

Abstract
For the nematode Caenorhabditis elegans, automated selection of animals of specific genotypes from a mixed pool has become essential for genetic interaction or chemical screens. To date, such selection has been accomplished using specialized instruments. However, access to such dedicated equipment is not common. Here we describe live animal fluorescence-activated cell sorting (laFACS), a protocol for automatic selection of live L1 animals using a standard FACS. We show that a FACS can be used for the precise identification of GFP-expressing and non-GFP-expressing sub-populations and can accomplish high-speed sorting of live animals. We have routinely collected 100,000 or more homozygotes from a mixed starting population within two hours and with greater than ninety-nine percent purity. The sorted animals continue to develop normally, making this protocol ideally suited for the isolation of terminal mutants for use in genetic interaction or chemical genetic screens.
Keywords: C. elegans, worm sorting, FACS, embryonic lethality, maternal-effect lethality, sterility
INTRODUCTION
Caenorhabditis elegans is an important model system for studying the fundamental genetic mechanisms underlying developmental and behavioral biology1. Among its many advantages as an animal model system is that it can be easily grown to very large populations and there is a spectacular infrastructure supporting this system that includes mutants affecting over half of its genes (2 and pers. comm. Donald G. Moerman and Robert Waterston), genome-scale RNAi reagents3–5, and an extensive collection of green fluorescent protein (GFP)-marked strains6. These features make this model system well suited for large-scale studies7–9.
It is estimated that 30% of the genes In C. elegans have a terminal phenotype such as embryonic lethality, maternal-effect lethality, or sterility10. While some terminal phenotypes (e. g. sterility) are viable, they do not produce viable offspring and thus it is impossible to grow them as a pure population of homozygotes. Balancer chromosomes are key tools for preventing the loss of terminal alleles in a segregating population. In a balanced heterozygote, the wild-type allele is carried by a balancer chromosome, which bears chromosomal aberrations such as inversions or translocations that prevent recombination events. Some balancers carry recessive lethal alleles, causing homozygous balancer embryos to be inviable, thus preventing the proliferation of animals that have lost the recessive terminal allele. Many balancers contain GFP transgenes (Table 1), enabling the easy identification of heterozygous animals. Since the balancer chromosome resists recombination, the GFP transgene is always associated with the wild-type allele and thus homozygous mutant individuals can easily be identified due to lack of GFP expression (see Figure 1 for an example of a balanced strain). However, manual isolation of homozygous individuals from a heterogeneous population is too labor intensive to be practicable for large-scale analyses.
Table 1.
GFP-marked balancers in C. elegansa
| Name of Balancer |
Homo. Lethal? |
Region Balanced | Type of Balancer | GFP Marker |
|---|---|---|---|---|
| nT1b[qIs51] | yes | right end of chromosome IV through unc-17, left end chromosome V through unc-76 |
reciprocal translocationc |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| hT2d,e[qIs48] | yes | chromosome I from left end through unc-101, chromosome III from right end through dpy- 17 |
reciprocal translocationc |
myo2::GFP, pes-10::GFP, ges-1::GFP |
| mIn1f[mIs14] | no | chromosome II between lin-31 and rol-1 |
inversion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| qC1 [qIs26]g | yes | left portion chromosome III between tra-1 and dpy-1 |
unknown | lag-2::GFP |
| eT1[nIs267] | no | chromosome V left end through unc-23, chromosome III right end through unc-36 |
reciprocal translocationc |
myo-2::GFP |
| mIs10h | no | chromosome V between unc- 60 and dpy-11. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| mIs11 | no | chromosome IV, unknown interval near dpy-20. This has been used successfully to balance deletions in cyb-1, syn- 4 tag-316, htp-1, mep-1, and tag-137. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| mIs12 | no | chromosome II ,unknown interval near unc-4. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| mIs13 | no | chromosome I, unknown interval near unc-54. This has been used successfully to balance deletions in tag-115, kin-1, npp-4, ero-1 and bbs-1. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| okIs57 | no | chromosome X, unknown interval near unc-3. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
| okIs59 | no | chromosome I, unknown interval between dpy-5 and unc-13. This has been used to balance deletions in air-2, dao- 5, npp-7, chn-1, tag-83 and gly- 2. |
insertion |
myo2::GFP, pes-10::GFP, F2B7.9::GFP |
nT1[qIs51] carries an uncharacterized lesion in daf-15. Some mutations lying within the balanced extents of nT1 are unstable over nT1[qIs51].
Reciprocal translocation balancers produce aneuploid inviable progeny20, and this reduces the expected yield of viable homozygotes.
The qIs48 element is sometimes lost by apparent rare recombination, leaving behind viable hT2 homozygotes marked with the original bli-4 mutation.
Some mutations lying within the balanced extents of hT2 are unstable over hT2[qIs48].
Some lethal alleles balanced by the original non-GFP mIn1[dpy-10] and lying near the left breakpoint are not balanced by mIn1[mIs14 dpy-10].
qIs26 also carries dominant rol-6 allele su1006.
Balancing activity of mIs10 should be confirmed.
Figure 1. Strain PF405 Genetics.
