

SUMMARY
Mass spectrometry-based proteomics now enables the absolute quantification of thousands of proteins in individual cell types. We used this technology to analyze the dynamic proteome changes occurring during human erythropoiesis. We quantified the absolute expression of 6,130 proteins during erythroid differentiation from late burst-forming units-erythroid (BFU-Es) to orthochromatic erythroblasts. A modest correlation between mRNA and protein expression was observed. We identified several proteins with unexpected expression patterns in erythroid cells, highlighting a breakpoint in the erythroid differentiation process at the basophilic stage. We also quantified the distribution of proteins between reticulocytes and pyrenocytes after enucleation. These analyses identified proteins that are actively sorted either with the reticulocyte or the pyrenocyte. Our study provides the absolute quantification of protein expression during a complex cellular differentiation process in humans, and it establishes a framework for future studies of disordered erythropoiesis.
In Brief
Gautier et al. use quantitative mass spectrometry to determine the absolute proteome composition of human erythroid progenitors throughout the differentiation process and the quantitative distribution of proteins between reticulocytes and pyrenocytes after enucleation.
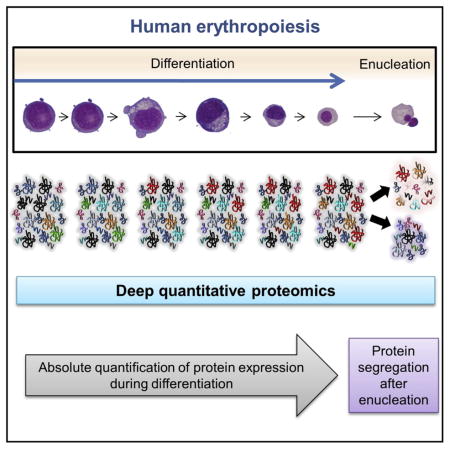
INTRODUCTION
Healthy humans produce around two million red cells each second of their lives. This tightly regulated process takes place in the bone marrow, and it begins with a restriction in the potency of multipotent hematopoietic stem cells to lineage-specific progenitor cells, such as progenitors strictly committed to the erythroid lineage. The second step is an amplification phase in which erythroid progenitors proliferate extensively under the control of several growth factors.
Although these cells are morphologically indistinguishable and their maturation process is continuous, two kinds of erythroid progenitors are successively distinguished. The first erythroid-committed progenitors are burst-forming units-erythroid (BFU-Es), which require stem cell factor (SCF), but not erythropoietin (EPO), for proliferation. In contrast, EPO is absolutely required for the survival and proliferation of the late erythroid progenitors called colony-forming units-erythroid (CFU-Es). The last phase of erythropoiesis is terminal differentiation. In this step, several morphologically recognizable precursors are successively produced: proerythroblast (ProE) cells and basophilic I and II (Baso1 and Baso2), polychromatophilic (Poly), and orthochromatic (Ortho) erythroblasts. During this process, the size of the cells gradually decreases, and they synthesize large amounts of hemoglobin (Hb) and reorganize their membrane with accompanying nuclear condensation. At the end of terminal erythroid differentiation, Ortho cells expel their nucleus, which is surrounded by plasma membrane with a small amount of cytoplasm, to generate a pyrenocyte, which is rapidly engulfed by macrophages of the erythroblastic niches, and a reticulocyte, which completes its maturation in the bloodstream. During this enucleation process, several proteins appear to be actively sorted between pyrenocytes and reticulocytes, although the extent of this active sorting process remains unclear.
Erythropoiesis is studied extensively both as a differentiation paradigm and because red blood cells are involved in many serious human diseases. Although several aspects are well understood at the molecular level, a global and integrated analysis of this differentiation process is required. Several transcriptomic analyses of erythropoiesis have been published, leading to the determination of the expression pattern of 8,500–12,000 genes at different differentiation stages (An et al., 2014; Kingsley et al., 2013; Li et al., 2014; Merryweather-Clarke et al., 2011; Shi et al., 2014). In contrast, a deep proteomic analysis of this differentiation process is still lacking. Because the relationship between mRNA and protein expression is far from straightforward (Vogel and Marcotte, 2012), a comprehensive characterization of the proteome of erythroid cells during their differentiation is now essential to better understand both normal erythropoiesis and the pathologies affecting this process.
Current proteomic methods allow the identification of several thousand proteins from microgram quantities of proteins. Robust comparative quantification methods were first developed by using differential labeling with stable isotopes. Until recently, the most widely used labeling method was the stable isotope labeling by amino acids in cell culture (SILAC) method (Mann, 2006). The major drawback of this powerful method is that primary cells usually do not proliferate enough in cell culture to allow the complete replacement of amino acids. Several methods to chemically label peptides, such as isobaric tags for relative and absolute quantitation (ITRAQ), have been developed to overcome this limitation. Today, efficient label-free quantification (LFQ) methods combine high-accuracy mass measurement of peptides using recently developed mass spectrometers with their reproducible retention time in the chromatographic step to quantitatively compare peptide populations in successive liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses. Key improvements to these methods were the development of highly efficient algorithms to align chromatograms from successive analyses (Cox et al., 2014) and the creation of absolute quantification procedures from label-free data (Wiśniewski et al., 2014).
In the present study, we analyzed the proteome of erythroid cells at different differentiation stages by an LFQ method. We used the recently described standardization method based on the MS signal of histone peptides to quantify the absolute copy number of more than 6,100 proteins, providing the most comprehensive proteomic database of erythropoiesis available to date. Moreover, we used an ITRAQ labeling method to quantify the segregation of 1,318 proteins between pyrenocytes and reticulocytes following enucleation. Cytoplasmic volumes and surface areas of pyrenocytes and reticulocytes were calculated from imaging flow cytometry, and they were used to estimate whether these proteins are passively or actively sorted into each cell type. The overall flowchart of the study design is presented in Figure 1.
Figure 1.

Experimental Design
RESULTS AND DISCUSSION
Erythroid Cell Populations
Our goal to decipher the modifications of the erythroid cell proteome during terminal differentiation required access to sufficient amounts of homogenous cell populations at different differentiation stages. Two potential strategies are available to obtain such material: either purification of cells at the different differentiation stages by flow sorting using specific cell surface markers or maintenance of a high level of synchrony in the cultures during the differentiation process. The latter strategy was chosen for the proteomic studies due to the limited ability of the sorting approach to generate sufficient amounts of stage-specific erythroid cell populations for analysis.
