. Unlike cholesterol, 20% and 40% ent-cholesterol had little effect on the ethanol sensitivity of BK channels [19]. The discrepancy with our findings could be explained by the simplicity of the artificial lipid bilayers. In the cited work, bilayers consisted of two phospholipid species, while native membranes are estimated to be composed of > 1000 lipids [23]. In an artificial lipid bilayer system, many natural protein and lipid partners of the BK channel complex are most likely absent.
One of the protein partners of the BK channel in native vascular smooth muscle tissue is the accessory β1 subunit. BK β1 subunits are unable to form functional channels themselves, yet they drastically modify BK current phenotype and channel pharmacology [10,20, 24–25]. BK channels that contain BK β1 subunits (heteromeric channels) respond to cholesterol differently when compared to homomeric BK channels consisting of only a BK α protein. Our recent patch-clamp data obtained from native (β1 subunit-containing) BK channels in membrane patches from freshly isolated vascular myocytes show that cholesterol and ent-cholesterol enrichment similarly enhanced BK channel function [26]. This result differs from our earlier data reporting inhibition of β1 subunits lacking BK channels in the presence of cholesterol but not ent-cholesterol in artificial lipid bilayers [18,27]. The discrepancy clearly points out the unique role of cholesterol and ent-cholesterol in tuning BK current in the presence of β1 subunits as opposed to channels lacking β1 subunits.
In the case of β1 subunit-containing BK channels, the similar ability of cholesterol and ent-cholesterol to enhance BK channel function may hold the key to unraveling the precise mechanism by which both steroids blunt alcohol-induced BK channel inhibition and the resulting cerebral artery constriction (Figs. 3–4).
Ent-cholesterol modifies lipid membrane properties similarly to cholesterol, whereas ent-cholesterol cannot be recognized by cholesterol-specific protein sites [15–17]. Thus, the similar ability of ent-cholesterol and natural cholesterol to blunt ethanol’s effect likely indicates that cholesterol control over ethanol action does not involve recognition by a protein site that specifically senses natural cholesterol. If such a site is needed, it would have very lax structural requirements that enable recognition of both cholesterol and its synthetic enantiomer. Alternatively, cholesterol control over alcohol’s effect is enabled by a lipid bilayer-mediated mechanism(s). This probability is favored by the fact that both cholesterol and ent-cholesterol affect bilayer physical properties similarly, consistent with the similar ability to blunt alcohol-induced BK channel inhibition and resulting vasoconstriction.
Another possible explanation for the cholesterol/ent-cholesterol antagonism of alcohol action on BK channel function and resulting vasoconstriction arises from the ability of cholesterol to diminish the lipid/membrane partition coefficient of a variety of small anesthetics, such as halothane [28], uncharged pentobarbitone [29], and benzyl alcohol [30]. Isothermal titration calorimetry data show that ethanol partitioning into phosphatidylcholine liposomes is reduced by cholesterol when present in the bilayer at concentrations exceeding 10 mol% [31]. An increase in cholesterol content from 10 to 40 mol% leads to a 3-fold decrease in ethanol partition coefficient into the liposome [31]. Considering that the cholesterol level in mammalian plasma membranes ranges between 20 and 50 mol% [32], it is expected that cholesterol enrichment would make it difficult for ethanol to partition into the membrane and thus reach effective concentrations to modulate relevant molecular targets. However, in our experiments, ethanol was probed at 50 mM. Our earlier work demonstrated that the effect of ethanol was concentration-dependent: ethanol-induced BK inhibition and resulting vasoconstriction were observed at ethanol concentrations as low as 3–10 mM [4,33]. Thus, a 3-fold drop of ethanol concentration able to reach into the bilayer would still render ethanol levels at which ethanol-induced constriction could be observed (albeit to a much lesser extent than seen when ethanol is probed at 50 mM). Interestingly, we have recently described an ethanol-sensing site in the cytosolic tail domain of the BK pore-forming α subunit [34]. The site recognizes ethanol molecules and mediates ethanol-induced BK channel activation that is observed in homomeric (only α subunits) channels at calcium levels below 30 μM [34,35]. In our current work, ethanol was applied to the inner leaflet of the myocyte membranes with the cytosolic tail domain of the channel being readily exposed to the ethanol-containing solution. If a cholesterol-driven decrease in ethanol partition coefficient into the membrane underlies cholesterol antagonism of ethanol’s effect on BK channel function and cerebral artery diameter, then the described ethanol-sensing site seems unlikely to play a major role in the ethanol-induced BK channel inhibition that was observed in our experiments and that is routinely reported in the presence of β1 subunits and high levels of Ca2+i [5,35].