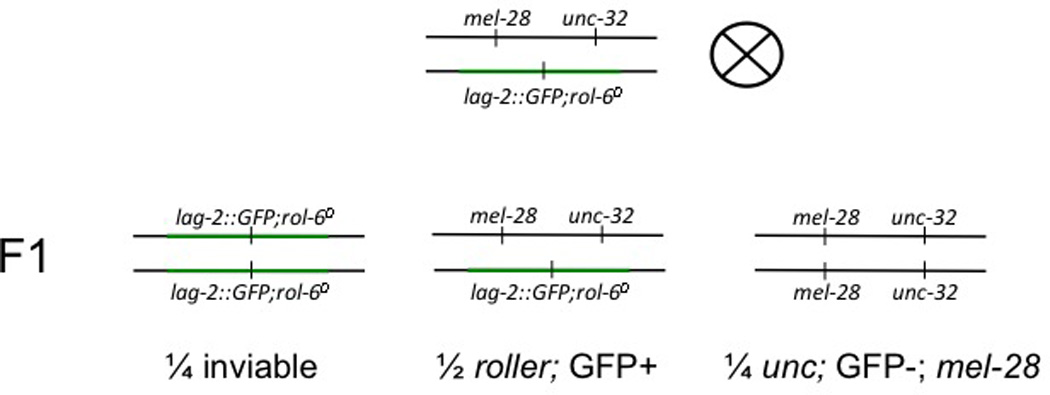
The qC1-balanced region of chromosome is shown in green. We used a version of qC1 with the qIs26 insertion that has the lag-2::GFP transgene and the dominant rol-6 allele su1006. The qIs26 insertion site created a recessive lethal allele such that qC1[qIs26] homozygotes are inviable. Heterozygotes have the Rol-6 phenotype and express GFP. Animals that have lost the balancer do not express GFP, are uncoordinated, and are mel-28 (and thus produce only dead embryos).
The microscopic size of C. elegans facilitates the use micro-fluidic and flow cytometric systems to analyze or sort and collect individual animals with specific optical properties. The COPAS (complex object parametric analysis and sorting) biosorter is expert apparatus developed especially for the optical analysis and sorting of nematodes11. This specialized sorter is well suited to the fluorescence-based isolations of sub-populations in a heterogeneous batch11 and is capable of much more, including stage-specific sorting and analysis of fluorescence within specific regions of the animals11–13. However, these dedicated machines are not widely available and therefore accessibility to high-throughput techniques for isolation of sterile/lethal mutants is restricted.
Recently, it has been reported that a fluorescence activated cell sorter (FACS), a more prevalent piece of equipment, can be used for automated sorting of live C. elegans larvae14 as well as fixed embryos15. We have successfully used this technique, dubbed laFACS (live animal FACS), to isolate over one hundred thousand larvae for RNAi genetic interaction screening of a maternal lethal mutant14. We used L1-stage larvae because 1) their small size (about 250 µm long and 15 µm wide) allows them to run through the FACS fluidics system and 2) starvation arrests growth at this stage and enables easy accumulation of developmentally synchronized animals. We employed the GFP-negative balancer signature to rapidly identify homozygous mutant larvae in a mixed population and sort them to high purity.
Such isolated larvae can be used for genetic interaction screens, chemical genetic screens, or any other application where large numbers of animals of a particular lethal or sterile genotype are required. There could conceivably be additional applications of laFACS. For example, it could be used to screen large populations for reporter gene activation. However, FACS machines and flow cytometers are designed to quantify fluorescence in relatively small particles and will not give a highly accurate quantitative readout of fluorescence in large objects such as C. elegans larva. Unlike The COPAS biosorter that is specifically designed to sort worms of all stages based on size and/or degree of fluorescence, we have used a FACS only to sort L1 larvae on the basis of presence or absence of fluorescence. Case-by-case trials will have to be undertaken to investigate whether laFACS is suitable for alternative applications. Furthermore, due to size restrictions, the use of laFACS is limited to L1-stage larvae (and embryos). Nonetheless, the ability to use a FACS to sort live C. elegans greatly increases the accessibility and feasibility of high throughput analyses in this model organism.
EXPERIMENTAL DESIGN
Strain construction
In the example presented here, we used laFACS to collect a pure population of maternal-effect lethal (mel-2816,17) homozygotes that we then subjected to an RNAi-based genetic interaction screen14. First, we generated a GFP-balanced mel-28 strain called PF405. In this strain, the mel-28 allele and a cis-linked unc-32 allele were carried on an otherwise wild-type chromosome, and were balanced by the qC1 balancer containing the qIs26 insertion (Figure 1). The qIs26 insertion includes both a lag-2::GFP reporter and the dominant rol-6 allele su100618, and gives rise to recessive lethality. PF405 heterozygotes produce homozygous qC1[qIs26] animals that die as embryos, heterozygous mel-28 unc-32/qC1[qIs26] animals that have a Rol-6 phenotype and express GFP in the distal tip cells of the gonads, and mel-28 unc-32 homozygotes which are uncoordinated, do not express GFP, and produce only dead eggs (Figures 1 and 2).
Figure 2. Strain PF405 Phenotypes.