Starting from purified CD34+ cell populations from cord blood, a two-step culture method was modified to obtain highly homogenous erythroid cell populations at different differentiation stages (see Experimental Procedures and Figure 2A). First, CD34+ cells were cultured for 7 days in the presence of IL3, IL6, and SCF to allow the proliferation of early progenitors. Erythroid cells were then purified using the CD36 marker to generate a cell population essentially composed of progenitors, because, without EPO, the erythroid cells failed to survive to the CFU-E stage. Starting from this step, cells were cultured in the presence of EPO and characterized each day both phenotypically by flow cytometry and morphologically by May-Grünwald-Giemsa (MGG) staining (Figures 2B, 2C, and S1). The cell populations were sampled daily for proteome analysis, although daily sampling of cells was not feasible in all experiments at the progenitor stage due to a scarcity of material (Figure 2D).
Figure 2. Erythroid Cell Cultures.

(A) Culture protocol is shown.
(B) Amplification rate of a typical culture is shown.
(C) Characterization of the differentiation stages in a typical culture. Upper panel: MGG staining of cytocentrifuged cells is shown; middle panel: flow cytometry analysis of integrin alpha 4 (CD49d) and band 3 expression is shown; lower panel: phenotypic characteristic features of the cell populations are shown.
(D) Cell sample grouping for data analysis. Cell samples from the four experiments were regrouped in seven categories according to the phenotypes described in (C) and MGG staining. Up to day 3, the unanalyzed samples did not contain enough material for analysis and two samples of experiments E1 (D6 and D9) that presented an equal mixture of two differentiation stages were not analyzed.
(E) Cell composition of each cell category. For each category resulting from the grouping presented in (D), the mean composition ±SD was calculated according to the erythroblast morphology determined by MGG staining.
Overall, terminal differentiation from the CD36+ cell purification to the Ortho stage lasted 10 ±1 days, and the amplification level was around 300-fold (Figure 2B). We distinguished two progenitor populations (Prog1 and Prog2) based on the expression levels of CD71 and CD34. The Prog1, but not Prog2, population expressed CD34 (Figure S1). Clonogenic cultures of CD36- sorted cells showed that the Prog1 population was mainly composed of late BFU-Es (Figure S1), in agreement with their overall amplification potential and the simultaneous expression of both CD34 and CD36 markers. Overall, we identified cell populations at seven distinct differentiation stages for data analysis (Figure 2C). Transition from one stage to the next took around 24 hr, although the differentiation process seemed to slow down slightly at later stages, leading to the grouping of 2 consecutive culture days to the same differentiation stage (Figure 2D). Four independent cultures established from four different cord blood samples were used for the proteomic analysis. Due to some biological variability, the duration of terminal differentiation ranged from 9 to 11 days. Each daily sampled cell population was analyzed separately by MS, and data from the four experiments were grouped according to Figure 2D for analysis. After this grouping, each cell population contained more than 50% of cells at the main differentiation stage, and more than 90% of the cells corresponded to the main differentiation stage and the stages just before and after this stage. As such, the set of samples used for the analysis accurately represents the entire erythroid differentiation process (Figure 2E).
Proteomic Analysis of Erythroid Cell Populations
To improve proteome coverage using the match between run (MBR) algorithm of MaxQuant software (Deshmukh et al., 2015), extracts from two cell lines with erythroid characteristics, UT7 and K562, were analyzed simultaneously with primary erythroid cells (validation of the MBR algorithm is presented in the Supplemental Experimental Procedures and Figure S2). This led to the identification of 7,361 proteins expressed in primary erythroid cells with a false discovery rate below 1% (Table S2). The mean sequence coverage of these proteins was 26.4% ±2.4%. We applied the recently described proteomic ruler to determine the absolute expression of these proteins, using the total MS signal of all core and linker histones and a histone content of 6.5 pg/human cell. This method requires that at least 12,000 peptides be identified in each experiment and that MS signals of at least two different peptides be measured for each protein to be quantified (Wiśniewski et al., 2014). Between 15,566 and 40,626 peptides were identified for each sample, enabling us to apply this method to all cell samples in each experiment and leading to the absolute quantification of 6,160 proteins. All subsequent calculations and their discussion are restricted to these quantified proteins.
Around 5,000 proteins were quantified at each differentiation stage, although this value was slightly lower at the final stages of differentiation (Figure 3A). This could be either due to a decreased number of proteins expressed in the late stages of differentiation and/or the high amounts of Hb that accumulate at these stages, which could reduce the identification efficiency of minor proteins via an ionization suppression effect. Most proteins were quantified in all of the seven differentiation stages, and only 550 proteins were observed in a single cell population (Figure 3B). Pearson correlation analysis revealed high homologies between the different populations (Figure 3C), indicating both the reproducibility of these analyses and the relative stability of the expression of many proteins during differentiation, despite strong modifications of the cells. Thus, these results confirm the observation realized at the transcriptomic level that most genes active during terminal differentiation are already expressed at the ProE stage (Kingsley et al., 2013). Both hierarchical clustering analysis (Figure 3D) and Pearson correlation analysis (Figure 3C) of these quantitative data confirmed the continuous evolution of the erythroblast proteomes according to the differentiation stages of erythroid cells. Expression of classical erythroid differentiation markers (CD36, TFRC, band 3, GPA, CD44, CD45, ITGα4, and c-KIT) followed the expected patterns (Figure S3A). The expression pattern of several selected proteins also was verified by western blot analysis (Figure S3B). All of these results show that the proteomic analyses accurately reflected the evolution of erythroid cells during terminal differentiation.
Figure 3. Absolute Proteome Quantification during Terminal Differentiation of Erythroid Cells.

(A) Total number of quantified proteins in each differentiation stage is shown.
(B) Number of quantified proteins according to the number of differentiation stages in which they have been quantified is shown.
(C) Pearson correlation analysis of quantified protein expression between the differentiation stages is shown.
(D) Hierarchical clustering analysis of quantified proteins at the different differentiation stages is shown.
(E) Determination of the total protein content per cell during differentiation. BCA curve: the amount of protein per cell was calculated by dividing the total amount of protein determined by a bicinchoninic acid assay using BSA as a standard by the number of cells. MS curve: the amount of protein per cell was calculated as the sum of the amount of each protein quantified by MS after standardization to the histone MS signal.
(F) Protein copy number in stable multimeric complexes at the ProE stage. Selected multimeric complexes contain between 14 (20S proteasome) and 44 (ribosome large subunit) different proteins with a single copy for each of them in the complex.
(G) Comparison of protein expression values determined by LFQ with published expression values. Except for c-KIT and the glucocorticoid receptor, which were measured in erythroid progenitors (Dai et al., 1994; Mayeux et al., 1983), all published values corresponded to protein expression in erythrocytes (Burton and Bruce, 2011). They were compared with values determined in Ortho cells.
(H) Protein expression range. The overall curve was drawn using the highest expression value of each protein irrespective of the differentiation stage.
