

Abstract
Ribosome biogenesis in eukaryotic cells is a highly dynamic and complex process innately linked to cell proliferation. The assembly of ribosomes is driven by a myriad of biogenesis factors that shape pre‐ribosomal particles by processing and folding the ribosomal RNA and incorporating ribosomal proteins. Biochemical approaches allowed the isolation and characterization of pre‐ribosomal particles from Saccharomyces cerevisiae, which lead to a spatiotemporal map of biogenesis intermediates along the path from the nucleolus to the cytoplasm. Here, we cloned almost the entire set (∼180) of ribosome biogenesis factors from the thermophilic fungus Chaetomium thermophilum in order to perform an in‐depth analysis of their protein–protein interaction network as well as exploring the suitability of these thermostable proteins for structural studies. First, we performed a systematic screen, testing about 80 factors for crystallization and structure determination. Next, we performed a yeast 2‐hybrid analysis and tested about 32,000 binary combinations, which identified more than 1000 protein–protein contacts between the thermophilic ribosome assembly factors. To exemplary verify several of these interactions, we performed biochemical reconstitution with the focus on the interaction network between 90S pre‐ribosome factors forming the ctUTP‐A and ctUTP‐B modules, and the Brix‐domain containing assembly factors of the pre‐60S subunit. Our work provides a rich resource for biochemical reconstitution and structural analyses of the conserved ribosome assembly machinery from a eukaryotic thermophile.
Keywords: Chaetomium thermophilum, ribosome biogenesis, interaction map, UTP‐A, UTP‐B, Brix proteins
Short abstract
Abbreviations
- BAP
Brix associated protein
- Brix
biogenesis of ribosomes in Xenopus
- EM
electron microscopy
- nsEM
negative stain electron microscopy
- RMSD
Root mean square deviation
- SEC
size exclusion chromatography
- UTP
U three proteins (U3 snoRNA associated proteins)
- TAP
tandem affinity purification
- Y2H
yeast 2‐hybrid
Introduction
Ribosome biogenesis in eukaryotes is a highly dynamic process that includes the coordinated assembly of four ribosomal RNAs (rRNA) and about 80 ribosomal proteins (r‐proteins). This process starts with the RNA polymerase I mediated transcription of a large precursor rRNA (pre‐rRNA). This 35S pre‐rRNA is co‐transcriptionally modified by snoRNPs and subsequently assembles with r‐proteins and biogenesis factors to form the early 90S pre‐ribosomal particles. During subsequent rRNA processing events, among them several endo‐ and exonucleolytic cleavages, the 35S rRNA gets cleaved at position A2, which separates the pathway of the small and large subunit. Within the pre‐40S and pre‐60S particles, the rRNA is further matured and additional r‐proteins are recruited. During these maturation events the associated biogenesis factors change dynamically. The pre‐ribosomes are then eventually exported through the nuclear pore complex (NPC) into the cytoplasm, where the final biogenesis steps yield the mature subunits (for review of ribosome biogenesis see1, 2, 3, 4, 5, 6, 7). Recently, there is increasing evidence that ribosome biogenesis is a promising target for cancer therapy,8, 9 as many chemotherapeutic drugs also target ribosome biogenesis.10 Therefore, the functional and structural understanding of ribosome assembly is expected to also facilitate the development of new small molecule inhibitors.
Most of the current knowledge about ribosome assembly has been obtained using the model organism Saccharomyces cerevisiae or human cells lines, using a broad range of established genetic and proteomic tools. In particular, the tandem‐affinity‐purification (TAP) method coupled with mass spectroscopy, Western blot analysis and Northern blot analysis were successfully used to analyse the protein and RNA composition of pre‐ribosomal complexes. Such a characterization of a large number of pre‐ribosomal particles uncovered a road map of the ribosome assembly pathway.11, 12, 13, 14, 15, 16 For a few of these particles a cryo‐EM structure has been determined.17, 18, 19, 20, 21, 22, 23, 24
Recently, an increasing number of studies utilized thermophilic proteins derived from the eukaryote Chaetomium thermophilum to achieve structural and mechanistic insights into various cellular processes.25, 26, 27, 28, 29, 30, 31, 32 Here, we exploited the biochemical properties of ribosome biogenesis factors from C. thermophilum to extend their structural and functional characterization. First, we annotated and cloned >180 ribosome biogenesis factors and performed a systematic analysis of expression, purification and crystallization of about 80 of these proteins. In parallel, we used the complete collection to perform a large‐scale Y2H screen. This approach analysed more than 32.000 individual protein pairs and revealed more than 1000 protein–protein interactions, including multiple interactions, which were not known so far. Based on these findings, we have selected a subset of proteins and validated the identified interactions in vitro, by reconstituting the direct protein‐protein interactions within the ctUTP‐A and ctUTP‐B complexes. Further, we were able to identify the binding partners of the Brix domain proteins and reconstitute these dimeric complexes in vitro. Thus, our work provides a solid basis and rich source for an indepth characterization of individual proteins and complexes involved in ribosome biogenesis.