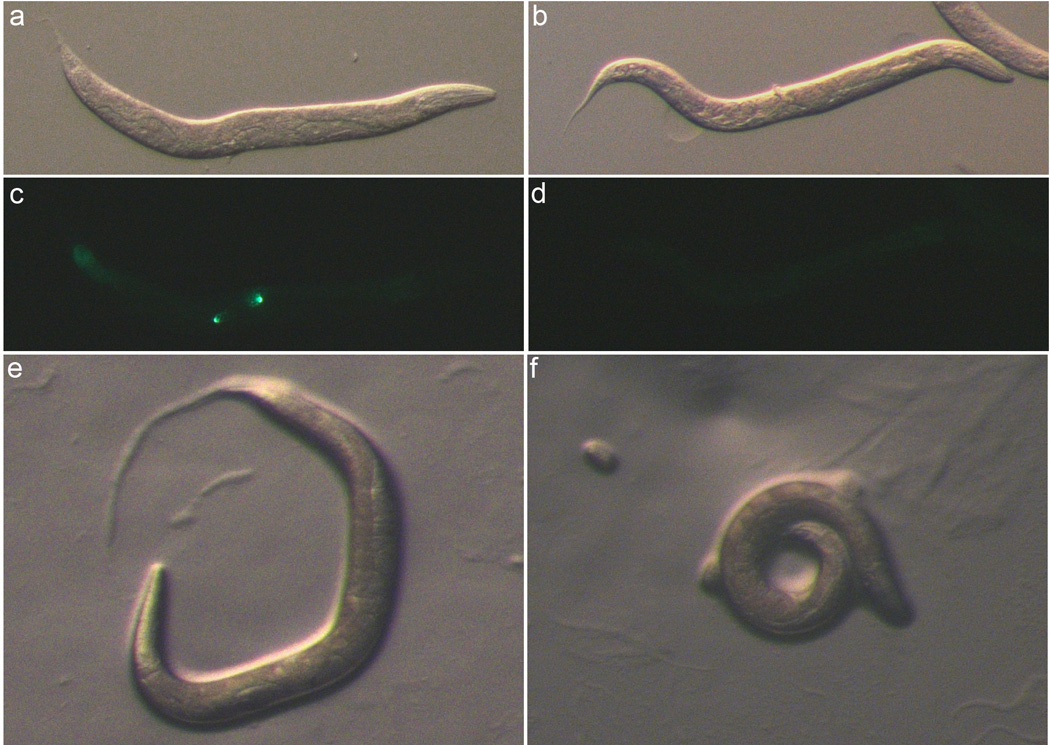
(a, c, e) Heterozygous mel-28 unc-32/qC1[qIs26] adults (Rol-6 phenotype). (b, d, f) Homozygous mel-28 unc-32 adults from the PF405 strain. Images were captured in brightfield (a, b, d, f), and with GFP filters (c–d). (a–d) Animals were anesthetized with levamisole and immobilized on an agar pad. (e–f) characteristic behavior of Rol-6 (e) and Unc (f) adults on an NGM plate .
Proper strain construction is critical for the successful execution of laFACS. We used a balancer that carries, in addition to the GFP transgene, a dominant visible marker (rol-6(su1006)) and a recessive lethal lesion. The visible marker enables distinction of heterozygotes without need of an epifluorescence microscope. If there are no appropriate balancers with this feature, it is recommended that the terminal allele of interest be kept in cis with a recessive visible marker. In our PF405 strain, the mel-28 chromosome also carries an unc-32 allele, thus mel-28 homozygotes from this strain are always uncoordinated (Figures 1 and 2E).
Using a balancer that confers recessive lethality is preferable, but not required. To grow worms for this protocol, we chose heterozygous animals to start the initial stocks, and then expanded them for two generations without specifically picking heterozygotes. Thus the L1’s we sorted were the F2 of the heterozygotes initially picked. Since our balancer was homozygous lethal and mel-28 homozygotes do not produce larvae, all of these L1’s arose from a heterozygous carrier and thus one out of three are expected to be mel-28 homozygotes (Figure 1). When using homozygous viable balancers, with each generation there will be balancer homozygotes that have lost the allele of interest. This lowers the expected yield of desired animals homozygous for the recessive terminal allele. However, because this protocol allows for the sorting of hundreds of thousands of animals, it should still be possible to use this protocol to isolate substantial numbers of homozygotes using a balancer that is not homozygous lethal.
Sorting larvae
To prepare the larvae for sorting, we used sodium hypochlorite treatment to isolate embryos from PF405 adults and allowed these to hatch without food so the animals were arrested at the L1 stage (Figure 3). After filtering the hatched L1’s to remove large debris, we used laFACS to isolate GFP-negative homozygous mel-28 L1 animals from the population. To prime the FACS for larval sorting we installed a 100 µm nozzle and we set up a stream at a lower than usual drop-drive frequency (~16 kHz as opposed to ~38 kHz; see Figure 4a) in order to optimize worm viability while passing through the apparatus.
Figure 3. Preparation of C. elegans for laFACS.