To verify the accuracy of the quantitative analysis, the total amount of protein content per cell was calculated from the proteomic analysis and compared with the amount directly determined from protein quantification and cell counting. Although a slight systematic difference between the two determinations was observed that could be due to the use of BSA as a protein standard for protein quantification, the evolution of cell protein contents along erythroid differentiation was very similar when calculated by either of the two methods (Figure 3E). We checked the relative expression of proteins involved in stable protein complexes containing one copy of each protein subunit, such as ribosome subunits or proteasome. For each complex, very similar numbers of each protein of the complex were quantified (Figure 3F). Lastly, we compared our quantification results with previously published data; however, except for a few proteins regulating erythropoiesis, such as hormone and growth factor receptors, most available data deal exclusively with mature erythrocyte proteins. To increase the number of values for the comparison, we made the assumption that most proteins of the circulating red cells would have been synthesized already by the Ortho stage. For this comparison, protein expression extended to nearly five logs, from 3,000 copies per cell for the glucocorticoid receptor to up to 270 × 106 copies for Hb. This analysis confirms the accuracy of the quantitative proteomic data (Figure 3G). Altogether, these controls strongly suggest that the data constitute an accurate determination of the absolute concentrations of most proteins expressed by erythroid cells along their terminal differentiation, and they confirm the accuracy of quantification methods based on histone signal standardization. These findings also show that the nuclear condensation occurring during erythroid differentiation does not significantly affect the overall histone content of the cells.
The expression range of identified proteins encompassed more than seven logs, ranging from tens of copies per cell for proteins such as MYB to several hundreds of millions for HBA or HBB. As expected, because the size of erythroblasts decreases during differentiation, the median number of protein copies per cell decreased from 30,000 copies/cell at the progenitor stage to 6,000 copies at the Ortho stage (Figure 3H).
Evolution of Gene and Protein Expression during Erythroid Differentiation
The total protein content of erythroblasts decreased during differentiation, with the cells simultaneously accumulating Hb. At the Ortho stage, Hb chains and AHSP, the molecular chaperone of Hbα chains, constituted 50% of the total cellular protein content (Figure 4A). At the progenitor or ProE stage, the 25 most abundant proteins constituted around 25% of the total protein mass of the cell, whereas at the Ortho stage they constituted more than 60%. This is due to Hb accumulation because, if its mass was subtracted, the contribution of the 10, 25, or 50 most abundant proteins remained constant throughout the cell differentiation process. Consequently, the real decrease in protein amounts per cell along erythroid differentiation reached 75% at the Ortho stage compared with the ProE stage (Figure 4B). This value was used to compare the evolution of the erythroid cell proteome during the differentiation process.
Figure 4. Evolution of the Proteome during Erythroid Terminal Differentiation.

(A) Contribution of the 10, 25, and 50 most abundant proteins to the entire protein content of the cell. The calculations were done with all proteins (Hb+) or after removal of the contributions of Hb chains and AHSP (Hb−). The Hb curve shows the contribution of hemoglobin chains and AHSP to the overall cell protein content.
(B) Evolution of the cellular protein content after subtraction of hemoglobin and AHSP contributions is shown.
(C–E) Quantitative expression of erythroid-specific transcription factors (C), splicing complexes proteins (D), and translation machinery proteins (E). Medians of protein expression are indicated for multiproteinic translation complexes (asterisks).
Proteins differentially expressed during erythroid differentiation were extracted by a multi-sample ANOVA test and submitted to k-means clustering analysis. As expected, most differentially expressed proteins decreased during erythroid differentiation (see Figure S4 and Table S3, clusters 1 and 2). Functional annotation analysis of these two clusters showed a significant enrichment of proteins related to general cellular processes, such as RNA processing, translation, metabolism, intracellular transport, proteasome degradation, and DNA replication and repair. Cluster 3 regrouped 40 proteins that significantly increased during differentiation. In addition to proteins involved in erythroid differentiation, such as Hb, band 3, SLC2A1 (GLUT1), SPTB, AHSP, or HEMGN (EDAG), this cluster also contained proteins such as optineurin (OPTN), an autophagy inducer; histone H1F0, a variant histone linker; and PHOS-PHO1, a phosphatase involved in bone mineralization. A fourth cluster showed proteins such as LEPR or IRF6, which peaked at the ProE/Baso1 stage and then decreased. The roles of these proteins in erythroid differentiation remain unknown.
Gene ontology (GO) analysis revealed a classical repartition of proteins among the different cellular compartments, including the presence of many membrane and secreted proteins (Figure S4). In total, 428 proteins with sequence-specific DNA-binding transcription factor activity or transcription cofactor activity according to GO were identified among the quantified proteins, including all specific transcription factors regulating erythropoiesis (Figure 4C).
The two STAT5 isoforms were expressed to different levels, with STAT5A being significantly more highly expressed than STAT5B at all differentiation stages. It is currently unknown whether each STAT5 isoform has different activities in erythroid cells, as has been reported recently in chronic myeloid leukemia cells (Casetti et al., 2013). Except for NFE2, expression of erythroid-specific transcription factors was already maximal at the progenitor or ProE stage, ranging from 10,000 to 50,000 copies per cell at these stages. GATA1, KLF1, and Tal1 are considered the master regulators of erythroid terminal differentiation. Their expression patterns were very similar, with a decrease at the end of differentiation that roughly followed the 4-fold decrease in the whole protein content of erythroblasts during their differentiation. GATA1 and KLF1 were both expressed at around 50,000 copies per cell at the ProE stage. These values should be compared with those of their DNA-binding sites determined by chromatin immunoprecipitation sequencing (ChIP-seq) or ChIP followed by 5′-to-3′ exonuclease treatment and then massively parallel DNA sequencing (ChIP-exo) studies, although the published determinations present variations, leading to some uncertainty as to the true number of DNA targets. Around 15,000 and between 5,700 and 10,000 DNA-binding sites have been identified for KLF1 and GATA1, respectively (Fujiwara et al., 2009; Han et al., 2015; Kassouf et al., 2010; Pilon et al., 2011). This suggests that enough molecules of these transcription factors should be present at each differentiation stage to regulate the entire set of their target genes. TAL1 follows the same expression kinetics as that of GATA1 and KLF1, but its expression level appears to be around 5-fold lower at all stages of differentiation. At several differentiation stages, TAL1 expression was close or even below the 15,000 DNA-binding sites identified in erythroid cells by ChIP-exo (Han et al., 2015), although other studies identified less than 3,000 DNA-binding sites for TAL1, suggesting that these sites have different affinities (Kassouf et al., 2010). The expression levels of ETO2, LMO2, STAT5A, and STAT5B were strongly decreased at the late stages of differentiation compared with the overall decrease in cell protein content. In contrast, NFE2 expression increased, and NFE2 appeared to be more highly expressed than other erythroid transcription factors, such as GATA1 and KLF1, at the end of the differentiation process.