Results and Discussion
Creating a resource of thermophilic ribosome biogenesis factors
In order to exploit the proteome of a thermophilic eukaryote to study ribosome assembly, we sought to clone all ribosome biogenesis factors from Chaetomium thermophilum (ct) [Fig. 1(A)]. The ribosome assembly factors of C. thermophilum were identified by blast searches (NCBI BLAST+) using the annotated yeast S. cerevisiae assembly factors (SGD annotation, GO term) against the translated genome of C. thermophilum.25, 33 We identified in total 181 putative orthologues (see Supporting Information Table S1) and validated them by multiple sequence alignment. For a few yeast ribosome biogenesis factors, including Fyv7, Lrp1, Nop19, YBL028c, Rlp7, and Alb1, no clear orthologue could be found by simple blast searches. However, most of these missing factors except Rlp7 and Nop19 are nonessential in yeast. Moreover, in yeast some biogenesis factors are paralogous, including Fpr3 and Fpr4, Ssf1 and Ssf2, Npa1 and Npa2. However, only one orthologue appears to be present in the thermophilic genome. Consistent with this observation, Ssf1 and Ssf2 are redundant for ribosome assembly in yeast.14, 34
Figure 1.
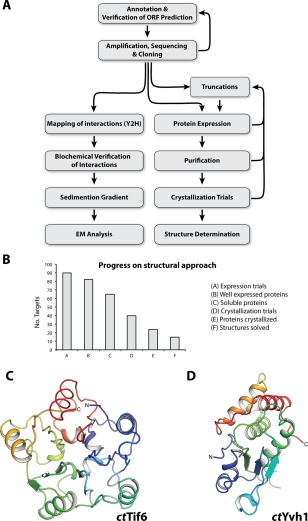
From gene identification to structure determination of ribosome biogenesis factors. (A) Scheme depicts the workflow from in silico ORF annotation to cloning, expression, determination of the interactome, biochemical characterization and structural analysis by cryo EM or crystallography. (B) Statistics of the structural approach. From 90 targets that were tested for expression in E. coli (column A), 77 were well expressed (column B) and 52 were soluble (column C), which went into large‐scale purification. Subsequently, 40 proteins were suitable for crystallization trials (column D), of which 24 yielded crystals (column E), and finally allowed determination of 14 structures (column F). (C,D) Crystal structure of the phosphatase domain (PD) of ctTif6 (C, pdb: 5M3Q) and ctYvh1 (D, pdb: 5M43) both colored from N‐ to C‐terminus (blue to red). The overall structure of ctTif6 is highly similar to that of scTif6 (RMSD 0.67 Å over 216 residues), with an internal fivefold symmetry. The phosphatase domain of ctYvh1 belongs to the family of dual‐specificity phosphatases, able to hydrolyze phosphate from phosphorylated serine/threonine as well as tyrosine residues.
Multisequence alignments35 of these 181 orthologous ribosome assembly factors from C. thermophilum confirmed >85% of the computer based gene predictions,25, 33 but also revealed some erroneous annotations, such as incorrect prediction of the start codon (e.g., Nmd3, Rsa4, Cbf5, Rei1) [Supporting Information Fig. S1(A)], or inaccurate prediction of intron boundaries (e.g., Sda1, Rli1, Noc1) [Supporting Information Fig. S1(B)]. In this way, 25 genes encoding these biogenesis factors, including ctCBF5, ctCMS1, ctDBP2, ctDIM1, ctDIM2, ctENP2, ctFPR4, ctFUN12, ctMRT4, ctMTR4, ctNMD3, ctNOC1, ctNOG1, ctNOP12, ctNSR1, ctREI1, ctRLI1, ctRRP8, ctRSA3, ctRSA4, ctRSP5, ctSDA1, ctUTP11, and ctXRN1 could be corrected and updated in the Uniprot database and the C. thermophilum genome resource.
Based on these annotations, we prepared cDNA and genomic DNA from C. thermophilum,25 PCR‐amplified the corresponding ORFs, cloned 181 factors into Y2H plasmids using NdeI and BamHI restriction sites and sequenced them (see “Materials and Methods”). The standardized cloning procedure allowed an easy and rapid transfer of the thermophilic ORFs into various plasmids for expression in E. coli or yeast, which carried different affinity tags and selection markers (see below). Together, this provides an exhaustive collection of thermophilic ribosome assembly factors involved in various stages of ribosome formation that could serve as a versatile source for biochemical and structural studies.