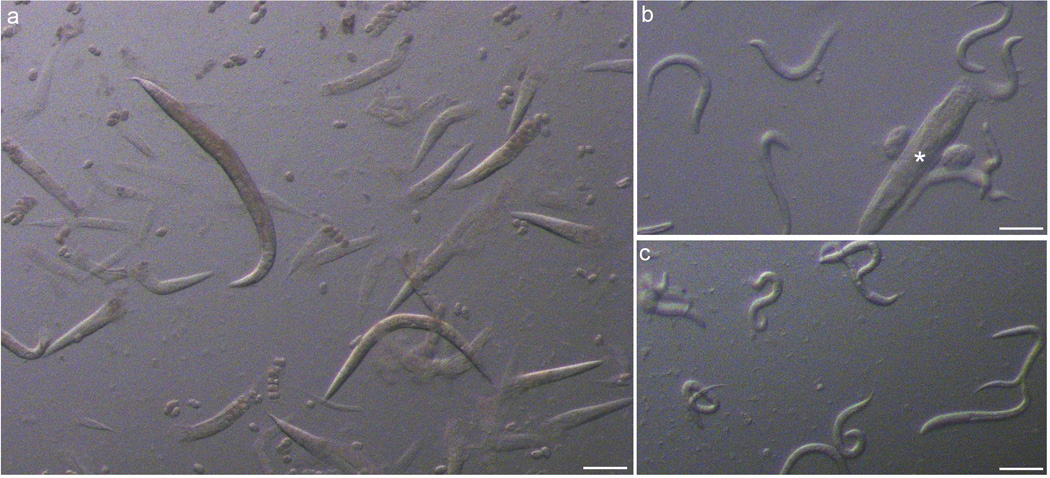
(a) After a ~4 minute treatment with sodium hypochlorite and agitation, most adults have burst and freed their embryos (arrowhead). (b) After overnight incubation in M9, many L1 larvae have hatched but large segments of adult corpses (asterisk) remain. (c) After filtering, only small debris and L1 larvae remain. Scale bar = 200 µm (a).100 µm (b–c).
Figure 4. Sorting C. elegans with laFACS.
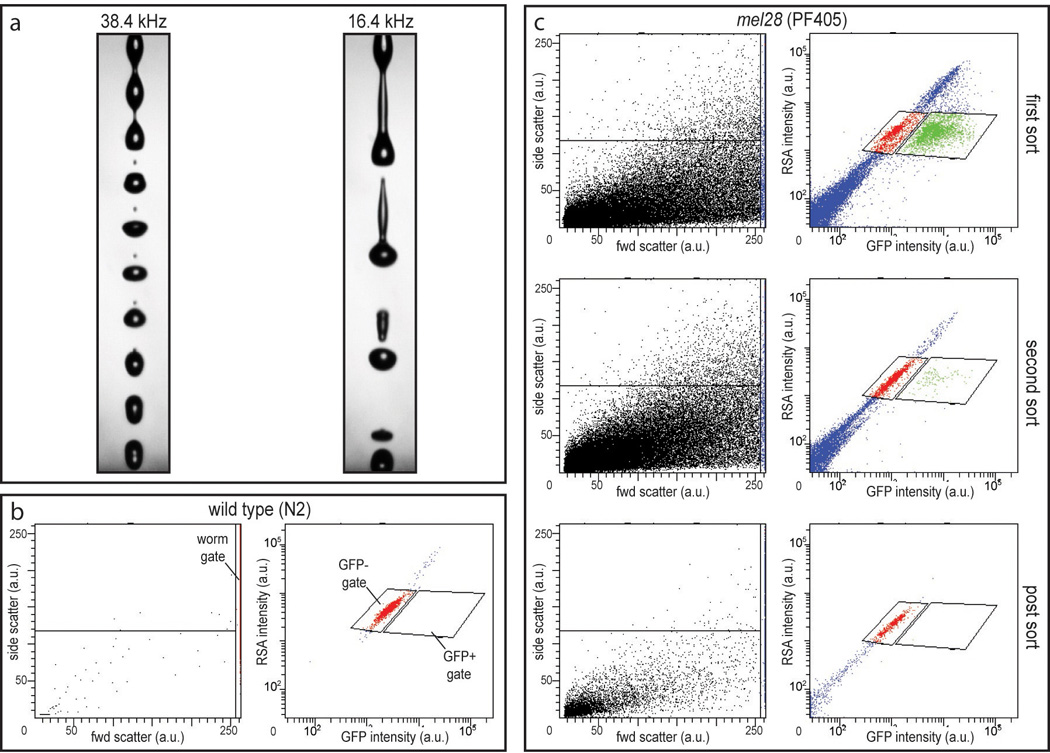
(a) Low-frequency drop-drive stream set up on the FACS facilitates larval survival. (b) Wild-type (N2) L1 larvae were assessed to visualize purely GFP-negative larvae. A gate was set to distinguish larvae from debris in the forward scatter vs. side scatter plot (worm gate; left panel) and two gates were set to identify GFP-negative and GFP-positive events in the GFP intensity (488 nm excitation, 530/30 emission) vs. red spectrum autofluorescence (RSA; 488 nm excitation, 610/20 nm emission) plot (GFP− and GFP+ gates, respectively; right panel). (c) Successive sorts showing the purification of GFP-negative larvae from a PF405 L1 population.
To determine FACS parameters and establish a signature for worms that do not express GFP we ran wild-type (N2) L1’s. A gate was set up in a forward scatter versus side scatter plot in order to distinguish worm-sized events from debris. In effect, larvae were so large that they were off-scale (even at the lowest photo multiplier tube voltage settings) and this “worm gate” encompassed only the events with the highest forward scatter values (Figure 4b left panel). This gate is especially useful when sorting strains that produce a lot of small debris, e.g. remnants of the inviable embryos present in the mel-28 population (see Figures 3c and 4c left panels). Only events that passed the “worm gate” were evaluated for green versus red fluorescent emission (emission around 530 and 610 nm, respectively; Figures 4b and c right panels). Voltage settings were adjusted so that GFP-negative larvae showed an equal ratio of green to red (auto)fluorescence and could be distinguished from GFP-positive larvae which showed relatively higher green than red fluorescence (Figures 4b and c). Gates were set to identify both GFP-positive and GFP-negative populations.