Several of these proteins associate with each other to constitute transcription complexes of different compositions and functions. Our data should allow the determination of which of these proteins is the limiting factor for the constitution of these complexes at the different differentiation stages. Indeed, FOG, which associates with GATA1 to constitute either repressive or activating transcription complexes, appeared to be 10-fold less expressed than GATA1. Cell fractionation analyses confirmed that all GATA1 proteins and most FOG1 proteins were localized to the nucleus of erythroblasts, showing that their relative nuclear concentrations could be deduced from their expression in whole cells (data not shown). Similarly, TAL1 should be the limiting element for the constitution of the LDB1 complexes. Thus, our data should contribute to the development of improved models of transcriptomic regulation during erythropoiesis by providing the quantitative expression pattern of most transcriptional regulators.
A high level of alternative splicing occurs during the late stages of differentiation, which has been suggested to significantly contribute to the proteome diversity during erythropoiesis (Pimentel et al., 2014). Our results show that the main splicing complexes followed the generally decreased protein expression between the ProE and Ortho stages (Figure 4D).
Most proteins involved in the translation machinery undergo the same 4-fold decrease (Figure 4E), despite the huge amount of Hb synthesis. Interestingly, erythroid cells expressed 4E-BP proteins at a level similar to that of eIF4E at the early differentiation stages, whereas expression of the 4E-BP proteins decreased more strongly than the expression of eIF4E at the end of differentiation. This suggests that a strong regulation of protein translation through mTORC1 could occur at the early stages of differentiation, whereas, in late stages, only a limited fraction of protein translation should be subjected to regulation through this pathway. This regulation could especially target globin synthesis, as previously reported (Chung et al., 2015). On the other hand, EIF2AK1 (HRI) expression was below the detection threshold at the progenitor stages and increased strongly during differentiation, implying that the overall protein synthesis is primarily regulated by heme availability in terminally differentiating cells.
Relation between the Transcriptome and Proteome during Erythropoiesis
We used the recently published transcriptomic analyses of differentiating erythroid cells from human cord blood (An et al., 2014; Li et al., 2014) to compare mRNA and protein expressions. In these studies, BFU-E cells were purified according to the lack of CD36 expression and corresponded to young BFU-Es (Li et al., 2014). Thus, the Prog1 population that we purified was more mature than the BFU-E cells analyzed by Li et al. (2014), whereas the Prog2 population could be equated to CFU-E cells. Li et al. (2014) performed the transcriptomic studies with polyadenylated RNA. Because histone mRNA is not polyadenylated, histones were removed from the comparison. Transcriptomic and proteomic data revealed, as usual, a limited correlation with Spearman’s rank correlation coefficients varying from 0.410 to 0.676 (Figure 5A). Few of the 20 most expressed proteins correlated with the 20 most expressed mRNA, except for globins at the last stages of differentiation (Table S1). Figure 5B shows two examples of highly expressed proteins whose expression modifications during differentiation followed (GAPDH) or not (TUBB4B) the pattern of mRNA expression, confirming that protein expression cannot be deduced directly from mRNA expression (Vogel and Marcotte, 2012).
Figure 5. Protein versus mRNA Expression Analysis along Erythroid Differentiation.

Transcriptomic data from An et al. (2014) and Li et al. (2014) were compared with quantified proteins.
(A) Spearman rank correlation analysis is shown.
(B) mRNA and protein expression of GAPDH and TUBB4B in erythroid cells during differentiation is shown.
(C) Contribution of the ten most expressed mRNA to the whole mRNA expression is shown.
(D) Spearman rank correlation analysis after mRNA/protein standardization. A correction factor was calculated for each gene expression value by dividing the protein copy number at the ProE stage by its mRNA quantification value. These correction factors were used to calculate a modified mRNA value that integrated translation efficiency and protein stability. A new Spearman rank correlation analysis was performed with these modified values.
We selected the genes with the most divergent patterns of mRNA and protein expression using Spearman’s correlation analysis. This led to the identification of 302 genes presenting an opposite evolution of mRNA and protein expression during differentiation. All of these genes except one showed the same increased mRNA expression associated with decreased protein expression (Table S4). Analysis of this set of genes suggested neither their regulation by common transcription factors according to TRANSFAC nor a biased representation of housekeeping genes as previously defined (Kim et al., 2014a). Interestingly, this analysis revealed the highly significant enrichment of genes with intron-retaining mRNA that has been identified recently in erythropoiesis (Edwards et al., 2016; Pimentel et al., 2016). Moreover, searching for Kyoto Encyclopedia of Genes and Genomes (KEGG) and GO term enrichment showed an overrepresentation of genes involved in RNA processing (Table S4), which also has been suggested to be regulated by intron retention during erythropoiesis (Edwards et al., 2016; Pimentel et al., 2016). Altogether, our data strongly support the recent hypothesis that intron retention participates in the regulation of gene expression during erythropoiesis. Of note, we identified SF3B1, which plays a major role in sideroblastic anemia.
Overall, the cell composition appeared more stable at the protein than at the mRNA level, and many strong variations detected at the mRNA level were buffered at the protein level. For example, XPO7 expression increased more than 100-fold between the ProE and Ortho stages at the mRNA level but only 2-fold at the protein level. Some of this discrepancy could be due to an overestimation of the mRNA variations that are calculated from relative expression values and, thus, do not integrate the cell size decrease during the differentiation process. Modifications in the translation machinery and its controls also should participate in this discrepancy. At the ProE stage, the ten most abundant mRNA constituted only 9.6% of the whole mRNA content, whereas they constituted 67% of the mRNA content at the Ortho stage (Figure 5C). It is unclear how late erythroblasts manage the simultaneous translation of a few highly expressed mRNA encoding erythrocyte-specific proteins and of many others encoding housekeeping and regulatory proteins. Spearman’s correlation analysis showed that the correlation between protein and mRNA worsened for the last steps of differentiation (Figure 5B). To confirm this result, we calculated the protein/ mRNA ratio for each quantified protein at the ProE stage, and we applied these correction factors to mRNA expression levels for each differentiation stage. Spearman’s correlation analysis using these modified values showed that the relationship between mRNA and proteins remained roughly constant up to the Baso2 stage, with correlation coefficients above 0.9, and then strongly decreased (Figure 5D), suggesting that different mechanisms regulating mRNA translation and/or protein stability are in play in the late stages of differentiation.