Systematic screen for protein expression, purification, and crystallization
From our previous experience with C. thermophilum proteins used in biochemical reconstitutions such as subcomplexes of the nuclear pore complex,36, 37 the analysis of the guided entry of tail‐anchored proteins to the ER (GET pathway30) or the analysis of ribosome associated factors,31 we also expected that thermophilic ribosome assembly factors have superior properties for biochemical, enzymatic and structural studies. After the identification of approx. 180 targets within the C. thermophilum genome, we aimed to design a general protocol for the expression and purification of many of them in parallel [Fig. 1(A)]. Therefore, we started a systematic analysis in a small scale volume (mini‐screening) with 44 initial targets using two different E. coli strains [Rosetta2 and BL21(DE3)] for protein expression and standard buffers for a mini‐scale in batch purification step [Supporting Information Figs. S2(A) and S2(B)]. As Rosetta2 was superior to BL21 (DE3) [Supporting Information Fig. S2(C)], we continued our analysis with 35 further factors using only Rosetta2. Beside this systematic screen, additional factors were pursued individually or with their binding partner (e.g. ctMrt4, ctArx1, ctErb1‐ctYtm1). In total, we could overexpress 77 out of 90 targets, 52 were soluble and suitable for further purification and crystallization trials [Fig. 1(B)]. Overall, we could determine the structures of 14 different targets [Fig. 1(B)] including 12 published (e.g., ctAcl4, ctSyo1, ctArx1, ctSqt1, ctYtm1, ctErb1, ctMrt4, ctRio2, ctCrm1, ctMtr2, ctMex67, and ctRsa419, 32, 38, 39, 40, 41, 42, 43, 44, 45) as well as ctTif6, and ctYvh1 [Fig. 1(C); Supporting Information Fig. S2(D), S2(E)]. For additional factors we are currently optimizing crystallization conditions. To further improve the rate of success, construct optimization, adaptation of expression and purification protocols up to using special crystallization strategies, such as carrier driven crystallization38 will be performed.
In the past, structural genomics approaches have been applied to cytosolic bacterial proteins, thermophilic bacteria and archaea,46, 47 whereas structural genomics approaches on eukaryotic targets focussed on specific protein families with a conserved core structure, such as kinases48 and phosphatases.49 In contrast, ribosome biogenesis factors in our screen belong to different protein families with a variety of folds including β‐propeller, α‐solenoid proteins, GTPases, helicases, aminopeptidases, and others. In addition, the majority of ribosome biogenesis factors are RNA binding proteins, which are often difficult to handle due to unstructured or highly charged regions. Nevertheless, the crystal structures of 14 out of 90 expressed factors were determined, proving that the use of thermophilic ribosome biogenesis factors is a powerful tool for structure determination.
Interaction network of thermophilic ribosome biogenesis factors
To gain a first insight into the network of interactions between the thermophilic ribosome assembly factors, we sought to use the collection of C. thermophilum factors in a large‐scale yeast 2‐hybrid (Y2H) analysis (see “Materials and Methods”). It is considered that a Y2H interaction represents predominantly direct protein‐protein interactions.50 Thus, Y2H information could be complementary to data obtained from biochemical isolation of pre‐ribosomal particles, which cannot reveal whether or not two ribosome biogenesis factors directly contact each other. Since previous studies revealed that some ribosome biogenesis factors (e.g., Rrp5, Has1) are involved in the assembly of both the small and large subunit, we took an unbiased Y2H approach and tested all possible protein combinations (>32,000) of 60S and 40S assembly factors. In this way our screen comprised 181 proteins, which have been used as both bait and prey proteins. The Y2H screening approach was based on a mating procedure to combine bait and prey plasmids in diploid yeast cells [Fig. 2(A)], which were subsequently analyzed for growth on SDC–His (medium interaction) and SDC–Ade plates [strong interaction, see “Materials and Methods”, Fig. 2(B,C)]. For an initial validation of the Y2H results, we repeated the screen, using a different set of yeast transformants (see “Materials and Methods”).
Figure 2.