Controls
Going through the steps of this protocol, it is important to regularly inspect the samples microscopically. We looked for larval viability, genotypic purity (as determined by GFP-fluorescence as well as Rol and Unc phenotypic characteristics), debris content and clumping/coagulation. Synchronized and filtered wild-type larvae and a sample of pure GFP-positive larvae can be taken along as a negative- and positive control for the FACS run. These control samples are especially practical when first setting up the technique.
MATERIALS
REAGENTS
C. elegans strain carrying allele of interest balanced by a GFP-containing balancer chromosome.
OP50 strain of E. coli (Caenorhabditis Genetics Center, http://www.cbs.umn.edu/CGC/)
Nematode Growth Medium (US Biological, cat. no. N1000)
tryptone (Fisher Scientific cat. no. BP1421)
CaCl2 dihydrate (Fisher Scientific cat. no. BP510)
KH2PO4 (Fisher Scientific cat. no. BP362)
K2HPO4 (Fisher Scientific cat. no. BP363)
Na2HPO4 heptahydrate (Fisher Scientific cat. no. BP331)
NaCl (Fisher Scientific cat. no. BP358)
MgSO4 heptahydrate (Fisher Scientific cat. no. BP213)
NaOH (Fisher Scientific cat. no. BP359)
sucrose (Fisher Scientific cat. no. BP220)
household sodium hypochlorite solution (4–6%, w/v)
PBS (phosphate buffered saline, pH7.0)
100 mm plates (Fisher Scientific cat. no. 08-757-12)
150 mm plates (Fisher Scientific cat. no. 08-757-14)
Cell strainer (40 µm; BD Falcon, cat. no. 352340)
50 ml conical tubes (Fisher Scientific cat. no. 14-432-22)
15 ml collection tubes (Fisher Scientific cat. no. 05-527-90)
REAGENT SETUP
100 mm standard NGM plates
Prepare 1 M CaCl2 and 1 M MgSO4. Autoclave and allow to cool to room temperature. Prepare 1 M K2HPO4 (dibasic) and 1 M KH2PO4 (monobasic). Autoclave and allow to cool to room temperature. Prepare 1 M Potassium Phosphate buffer (pH 6.0) by mixing 132 ml of autoclaved 1 M K2HPO4 with 868 ml of autoclaved 1 M KH2PO4 in a sterile bottle.
For one liter of NGM, mix 23.005 g of Nematode Growth Medium in 975 ml of deionized water. Autoclave with a stir bar in the solution. After removal from the autoclave, stir the molten solution on a stir plate until it has cooled to ~55° C, then add 24.7 ml of 1 M Potassium Phosphate solution, 1 ml of 1 M CaCl2, and 1 ml of 1 M MgSO4. After mixing, pour 28 ml per each 100 mm plate. If pouring by hand, plan to get approximately 30 plates per liter. Allow plates to rest at room temperature for 1–3 days, to allow condensation to evaporate. Unseeded NGM plates may be stored tightly sealed in plastic sleeves and kept at 4° C for months. To seed plates, grow a 50 ml culture of OP 50 E. coli in LB without antibiotics using standard sterile microbiological techniques. This culture may be stored at 4° C for four weeks. Drop 200 µl of OP50 culture onto each poured NGM plate, and allow to rest at room temperature overnight. Seeded plates may be stored at 4° C for four weeks.
150 mm superseeded 5× peptone plates
For one liter of NGM, mix 23.005 g of Nematode Growth Medium and 10 g of tryptone in 975 ml of deionized water. The tryptone will not go into solution completely until autoclaved. Autoclave with a stir bar in the solution. After removal from the autoclave, stir the molten solution on a stir plate until it has cooled to ~55° C, then add 24.7 ml of 1 M Potassium Phosphate solution, 1 ml of 1 M CaCl2, and 1 ml of 1 M MgSO4 (solutions described in standard NGM recipe). Pour ~55 ml per 150 mm plate, if pouring by hand expect to get 18 plates per liter. Let plates sit at room temperature for 1–3 days, to allow condensation to evaporate. Unseeded plates may be stored tightly sealed in plastic sleeves and kept at 4° C for months. To superseed the plates, prepare a culture of OP-50 in LB, then pellet it and resuspend it in 1/10th the volume of M9 (e.g., a 200 ml solution of OP50 would be pelleted and resuspended into 20 ml of M9). Use a sterile bent glass rod to spread ~1 ml of the concentrated OP50 across each 150mm 5× peptone plate, making sure the bacteria covers the surface area of the plate. Seeded plates may be stored at 4° C for four weeks.
M9
Mix 3.0 g of KH2PO4, 6.0 g of Na2HPO4, and 5.0 g of NaCl in 1 liter of deionized H2O. Autoclave, then cool to room temperature. Using sterile technique, add 1 ml of sterile 1 M MgSO4 and swirl to mix. Prepared M9 may be stored at room temperature indefinitely.
Sodium Hypochlorite Solution
Mix 30 ml of household household sodium hypochlorite solution (bleach) with 60 ml deionized H2O and 10 ml 10N NaOH. (Store at room temperature in the dark for no more than one month.)
CAUTION: hypochlorite solutions lose activity over time. Use proper ventilation when working with sodium hypochlorite.