Quantitative Proteomics Reveals New Aspects of Erythropoiesis
Our analysis revealed the expression of several unexpected proteins. Although our study design did not favor the identification of secreted proteins, we identified several soluble proteins produced by erythroid cells, such as GDF15, but also several secreted proteins not previously reported. We detected the presence of IL18 and hemopexin (HPX) in erythroblasts. IL18 also was detected at the mRNA level throughout the differentiation process (An et al., 2014). Its role in this process remains unclear. In contrast, HPX was not detected at the mRNA level, suggesting that, similar to transferrin, exogenous HPX proteins specifically associate with erythroid cells. HPX is a plasma protein with a high affinity for free heme that targets it to the liver for degradation while neutralizing its deleterious oxidizing capacities. We confirmed the presence of HPX in erythroid cells by western blot analysis (Figure S3B). CD91 (LRP1), the hepatic and macrophage HPX receptor (Hvidberg et al., 2005), also was quantified in the proteomic analysis, and we confirmed its expression pattern by western blot analysis (Figure S3B). In addition to HPX, CD91 is involved in the uptake of several plasma proteins, including alpha macroglobulin, which also accumulated in erythroid cells at the same differentiation stages (Table S2). Interestingly, CD91 expression and HPX accumulation presented a very transient pattern, and these proteins were essentially expressed at the basophilic stages when globin expression started to rise (Figure 4A). These results suggest that the erythroblasts are able to internalize heme at the beginning of the hemoglobinization process to contribute to Hb production.
Overall, our results show that the basophilic stages appear to constitute a major milestone in the differentiation process, as revealed by the unsupervised clustering analysis that introduced the first dichotomy between the Baso1 and Baso2 stages (Figure 3D). Interestingly, the basophilic stages appear to constitute a major difference between murine and human erythropoiesis, because an extra cell division occurs in humans at this stage. At the Baso2 stage, the expression of globins, band 3, and GPA started to rise (Figures 4A and S3), whereas c-KIT expression dramatically decreased (Figure S3). At this stage, many proteins from the splicing machinery also started to decrease (Figure 4D). EPOR expression decreased strongly at the murine basophilic stage (Zhang et al., 2003). Although we could not quantify EPOR expression in these studies, likely because the presence of EPO induces the downregulation of its receptor, our data show that expression levels of STAT5A (Figure 4C) and TFR2 (Table S1), two proteins involved in EPOR signaling, also strongly decreased at the basophilic stages. A transient caspase activation process required for the achievement of terminal differentiation occurs at the basophilic stage (Zermati et al., 2001). It is tempting to speculate that the decreased intracellular signaling due to c-KIT and possibly to EPOR downregulation is responsible for these key modifications at this step.
Because of its function as an oxygen and carbon dioxide transporter, the erythrocyte is highly exposed to oxidative stress, with 0.5%–3% of its Hb content auto-oxidizing each day to form methemoglobin and a superoxide radical. Survival of the erythrocyte for up to 120 days in the bloodstream is thus highly dependent on the establishment of a robust machinery to both buffer the oxidizing species and repair damaged proteins and lipids. The erythrocyte redox system cannot be separated from glucose metabolism, which constitutes the only metabolic pathway able to both supply ATP and reduce pyridine nucleotides in these cells. Accordingly, mutations in several key enzymes of these pathways, such as PK or G6PD, are responsible for hereditary hemolytic anemia in humans.
Our results give new insights into the establishment of the enzymes of these pathways. Transcriptomic analyses showed that the expression of several genes of these pathways was strongly increased at the Ortho stage. Nevertheless, except for BPGM and GLUT1, a corresponding increase in protein expression was never observed (Figure 6). BPGM plays a key role in erythrocytes by regulating Hb affinity for O2 through 2-3BPG synthesis. It would be interesting to determine the pattern of BPGM expression in adult erythroblasts, because cord blood erythroblasts express 37.7% ±3.6% of fetal Hb, which presents a low affinity for 2-3BPG in contrast to adult Hb. Erythroid cells use different glucose transporters. In mice, fetal erythrocytes express GLUT1 and there is a postnatal switch from GLUT1 to GLUT4, whereas glucose enters human erythrocytes using GLUT1 transporter, which also allows dehydroascorbic acid (DHA) uptake after its association with stomatin (Montel-Hagen et al., 2008). Because stomatin is still highly expressed during the early stages of erythroid differentiation, this suggests that young progenitor cells already possess high DHA uptake potential and thereby buffer the oxidizing environment resulting from the high iron content at this stage. We found that GLUT3 (SLC2A3) was expressed at the early stages of human erythroid differentiation at levels similar to that of GLUT1 (SLC2A1) and that its expression decreased rapidly while GLUT1 expression increased dramatically.
Figure 6. Setting up of the Erythrocyte Glucose and Redox Metabolic Pathways during Erythropoiesis.

Except for SLC2A3, which is probably no longer detectable in mature erythrocytes, the figure is restricted to proteins expressed in mature erythrocytes. The main part of the figure shows their expression levels at the Ortho stage. Genetic defects of enzymes labeled in red are responsible for red cell pathologies in humans. The panels in the left part of the figure represent the evolution of mRNA and protein expression during the seven differentiation stages of terminal erythropoiesis defined in Figure 2. Transcriptomic expression values are from An et al. (2014) and Li et al. (2014).
Deficiency of the R isoform of PK is the most frequent enzyme abnormality of the Embden-Meyerhof pathway, causing hereditary hemolytic anemia. The M isoform of PK is expressed at higher levels than the R isoform in progenitor cells, but its expression dramatically decreases while the expression of the R isoform remains roughly constant throughout the differentiation process. Similar to most other proteins involved in the Embden-Meyerhof or the pentose phosphate pathways, G6PD roughly followed the same decrease as the overall cell protein content (Figure 6). In contrast, the levels of several proteins involved in redox regulation decreased less than the overall protein content during the differentiation process. This was especially true for CYB5R3, which uses NADH to reduce iron in methemoglobin, as well as for thioredoxin and catalase. Subsequently, these proteins were enriched and likely to increase the redox capacity of erythrocytes. Overall, our data show that only GLUT1 expression is hugely increased during erythroid differentiation at the protein level, suggesting that increasing glucose and DHA uptake capacities while fine-tuning some steps in the redox system appear sufficient to fulfill the specific needs of erythrocytes.
Protein Segregation during Enucleation
In mammals, erythroid cells expel their nucleus at the end of differentiation, giving rise to a pyrenocyte and a reticulocyte that ultimately matures to an erythrocyte. Due to a lack of an ATP-producing system, pyrenocytes are highly unstable and are rapidly engulfed by macrophages in vivo. They rapidly disintegrate in cell culture. To simultaneously compare the reticulocytes and pyrenocytes produced, we sampled the cells at the onset of the enucleation phase using an appropriate cell culture protocol (Giarratana et al., 2011). At this time, the cell culture contained erythroblasts, pyrenocytes, and reticulocytes (Figure 7A). The cells were separated by fluorescence-activated cell sorting (FACS) according to size, DNA content, and surface expression of GPA (Figure 7B).