Remarkably, cholesterol blunting of BK channel-mediated sensitivity of cerebral artery to ethanol is not the only case when cholesterol enrichment of the artery results in loss of pharmacological modulation by BK channel-targeting pharmacological agents. In our recent work, we reported a significant decrease in cerebral artery constriction by selective BK channel blocker paxilline [8]. It has been proposed that paxilline should occupy the central cavity (pore) region of the BK α subunit tetramer in order to block BK current [36]. Thus, in the case of paxilline, the cholesterol-driven decrease in membrane partition coefficient would not play a significant role. At this point, it remains unclear whether cholesterol-driven blunting of ethanol and paxilline’s effect share a mechanism(s). The origin of this common, “non-bilayer” mechanism is speculative at this point but should likely involve a common downstream effector that effectively senses the presence of cholesterol and reduces the efficacy of several BK channel inhibitors.
Our data from arteries and myocytes of KCNMB1 KO mice clearly demonstrated that the BK β1 subunit was not necessary for cholesterol antagonism of ethanol’s effect (Figs. 6–8). Thus, the mechanism(s) that enables cholesterol control over ethanol does not require such BK regulatory subunit. One likely possibility is that such mechanism solely involves a non-specific steroid-recognizing protein site in the BK α subunit. The exploration of this possibility would require crystallographic studies and/or computational modeling of cholesterol and ent-cholesterol docking on this protein. Remarkably, we earlier identified seven cholesterol recognition amino acid consensus (CRAC) motifs within the BK α subunit’s cytosolic tail domain that contribute to cholesterol sensitivity of the homomeric BK channel in artificial lipid bilayers [27]. The contribution of each individual CRAC motif to cholesterol sensitivity and sensing of elevated cholesterol levels in the native BK channel environment remains to be explored. The most important question, however, is whether any CRAC is critical for cholesterol control over ethanol’s effect. The work addressing this question is currently ongoing.
5. Conclusions
For the first time, we showed that ethanol’s effect on BK channel inhibition and cerebral artery constriction are blunted by elevated cholesterol levels. Cholesterol’s control over ethanol’s effect is unlikely to be mediated by a protein site that is specific for natural cholesterol but rather involves a protein site with lax structural requirements towards the isomery of steroid ligand. Certainly, involvement of bilayer-mediated mechanisms cannot be ruled out. Disregarding the mechanisms by which an increased cholesterol level blunts ethanol’s effect on BK channel function and cerebral artery diameter, BK β1 subunits are not necessary for cholesterol antagonism of alcohol’s effect when the BK channel is studied in a native membrane environment (either in myocyte membranes or within cerebral arteries). A deep understanding of mechanisms that underlie cholesterol-ethanol interactions at the level of cerebral arteries is critical for developing novel therapeutics that would eliminate deleterious effects of episodic (binge) drinking on cerebral circulation.
Supplementary Material
Acknowledgments
We deeply thank Dr. Guruprasad Kuntamallappanavar (Department of Pharmacology, UTHSC) for maintaining the KCNMB1 KO mouse colony. Supported by NIH grants R37 AA11560 (AD) and R01 AA023764 (AB).
Abbreviations
- BK
voltage- and calcium-gated potassium channels of large conductance
- CLR
cholesterol
- CRAC
cholesterol recognition amino acid consensus
- ent-cholesterol
enantiomeric cholesterol
- I/O
inside-out (membrane patch)
- KO
knockout
- LDL
low-density lipoprotein
- MβCD
methyl-β-cyclodextrin
- Po
open probability
- PSS
physiological saline solution
- wt
wild type
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbalip.2016.08.013.
Footnotes
Conflict of interest
Authors declare no conflict of interest.
Transparency document
The Transparency document associated with this article can be found, in the online version.