EQUIPMENT
FACS equipped with a 488 nm excitation laser and 530/30 nm and 610/20 nm emission filters (BD FACSAria I)
Epifluorescence dissecting microscope suitable for imaging GFP (such as the Leica MZ16FA).
EQUIPMENT SETUP
FACS preparation
Clean and sterilize FACS fluidics system, if required, and use PBS as sheath fluid. Install a 100 µm nozzle and set the sheath pressure at 20 p.s.i. Set up a stable stream with low drop-drive frequency. Standard operating parameters for the FACSAria run FACS drop-drive frequency in the vicinity of 38 kHz when using the 100 µm nozzle, thereby creating a stable droplet break-off point at the correct distance from the nozzle. However, at this frequency we noted a low recovery rate (+/− 50%), indicating a large proportion of the larvae did not survive the sort. A stable break-off point can be obtained at lower frequency (+/− 16 kHz; Figure 4a). At this lower frequency it is also possible to set an accurate drop delay and stable side streams (as determined by Accudrop beads, BD Biosciences). Sorting with this setup led to a greatly improved recovery rate (+/−80%). Set the sample agitation function (if available) to ensure larvae don’t settle to the bottom of the sample tube during the sort.
Run a wild-type control (Figure 4b). Adjust the flow rate to get to approximately 100–300 events per second. Prepare a scatter-plot analysis of forward scatter area (FSC-A) versus side scatter area (SSC-A) (Figure 4b left panel). Set low power to the FSC photo multiplier tube (PTM); intact L1 larvae will give a FSC-A signal that is off-scale, debris will give relatively weaker signals. Set SSC-A PMT power to visualize the widest possible range of events. This scatter plot will make it possible to monitor debris content in your sample (Figures 4b and c left panels). Set a gate to isolate events with the highest FSC-A signal, these contain the larvae (Figure 4b left panel “worm gate”). Prepare a scatter-plot analysis of green fluorescence (GFP; 488 nm excitation and emission at 530/30 nm) and red fluorescence (red spectrum autofluorescence [RSA]; 488 nm excitation and emission at 610/20 nm) and display only the population that passes the worm gate set in the FSC-A vs. SSC-A plot (Figure 4b right panel). Adjust the GFP and RSA PMT settings to center the event population representing the GFP-negative, intact larvae (Figure 4b right panel). This scatter plot will make it possible to distinguish true GFP fluorescence from autofluorescence by determining the ratio of green to red fluorescence. Use compensation settings to adjust for spectral overlap between GFP emission and the RSA filter set (settings will depend on the equipment and reporter strain used). This will aid in the distinction between GFP-positive and GFP-negative populations.
Prepare a 15 ml collection tube in the sort block. The collected larvae will remain viable in the PBS that accumulates in the collection tube throughout the sort. Trial sorts of negative- and positive-control samples should be performed and inspected microscopically to assess recovery rate, viability and purity of the sorted animals.
PROCEDURE
Place 12 GFP-balanced heterozygous hermaphrodite young adults on each of 10 100 mm standard NGM plates seeded with OP50 and allow to grow at 20° C until plate is full of young adults but worms are not starved (there is OP50 remaining on the plate).
Wash worms from plates using M9 (1–3 ml per plate) and collect M9 worm suspension to a 50 ml conical tube.
Spin collected worms at 700 g for 5 minutes and carefully decant supernatant.
Add 25 ml of cold M9 and 25 ml of cold 60% sucrose to worm pellet and mix. (Store autoclaved solutions of M9 and 60% sucrose at 4° C for this purpose.)
Spin at 1500 g at 4° C for 5 minutes.
Remove gravid worms from the top and place in a new 50 ml conical tube.
Wash collected worms with M9 by filling tube with M9, spinning at 700 g for 5 minutes, then carefully decanting supernatant.
Resuspend worm pellet in ~25 ml of sodium hypochlorite solution.
Swirl the solution every minute or so. After about 4 minutes, the solution should have become cloudy, indicating that many adults have dissolved. Immediately fill the tube with M9 and spin at 1500 g at room temperature for 2 minutes. Carefully decant bleach solution without disturbing the worm embryo pellet.
Wash the embryo pellet. Fill the tube with M9 and vortex to resuspend pellet, spin at 1500 g for 2 minutes, and decant M9.
Repeat step 10 two more times.
Resuspend washed embryos is 5–10 ml of M9.
Allow collected embryos to hatch in M9 by rotating at 25° C for 14–20 hours.
Plate synchronized larvae to 150mm 5× peptone plates superseeded with OP50. Place 25,000–50,000 L1 larvae on each plate. Incubate at 20° C until most plated worms are gravid adults (3–6 days, depending on the strain). There should be plenty of embryos present on the plate at the time of harvest. ?TROUBLESHOOTING
Although the population will be mostly synchronized at this step, purify it further by performing a sucrose float (as in steps 2–7) to separate gravid adults from any remaining larvae. Use one 50 ml conical per five full plates of worms.
Treat collected worms with sodium hypochlorite solution to isolate embryos (as in steps 8–11). CRITICAL STEP: The embryos from this step will hatch to form the L1's that will be sorted the following day, so it is important for these embryos to be as clean as possible to avoid putting extra particulate matter through the FACS. Ideally at this stage the adult worm bodies are dissolved completely by the bleach leaving only a pure solution of embryos. However, overbleaching at this step will kill embryos, lowering the L1 yield. Different bottles of bleach have different potencies, thus the amount of time spent bleaching these worms must be determined empirically. After each minute in bleach solution, mix the worm suspension and remove one microliter to examine using a stereoscopic microscope. When 75%-90% of the gravid adults have burst open and released their embryos (Figure 3a), proceed to M9 washes.