Figure 7. Protein Sorting during Enucleation.

(A) Pyrenocytes and reticulocytes were purified from late erythroid cultures by FACS through DNA (Hoechst 33342) and GPA quantification and cell size.
(B) Flow cytometry analysis of late erythroid culture is shown.
(C) Results from ITRAQ analysis. The proteins were separated to their main subcellular compartment according to Gene Ontology cellular component (GOCC) annotations.
(D) Imaging flow cytometry. Images from pyrenocytes and reticulocytes were selected and masks were drawn to delineate the limits of the cells and the nucleus in the pyrenocyte. The radii of these objects were calculated by assuming a spherical shape for all of these objects and used to determine the volume and surface of the objects (mean ±SD, n = 1,900 for pyrenocytes and 3,800 for reticulocytes).
(E) The percentage of each protein segregating with the reticulocyte was calculated and the proteins were classified according to their subcellular compartment using GOCC annotations. Protein lists for each cellular compartment are presented in Table S6. The shaded area represents the 95% confidence interval for the cytoplasm volume (green area) or the plasma membrane area (pink area) segregating with the reticulocyte, and the horizontal bars show the median repartition value for the proteins of each compartment. Erythroid complexes refer to the ankyrin and band 4.1 complexes of the erythrocyte.
To verify the quality of the cell preparation, equal numbers of sorted pyrenocytes and reticulocytes were mixed and analyzed by absolute LFQ (Table S2). From these data, we calculated that the sum of the protein amounts for one reticulocyte and one pyrenocyte was 44.1 pg, a value very similar to the protein content of an Ortho cell (39 ±9 pg; see Figure 3E). Moreover, Pearson correlation analysis showed a high similarity between the proteome of Ortho cells and of the 1:1 mixture of pyrenocytes and reticulocytes, with a correlation coefficient of 0.93. Because the protein composition of pyrenocytes and reticulocytes is very different, a label-free analysis strategy could not be used to quantify proteins associated with each cell, and their proteomes were compared after ITRAQ labeling. Cells from three independent cell cultures were analyzed to determine the relative distribution of 1,318 proteins (Figure 7C; Table S5). Although most nuclear proteins segregated with the pyrenocyte as expected, cytosolic and membrane proteins distributed differentially depending on the protein, with some of them completely segregating with either the pyrenocyte or the reticulocyte.
To determine whether a protein segregates evenly between the two cell types or whether it is actively sorted, we determined the distribution of the compartment where it was embedded. Although the reticulocyte volume and cell surface area have been determined previously (Da Costa et al., 2001; Kim et al., 2014b), these values are lacking for pyrenocytes. To determine them, we used an imaging flow cytometry approach. The cell surfaces of both cell types were labeled with anti-GPA antibodies, whereas nuclei were labeled with Hoechst 33342. Erythroblasts were identified according to their size and filtered out. Pyrenocytes, nuclei, and reticulocytes were considered spherical objects, leading to the determination of cytoplasmic volumes of 219 ±54 fl and 49 ±11 fl and plasma membrane surface areas of 175 ±29 μm2 and 110 ±12 μm2 for reticulocytes and pyrenocytes, respectively (Figure 7D). Although these values have been obtained using very different methods, they agree well with the surface and volume of reticulocytes recently determined using common-path diffraction optical tomography (Kim et al., 2014b). Thus, around 82% of the cytoplasm and 61% of the plasma membrane segregated with the reticulocyte during enucleation.
Several proteins showed unexpected localizations. Although all histones entirely segregated with the nucleus, the exception was H4, with 41% found in the reticulocyte. Based on the copy number of H4 determined in Ortho cells, a very large number of copies of H4, about 45 × 106 copies, were present in each reticulocyte. The reason why H4 is partly exported from the nucleus is unclear. It is tempting to suppose that this could favor nuclear condensation, although a specific role for H4 in the reticulocyte cannot be excluded. Whatever the role of H4 export to the reticulocyte, mature erythrocytes no longer contain histones (Lange et al., 2014). Our data show that 67% of the 112,000 20S proteasome particles segregated with the reticulocyte, indicating that the reticulocyte maintains a high capacity of protein degradation sufficient to manage the removal of proteins not to be included in erythrocytes. Overall repartition of 454 proteins with a nuclear localization according to GO was quantified, and the median value of their repartition was 84.4% toward the pyrenocyte (Figure 7E). Thus, although some proteins such as H4 are specifically exported to the cytoplasm and segregate with the reticulocyte, our data do not support the previously reported massive export of nuclear proteins at the end of the erythroid differentiation process in mice (Hattangadi et al., 2014).
Overall, our results show that many cytoplasmic and membrane proteins appear to be specifically sorted to one or the other cell type, because their distribution does not agree with the overall distribution of the cell compartment in which they should be located (Figure 7E; Table S6). Particularly, proteins of the red cell membrane seem to be actively sorted to the reticulocyte, whereas other membrane proteins, such as TFRC or SLC2A1, most likely follow a random distribution or are even specifically sorted to the pyrenocyte, such as ITGB1 or CD36.
Conclusions
Our data provide a detailed description of the entire proteome of differentiating erythroid cells, demonstrating the capacity of current proteomic approaches to provide the absolute quantification of most cellular proteins during a complex differentiation process using limited amounts of biological material. Absolute quantification is a key improvement to the whole proteomic analyses required to fuel the development of theoretical models describing and simulating cell behavior. Several approaches to achieve this goal recently have been initiated. Beck et al. (2011) used spiked heavy peptides to quantify selected proteins and calculated the expression of other proteins from the quantified protein markers, whereas Wiśniewski et al. (2014) used the invariant histone levels to internally standardize the quantification data. Our results confirm the accuracy of this method (see Figures 3E–3G). As reported by Wiśniewski et al. (2014), we observed that the dynamic range of protein expression encompasses seven logs, although it is unclear whether this represents the current technical limitation of protein quantification in these global proteomic studies or the true range of protein expression.