References
- 1.Puddey IB, Rakic V, Dimmitt SB, Beilin LJ. Influence of pattern of drinking on cardiovascular disease and cardiovascular risk factors–a review. Addiction. 1999;94:649–663. doi: 10.1046/j.1360-0443.1999.9456493.x. [DOI] [PubMed] [Google Scholar]
- 2.Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. JAMA. 2003;289:579–588. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- 3.Altura BM, Altura BT. Alcohol, the cerebral circulation and strokes. Alcohol. 1984;1:325–331. doi: 10.1016/0741-8329(84)90056-9. [DOI] [PubMed] [Google Scholar]
- 4.Liu P, Xi Q, Ahmed A, Jaggar JH, Dopico AM. Essential role for smooth muscle BK channels in alcohol-induced cerebrovascular constriction. Proc Natl Acad Sci U S A. 2004;101:18217–18222. doi: 10.1073/pnas.0406096102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukiya AN, Liu J, Dopico AM. The BK channel accessory beta1 subunit determines alcohol-induced cerebrovascular constriction. FEBS Lett. 2009;583:2779–2784. doi: 10.1016/j.febslet.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- 7.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 8.Bukiya AM, Dopico CW, Leffler A. Fedinec, dietary cholesterol protects against alcohol-induced cerebral artery constriction. Alcohol Clin Exp Res. 2014;38:1216–1226. doi: 10.1111/acer.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bukiya AN, Vaithianathan T, Kuntamallappanavar G, Asuncion-Chin M, Dopico AM. Smooth muscle cholesterol enables BK β1 subunit-mediated channel inhibition and subsequent vasoconstriction evoked by alcohol. Arterioscler Thromb Vasc Biol. 2011 Nov;31(11):2410–2423. doi: 10.1161/ATVBAHA.111.233965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bukiya AN, McMillan JE, Fedinec AL, Patil SA, Miller DD, Leffler CW, Parrill AL, Dopico AM. Cerebrovascular dilation via selective targeting of the cholane steroid-recognition site in the BK channel β1-subunit by a novel nonsteroidal agent. Mol Pharmacol. 2013;83:1030–1044. doi: 10.1124/mol.112.083519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 1768. 2007:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belani JD, Rychnovsky SD. A concise synthesis of ent-cholesterol. J Organomet Chem. 2008;73:2768–2773. doi: 10.1021/jo702694g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pérez GJ, Bonev AD, Nelson MT. Micromolar Ca(2+) from sparks activates Ca(2+)-sensitive K(+) channels in rat cerebral artery smooth muscle. Am J Phys Cell Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 14.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alakoskela JM, Sabatini K, Jiang X, Laitala V, Covey DF, Kinnunen PK. Enantiospecific interactions between cholesterol and phospholipids. Langmuir. 2008;24:830–836. doi: 10.1021/la702909q. [DOI] [PubMed] [Google Scholar]
- 16.Crowder CM, Westover EJ, Kumar AS, Ostlund RE, Jr, Covey DF. Enantiospecificity of cholesterol function in vivo. J Biol Chem. 2001;276:44369–44372. doi: 10.1074/jbc.C100535200. [DOI] [PubMed] [Google Scholar]
- 17.Westover EJ, Covey DF. The enantiomer of cholesterol. J Membr Biol. 2004;202:61–72. doi: 10.1007/s00232-004-0714-7. [DOI] [PubMed] [Google Scholar]
- 18.Bukiya AN, Belani JD, Rychnovsky S, Dopico AM. Specificity of cholesterol and analogs to modulate BK channels points to direct sterol-channel protein interactions. J Gen Physiol. 2011;137:93–110. doi: 10.1085/jgp.201010519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan C, Chen M, Covey DF, Johnston LJ, Treistman SN. Cholesterol tuning of BK ethanol response is enantioselective, and is a function of accompanying lipids. PLoS One. 2011;6:e27572. doi: 10.1371/journal.pone.0027572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 21.Rehberg B, Urban BW, Duch DS. The membrane lipid cholesterol modulates anesthetic actions on a human brain ion channel. Anesthesiology. 1995;82:749–758. doi: 10.1097/00000542-199503000-00017. [DOI] [PubMed] [Google Scholar]