Allow collected embryos to hatch in at least 5–10 ml of M9 in a 50 ml conical while rotating at 25 degrees for 14–20 hours (Figure 3b).
Dilute the L1’s by filling each conical to the top with M9 and filter each tube of L1’s 2× using a 40 µM nylon cell strainer (Figure 3c).
Concentrate the larvae by spinning at 750 g for 5 minutes and then resuspend them in M9 to a concentration of 200–300 L1’s/µl. Transfer the larval solution to a tube suitable for loading onto the FACS.
Inspect the suspension microscopically and examine debris content, viability and purity.
Run sample through the FACS and adjust flow rate to approximately 100–300 events per second. ?TROUBLESHOOTING
Set gates encompassing the GFP-positive and GFP-negative populations (Figures 4b and c).Larvae with GFP expression will form a population of events off the diagonal in the GFP vs. RSA plot (with a high GFP to RSA ratio) not seen in the wild-type control sample (Figures 4b and c). ?TROUBLESHOOTING
Sort the GFP-negative larvae into a 15 ml collection tube.
Concentrate the sorted larvae by spinning at 750 g for 5 minutes and resuspend them in PBS to a concentration of 200–300 L1’s/µl.
Inspect the suspension microscopically and examine recovery-rate, viability and purity. ?TROUBLESHOOTING
Re-sort the larval suspension until sufficient purity is obtained (repeat steps 21–23, Figure 4c middle panels).
Cytometrically re-analyze a portion of the final sort to verify purity (Figure 4c lower panels) and inspect the suspension microscopically and examine recovery-rate, viability and purity.
Concentrate the larvae by spinning at 750 g for 5 minutes and resuspend in a medium suitable for the final application. (We used the sorted homozygous mutants in an RNAi-based genetic interaction screen performed in 96-well plates14).
Clean and sterilize the FACS fluidics system.
TIMING
The total timing of the protocol varies depending on the strain used, ranging from one week to two weeks. After a sufficient population of the strain has been grown and the L1's isolated, FACS set-up takes 30 minutes and the actual sort takes about 2 hours.
Step 1: 3–6 days
Steps 2–12: ~2 hours
Step 13: 14–20 hours
Step 14: 3–6 days
Steps 15–16: ~2 hours
Step 17: 14–20 hours
Steps 18–20: ~1 hour
Steps 21–29: ~2 hours
TROUBLESHOOTING
| step | problem | possible reason | solution |
|---|---|---|---|
| 14 | Many eggs do not hatch |
1. Did not allow enough time to hatch 2. Overbleaching 3. Not enough M9 was added or tube was not agitated. 4. Balancer itself gives rise to embryonic lethality |
1. Allow longer time for eggs to hatch, some strains might require a longer incubation. 2. Bleach for a maximum of four minutes. 3. Use at least 5 ml of M9, have worms in a 50 ml conical, and be sure they are rotating during the entire incubation. 4. Homozygous lethal balancers and reciprocal translocation balancers produce some inviable embryos. Some proportion of embryos will not hatch, and this should not disrupt further applications provided there are enough live L1's left for sorting. |
| 21 | Recurrent clogged nozzle on the FACS |
1. Did not bleach stock well enough, leading to excess of debris 2. Filtering insufficient 3. Worm suspension too concentrated |
1. Bleach for a longer amount of time to reduce debris from adult worm bodies. 2. Dilute worm solution more before filtering or filter an additional time. 3. Dilute worm suspension. |
| 22 | No clearly distinct GFP- negative population visible |
1. Samples with high debris content will have more debris that passes through the FSC vs SSC worm gate (Figure 4c left panels). Such samples will display events the GFP vs. RSA plot with a wide range of autofluorescent intensity, visible as a diagonal smear obscuring the GFP-negative population (Figure 4c right panels) |
1. Use the upper and lower limits of RSA- intensity of the GFP-positive gate to guide the placement of the GFP-negative gate (Figure 4c right panels). |
| 25 | Collected GFP- larvae do not have desired genotype |
1. Balancer unstable | 1. Check GFP- animals from balanced stock to determine if they have the desired genotype. If not, the balancer may have broken down and permitted recombination. Thaw a frozen stock of the strain to recover the original balanced line. If this is a recurrent problem, then try a different balancer. |
| 25 | Some inviable larvae recovered |
1. Reciprocal translocation balancer used |
1. In GFP-marked reciprocal translocation balancers the GFP insertion is present on just one of the translocated chromosome arms. Depending on the location of the terminal allele, there could be GFP- half-translocation (aneuploid) animals that are not homozygous for the terminal allele. If the recovered animals are to be used for a chemical or genetic screen, then the presence of some GFP- inviable larvae of the wrong genotype should not disrupt the screen provided there are enough viable larvae collected. To avoid aneuploidy, use a balancer that is not a reciprocal translocation. |
ANTICIPATED RESULTS
We generally achieved an 80% or greater recovery rate (larvae recovered/events sorted). The unrecovered fraction of larvae disintegrates during the sort and is seen in subsequent re-sorts as debris (Figure 4c). Sorted larvae were viable and showed no reduced growth or survival in subsequent growth assays (compared to an unsorted control; data not shown). The purity of the sorted larvae (GFP-negative larvae/total larvae) was generally 90% or higher after the initial FACS run in our example sort of mel-28 larvae (this will depend on the initial ratio of GFP-positive to GFP-negative larvae and debris content). A subsequent re-sort of the collected larvae typically gave a final purity of >99% GFP-negative larvae (Figure 4c).