We could detect several proteins expressed at levels of a few hundred copies per cell, such as MYC or MYB, although several erythroid regulators such as EPOR or JAK2, although detected, could not be precisely quantified. Scatchard analysis using radiolabeled EPO has shown that erythroid progenitors express around 500 EPORs per cell (Mayeux et al., 1987). Overall, this suggests that the coverage of proteins expressed below a few thousand copies per cell is incomplete in our study. Of the 10,000 genes shown to be expressed in the previous transcriptome analysis, our proteomic study detected around 75% of the erythroid proteins encoded by these genes, with more than 60% of these proteins quantified. Although further technical improvements are still needed to cover the whole erythroid proteome, especially at the quantitative level, our data constitute a significant improvement in the global description of erythropoiesis, and they provide an important toolbox for the understanding of the erythroid differentiation process and its deregulations in pathologies leading to erythroid deficiencies, such as myelodysplastic syndromes, thalassemia, and Diamond-Blackfan anemia. The protein expression values are freely available in a user-friendly web site dedicated to red cell physiology and pathologies (http://www.dsimb.inserm.fr/respire/).
EXPERIMENTAL PROCEDURES
A detailed version of the experimental procedures is available as the Supplemental Experimental Procedures.
Erythroid Cell Culture and Characterization
A two-step culture method was adapted from Freyssinier et al. (1999) to obtain highly stage-enriched erythroblast populations. Briefly, CD34+ progenitors were purified from human cord blood and cultured for 7 days with IL6, IL3, and SCF to expand hematopoietic progenitors. Then, CD36+ erythroid progenitors were purified and cultured for 3 days with EPO, IL3, SCF, and dexamethasone; for 3 days with EPO, human serum, and decreasing concentrations of SCF, then for 5 days with EPO and fetal calf serum. Each day cells were characterized by flow cytometry for CD34, CD36, GPA, CD71, CD49d, and band 3, by MGG staining and by benzidine staining for hemoglobinization, and they were sampled for proteomic analysis. To optimize the enucleation rate, a slightly modified culture protocol was used for reticulocyte and pyrenocyte sorting (Giarratana et al., 2011).
LFQ Proteomic Analyses
Peptides were prepared from whole-cell lysates, using the filter-aided sample preparation (FASP) method essentially as previously described (Wiśniewski et al., 2009), and fractionated by strong cationic exchange (SCX) StageTips (Kulak et al., 2014). MS analyses were performed on a Dionex U3000 RSLC nano-LC-system coupled to either a Q-Exactive or an Orbitrap-Velos mass spectrometer (Thermo Fisher Scientific). Peptides were separated on a C18 reverse-phase resin (75-μm inner diameter and 15-cm length) with a 3-hr gradient. The mass spectrometer acquired data throughout the elution process and operated in a data-dependent scheme.
LFQ Data and Statistical Analyses
The MS data were processed by MaxQuant version 1.5.2.8 (Cox et al., 2014) using human sequences from the Uniprot-Swiss-prot database (Uniprot, release 2015-02) with a false discovery rate below 1% for both peptides and proteins. Label-free protein quantification was done using unique and razor peptides.
For bioinformatic analysis, LFQ results from MaxQuant were imported into Perseus software (version 1.5.1.6). Protein copy numbers per cell were then calculated using the Protein ruler plugin of Perseus by standardization to the total histone MS signal, as recently described (Wiśniewski et al., 2014).
Each experiment corresponding to a whole culture from a single cord blood sample was analyzed separately in MaxQuant. Mean ±SD and coefficient of variation values were calculated on the basis of the absolute quantification values determined using Perseus, as described above. The type of statistical analysis performed is indicated with each result; ANOVA, Spearman’s rank correlation, Pearson correlation, and hierarchical clustering analyses were done using Perseus. Over-/underrepresentation analyses (ORA) were done using GeneTrail with false discovery rate adjustment according to Benjamini and Hochberg with p < 0.01 and an observed/predicted ratio > 2.
ITRAQ Quantification
Proteins from whole-cell lysates were digested using the FASP method and labeled with ITRAQ reagents according to the manufacturer’s instructions. After mixing, the peptides were separated by isoelectric focalization using an Agilent 3100 Off-Gel fractionator and analyzed by LC-MS/MS using an Orbitrap Velos mass spectrometer. The mass spectrometer acquired data throughout the 2-hr elution process with full MS scans acquired with the Orbitrap, followed by up to ten linear trap quadrupole (LTQ) MS/MS collision-induced dissociation (CID) spectra and ten MS/MS higher-energy collisional dissociation (HCD) spectra of the most abundant ions detected in the MS scan. The data were first analyzed using Proteome Discoverer 1.3 to produce mgf files after merging the HCD and CID scans. The files were then analyzed using Protein Pilot v4 using the Uniprot human database.
Supplementary Material
Highlights.
Absolute quantification of the cell proteome during human erythroid differentiation
Comparison of transcriptome and proteome modifications during erythropoiesis
Distribution of 1,300 proteins between reticulocytes and pyrenocytes after enucleation
Acknowledgments
This study was supported by grants from the Laboratory of Excellence GR-Ex, reference ANR-11-LABX-0051. The Laboratory of Excellence GR-Ex is funded by the program “Investissements d’avenir” of the French National Research Agency, reference ANR-11-IDEX-0005-02. We thank Dr. I. Dusanter and the CRB/Cell Therapy Unit, Saint-Louis Hospital for cord blood samples; the Cybio Platform for FACS; and Dr. J. Bodin (Arkesys Company) and M. LeGall (3P5) for their help with the statistical and bioinformatic analyses, respectively. We thank A. Pandey and S.S. Manda for their sharing of the housekeeping gene file and Dr. F. Bouillaud (Cochin Institute) for helpful discussions. We thank Dr. C. Etchebest (INTS Paris) and Dr. S. Teletchea (University of Nantes) for integrating the protein expression values in their red cell-dedicated website.
Footnotes
ACCESSION NUMBERS
The accession numbers for the rawmass spectrometric data reported in this paper are from ProteomeXchange Consortium (http://www.proteomexchange.org/): PXD004313, PXD004314, PXD004315, and PXD004316.
Supplemental Information includes Supplemental Experimental Procedures, four figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.06.085.
AUTHOR CONTRIBUTIONS
Methodology, F.G., L.D., F.V., Y.Z., and P.M.; Investigation, E.-F.G., S.D., M.L., V.S., M.D., M.-C.G., A.R., F.V., Y.Z., and P.M.; Formal Analysis, E.-F.G., S.D., M.L., F.G., M.D., J.H., N.M., F.V., Y.Z., and P.M.; Original Draft, C.L. and P.M.; Review & Editing, all authors; Conceptualization, E.-F.G., S.D., M.L., F.G., C.L., F.V., Y.Z., and P.M.; Supervision, P.M.