- 22.Sooksawate T, Simmonds MA. Influence of membrane cholesterol on modulation of the GABA(A) receptor by neuroactive steroids and other potentiators. Br J Pharmacol. 2001;134:1303–1311. doi: 10.1038/sj.bjp.0704360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sud M, Fahy E, Cotter D, Brown A, Dennis E, Glass C, Merrill A, Murphy R, Raetz C, Russell D, Subramaniam S. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35:D527–D532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bukiya AN, Liu J, Toro L, Dopico AM. Beta1 (KCNMB1) subunits mediate lithocholate activation of large-conductance Ca2+-activated K+ channels and dilation in small, resistance-size arteries. Mol Pharmacol. 2007;72:359–369. doi: 10.1124/mol.107.034330. [DOI] [PubMed] [Google Scholar]
- 25.Bukiya AN, Vaithianathan T, Toro L, Dopico AM. Channel beta2–4 subunits fail to substitute for beta1 in sensitizing BK channels to lithocholate. Biochem Biophys Res Commun. 2009;390:995–1000. doi: 10.1016/j.bbrc.2009.10.091. [DOI] [PubMed] [Google Scholar]
- 26.Bisen S, Belani J, Rychnovsky S, Dopico A, Bukiya A. Cholesterol and ent-cholesterol enrichment similarly enhances BK channel function and blunts alcohol effect in rat cerebral artery. Research Society on Alcoholism 39th Annual Meeting. 2016 [Google Scholar]
- 27.Singh AK, McMillan J, Bukiya AN, Burton B, Parrill AL, Dopico AM. Multiple cholesterol recognition/interaction amino acid consensus (CRAC) motifs in cytosolic C tail of Slo1 subunit determine cholesterol sensitivity of Ca2+- and voltage-gated K+ (BK) channels. J Biol Chem. 2012;287:20509–20521. doi: 10.1074/jbc.M112.356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lechleiter J, Wells M, Gruener R. Halothane-induced changes in acetylcholine receptor channel kinetics are attenuated by cholesterol. Biochim Biophys Acta. 1986;856:640–645. doi: 10.1016/0005-2736(86)90159-8. [DOI] [PubMed] [Google Scholar]
- 29.Miller KW, Yu SC. The dependence of the lipid bilayer membrane: buffer partition coefficient of pentobarbitone on pH and lipid composition. Br J Pharmacol. 1977;61:57–63. doi: 10.1111/j.1476-5381.1977.tb09739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colley CM, Metcalfe JC. The localisation of small molecules in lipid bilayers. FEBS Lett. 1972;24:241–246. doi: 10.1016/0014-5793(72)80364-8. [DOI] [PubMed] [Google Scholar]
- 31.Trandum C, Westh P, Jorgensen K, Mouritsen OG. A thermodynamic study of the effects of cholesterol on the interaction between liposomes and ethanol. Biophys J. 2000;78:2486–2492. doi: 10.1016/S0006-3495(00)76793-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bloch K. Cholesterol: evolution of structure and function. In: Vance DE, Vance JE, editors. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier; Amsterdam, The Netherlands: 1991. pp. 363–381. [Google Scholar]
- 33.Dopico AM. Ethanol sensitivity of BK(Ca) channels from arterial smooth muscle does not require the presence of the beta 1-subunit. Am J Phys Cell Physiol. 2003;284:C1468–C1480. doi: 10.1152/ajpcell.00421.2002. [DOI] [PubMed] [Google Scholar]
- 34.Bukiya AN, Kuntamallappanavar G, Edwards J, Singh AK, Shivakumar B, Dopico AM. An alcohol-sensing site in the calcium- and voltage-gated, large conductance potassium (BK) channel. Proc Natl Acad Sci U S A. 2014;111:9313–9318. doi: 10.1073/pnas.1317363111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Vaithianathan T, Manivannan K, Parrill A, Dopico AM. Ethanol modulates BKCa channels by acting as an adjuvant of calcium. Mol Pharmacol. 2008;74:628–640. doi: 10.1124/mol.108.048694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Tang QY, Xia XM, Lingle CJ. Glycine311, a determinant of paxilline block in BK channels: a novel bend in the BK S6 helix. J Gen Physiol. 2010;135:481–494. doi: 10.1085/jgp.201010403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.