In order to ensure a yield of at least 100,000 pure, homozygous mel-28 larvae we started out with ~660,000 larvae. With the PF405 strain, one in three of the prepared L1 larvae are homozygous mutants and therefore GFP-negative (~220,000 larvae). The expected proportion of homozygous larvae will vary depending on the balancer used. With an 80% recovery rate (~176,000 successfully sorted larvae) and 90% purity (~158,000 GFP-negative larvae) this first sort was re-sorted to obtain ~127,000 GFP-negative larvae with >99% purity. This isolation required one week of growth and larval preparation time and 3 hours of reserved FACS time. We used a FACSAria I (BD Biosciences) in a core facility shared by many other users and none of the subsequent applications were affected by sorting C. elegans larvae.
Acknowledgments
We thank Erik Andersen, Donald G. Moerman, and Robert Waterston for sharing unpublished data, Pui-leng Ip, Jessica Lucas and Katherine Erikson for technical assistance, and Scott D. Weatherbee for critically reviewing the manuscript. Funding sources included the NICHD (R01HD046236) and NHGRI (U01 HG004276) to FP, the NIH (R01GM078279-01) to KDB, and the NSF (0827858) and Fairfield University start-up funds to AGF. Nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR).
Footnotes
AUTHOR CONTRIBUTIONS STATEMENTS
FP and AF initially developed the idea, BB and AF developed the FACS conditions, and FP, AF, BB, EM, ME and KB contributed to the realization of the protocol.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moerman DG, Barstead RJ. Towards a mutation in every gene in Caenorhabditis elegans. Briefings in functional genomics & proteomics. 2008;7(3):195–204. doi: 10.1093/bfgp/eln016. [DOI] [PubMed] [Google Scholar]
- 3.Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421(6920):231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 4.Reboul J, et al. C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet. 2003;34(1):35–41. doi: 10.1038/ng1140. [DOI] [PubMed] [Google Scholar]
- 5.Lamesch P, et al. C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions. Genome research. 2004;14(10B):2064–2069. doi: 10.1101/gr.2496804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohler WA, Isaacson AB. Imaging embryonic development in Caenorhabditis elegans. Cold Spring Harbor protocols. 2010(3) doi: 10.1101/pdb.top71. pdb top71. [DOI] [PubMed] [Google Scholar]
- 7.Gunsalus KC, et al. Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature. 2005;436(7052):861–865. doi: 10.1038/nature03876. [DOI] [PubMed] [Google Scholar]
- 8.Sönnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434(7032):462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 9.Green RA, et al. A High-Resolution C. elegans Essential Gene Network Based on Phenotypic Profiling of a Complex Tissue. Cell. 2011;145(3):470–482. doi: 10.1016/j.cell.2011.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kemphues K The C. elegans Research Community. WormBook. [Google Scholar]
- 11.Pulak R. Techniques for analysis, sorting, and dispensing of C. elegans on the COPAS flow-sorting system. Methods in molecular biology (Clifton, N.J. 2006;351:275–286. doi: 10.1385/1-59745-151-7:275. [DOI] [PubMed] [Google Scholar]
- 12.Dupuy D, et al. Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nature biotechnology. 2007;25(6):663–668. doi: 10.1038/nbt1305. [DOI] [PubMed] [Google Scholar]
- 13.Doitsidou M, Flames N, Lee AC, Boyanov A, Hobert O. Automated screening for mutants affecting dopaminergic-neuron specification in C. elegans. Nature methods. 2008;5(10):869–872. doi: 10.1038/nmeth.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez AG, Mis EK, Bargmann BO, Birnbaum KD, Piano F. Automated sorting of live C. elegans using laFACS. Nature methods. 2010;7(6):417–418. doi: 10.1038/nmeth.f.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoeckius M, et al. Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression. Nature methods. 2009;6(10):745–751. doi: 10.1038/nmeth.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez AG, Piano F. MEL-28 is downstream of the Ran cycle and is required for nuclear-envelope function and chromatin maintenance. Curr Biol. 2006;16(17):1757–1763. doi: 10.1016/j.cub.2006.07.071. [DOI] [PubMed] [Google Scholar]
- 17.Galy V, Askjaer P, Franz C, Lopez-Iglesias C, Mattaj IW. MEL-28, a novel nuclear-envelope and kinetochore protein essential for zygotic nuclear-envelope assembly in C. elegans. Curr Biol. 2006;16(17):1748–1756. doi: 10.1016/j.cub.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 18.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. The EMBO journal. 1991;10(12):3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgley ML, Riddle DL. LG II balancer chromosomes in Caenorhabditis elegans: mT1(II;III) and the mIn1 set of dominantly and recessively marked inversions. Mol Genet Genomics. 2001;266(3):385–395. doi: 10.1007/s004380100523. [DOI] [PubMed] [Google Scholar]
- 20.Edgley MK, Baillie David L, Riddle Donald L, Rose Ann M. WormBook. 2006 The C. elegans research community. [Google Scholar]