References
- An X, Schulz VP, Li J, Wu K, Liu J, Xue F, Hu J, Mohandas N, Gallagher PG. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123:3466–3477. doi: 10.1182/blood-2014-01-548305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NM, Bruce LJ. Modelling the structure of the red cell membrane. Biochem Cell Biol. 2011;89:200–215. doi: 10.1139/o10-154. [DOI] [PubMed] [Google Scholar]
- Casetti L, Martin-Lannerée S, Najjar I, Plo I, Augé S, Roy L, Chomel JC, Lauret E, Turhan AG, Dusanter-Fourt I. Differential contributions of STAT5A and STAT5B to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013;73:2052–2058. doi: 10.1158/0008-5472.CAN-12-3955. [DOI] [PubMed] [Google Scholar]
- Chung J, Bauer DE, Ghamari A, Nizzi CP, Deck KM, Kingsley PD, Yien YY, Huston NC, Chen C, Schultz IJ, et al. The mTORC1/ 4E-BP pathway coordinates hemoglobin production with L-leucine availability. Sci Signal. 2015;8:ra34. doi: 10.1126/scisignal.aaa5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa L, Mohandas N, Sorette M, Grange MJ, Tchernia G, Cynober T. Temporal differences in membrane loss lead to distinct reticulocyte features in hereditary spherocytosis and in immune hemolytic anemia. Blood. 2001;98:2894–2899. doi: 10.1182/blood.v98.10.2894. [DOI] [PubMed] [Google Scholar]
- Dai CH, Krantz SB, Koury ST, Kollar K. Polycythaemia vera. IV Specific binding of stem cell factor to normal and polycythaemia vera highly purified erythroid progenitor cells. Br J Haematol. 1994;88:497–505. doi: 10.1111/j.1365-2141.1994.tb05065.x. [DOI] [PubMed] [Google Scholar]
- Deshmukh AS, Murgia M, Nagaraj N, Treebak JT, Cox J, Mann M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol Cell Proteomics. 2015;14:841–853. doi: 10.1074/mcp.M114.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CR, Ritchie W, Wong JJ, Schmitz U, Middleton R, An X, Mohandas N, Rasko JE, Blobel GA. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood. 2016;127:e24–e34. doi: 10.1182/blood-2016-01-692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyssinier JM, Lecoq-Lafon C, Amsellem S, Picard F, Ducrocq R, Mayeux P, Lacombe C, Fichelson S. Purification, amplification and characterization of a population of human erythroid progenitors. Br J Haematol. 1999;106:912–922. doi: 10.1046/j.1365-2141.1999.01639.x. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, Bresnick EH. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell. 2009;36:667–681. doi: 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giarratana MC, Rouard H, Dumont A, Kiger L, Safeukui I, Le Pennec PY, François S, Trugnan G, Peyrard T, Marie T, et al. Proof of principle for transfusion of in vitro-generated red blood cells. Blood. 2011;118:5071–5079. doi: 10.1182/blood-2011-06-362038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GC, Vinayachandran V, Bataille AR, Park B, Chan-Salis KY, Keller CA, Long M, Mahony S, Hardison RC, Pugh BF. Genome-wide organization of GATA1 and TAL1 determined at high resolution. Mol Cell Biol. 2015;36:157–172. doi: 10.1128/MCB.00806-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattangadi SM, Martinez-Morilla S, Patterson HC, Shi J, Burke K, Avila-Figueroa A, Venkatesan S, Wang J, Paulsen K, Görlich D, et al. Histones to the cytosol: exportin 7 is essential for normal terminal erythroid nuclear maturation. Blood. 2014;124:1931–1940. doi: 10.1182/blood-2013-11-537761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106:2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- Kassouf MT, Hughes JR, Taylor S, McGowan SJ, Soneji S, Green AL, Vyas P, Porcher C. Genome-wide identification of TAL1’s functional targets: insights into its mechanisms of action in primary erythroid cells. Genome Res. 2010;20:1064–1083. doi: 10.1101/gr.104935.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. A draft map of the human proteome. Nature. 2014a;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Shim H, Kim K, Park H, Jang S, Park Y. Profiling individual human red blood cells using common-path diffraction optical tomography. Sci Rep. 2014b;4:6659. doi: 10.1038/srep06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley PD, Greenfest-Allen E, Frame JM, Bushnell TP, Malik J, McGrath KE, Stoeckert CJ, Palis J. Ontogeny of erythroid gene expression. Blood. 2013;121:e5–e13. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Lange PF, Huesgen PF, Nguyen K, Overall CM. Annotating N termini for the human proteome project: N termini and Nα-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J Proteome Res. 2014;13:2028–2044. doi: 10.1021/pr401191w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Hale J, Bhagia P, Xue F, Chen L, Jaffray J, Yan H, Lane J, Gallagher PG, Mohandas N, et al. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood. 2014;124:3636–3645. doi: 10.1182/blood-2014-07-588806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- Mayeux P, Billat C, Felix JM, Jacquot R. Evidence for glucocorticosteroid receptors in the erythroid cell line of fetal rat liver. J Endocrinol. 1983;96:311–319. doi: 10.1677/joe.0.0960311. [DOI] [PubMed] [Google Scholar]
- Mayeux P, Billat C, Jacquot R. The erythropoietin receptor of rat erythroid progenitor lens. Characterization and affinity cross-linkage. J Biol Chem. 1987;262:13985–13990. [PubMed] [Google Scholar]
- Merryweather-Clarke AT, Atzberger A, Soneji S, Gray N, Clark K, Waugh C, McGowan SJ, Taylor S, Nandi AK, Wood WG, et al. Global gene expression analysis of human erythroid progenitors. Blood. 2011;117:e96–e108. doi: 10.1182/blood-2010-07-290825. [DOI] [PubMed] [Google Scholar]
- Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Pilon AM, Ajay SS, Kumar SA, Steiner LA, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, et al. Genome-wide ChIPSeq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood. 2011;118:e139–e148. doi: 10.1182/blood-2011-05-355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee S, Ghanem D, An X, Li J, Mohandas N, Pachter L, Conboy JG. A dynamic alternative splicing program regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2014;42:4031–4042. doi: 10.1093/nar/gkt1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2016;44:838–851. doi: 10.1093/nar/gkv1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Lin YH, Sierant MC, Zhu F, Cui S, Guan Y, Sartor MA, Tanabe O, Lim KC, Engel JD. Developmental transcriptome analysis of human erythropoiesis. Hum Mol Genet. 2014;23:4528–4542. doi: 10.1093/hmg/ddu167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Hein MY, Cox J, Mann M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics. 2014;13:3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zermati Y, Garrido C, Amsellem S, Fishelson S, Bouscary D, Valensi F, Varet B, Solary E, Hermine O. Caspase activation is required for terminal erythroid differentiation. J Exp Med. 2001;193:247–254. doi: 10.1084/jem.193.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102:3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.