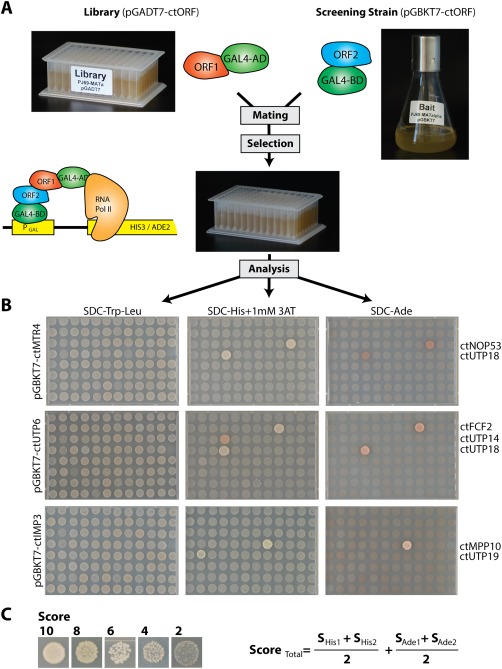
Illustration of the screening procedure for Y2H interactions. (A) Scheme of the experimental setup of the Y2H screen. Yeast strain PJ69‐4 MATa was transformed with 181 different Prey plasmids pGADT7 and a mix of five transformants was transferred to one position within two 96 deep well plates, representing the yeast‐two‐hybrid (Y2H) library. During the screening procedure, a liquid culture of the yeast strain (PJ69‐4 MATalpha) carrying the bait protein (pGBKT7) was mated against the refreshed library in a 96 deep well plate in liquid YPD medium. The next day, YPD was replaced with SDC‐Trp‐Leu medium to select for diploid cells carrying both plasmids. (B) After 2 day's incubation (30°C), the cells were spotted on SDC‐Trp‐Leu plates to control the mating efficiency, and on SDC‐Trp‐Leu‐His + 1 mM 3‐AT and SDC‐Trp‐Leu‐Ade plates to screen for medium and strong interactions. Plates were incubated at 30°C and documented after four and 7 days. Examples are shown in B. Bait proteins are listed on the left side and interacting prey proteins are indicated at the right side. (C) The strength of the Y2H interaction was ranked based on size and numbers of colonies. The total score was calculated from the mean value of 2 repetitions and the sum of the score derived from growth on SDC‐Trp‐Leu‐His11 mM 3‐AT and SDC‐Trp‐Leu‐Ade plates. The detected interactions are summarized in Supporting Information Tables S3 and S4 and can be viewed together with the primary data at http://y2h.embl.de.
In order to visualize this interactome of thermophile ribosome assembly factors and display it in an accessible way, we utilized the xiNET software.51 This software provides an interface through which the interaction network of a particular protein of interest can be viewed. In this 2‐hybrid network, the strength of the interactions was ranked based on colony size and numbers of colonies with a maximal score of 20 (Supporting Information Tables S3 and S4). A slide bar within the software allows a threshold to be set, which will hide all interactions with a score below that value. Finally, the software allows the separation of the interactome into SSU and LSU interaction maps. The software including the primary dataset can be accessed at http://y2h.embl.de.
From the approximately 32,000 combinations tested, we found a total of 1049 interactions (Supporting Information Table S3), of which 270 of them were very robust (Score ≧ 10, Supporting Information Table S4). No interactions were observed for the bait and the prey plasmid of 17 genes. Since several of these biogenesis factors are known to be stably associated with pre‐ribosomal particles (Arx1, Lsg1, Sof1, Enp2, and Mrt4), it is possible that they interact predominantly with rRNA or ribosomal proteins that were not included in the screen.
Among the identified Y2H pairs, we found several that were previously identified in other organisms (Supporting Information Table S4), including interactions between various 90S factors (ctMpp10–ctImp3 and ctMpp10–ctImp4,52, 53 ctRcl1–ctBms1,54 ctKrr1–ctFaf1,55, 56 ctNhp2–ctNop1057), late 40S factors (ctNob1–ctDim2, ctHrr25–ctLtv1), 60S factors (ctRrp5–ctNoc1,58 ctLas1–ctGrc359, 60), or the exosome (ctRrp46–ctRrp43, ctRrp45–ctRrp40, ctMtr3–ctRrp42, ctMtr3–ctRrp43, ctRrp45–ctRrp4, ctRrp45–ctRrp461). In addition, our screen revealed many interactions that have not been identified in related screens based on mesophilic ribosome assembly factors (Supporting Information Table S4). These novel interactions are found in the context of pre‐40S assembly (ctEsf1–ctRrp3, ctUtp2–ctEnp1, ctUtp6–ctFcf2, ctUtp18–ctMtr4,62 ctEfg1–ctDbp4) and pre‐60S assembly (ctNop53–ctMtr4,62 ctNpa1–ctRsa3, ctNog1–ctMak16). The fact that our screen detects interactions previously identified for mesophilic proteins supports the hypothesis that the novel interactions detected are also conserved in evolution. Thus, our thermophilic interaction map contributes to a better understanding of ribosome formation in eukaryotic cells.
Biochemical reconstitution of the thermophilic UTP‐A and UTP‐B complex
To confirm that the identified Y2H interactions represent direct protein–protein interactions, we focused on reconstituting the interactions within the ctUTP‐A and ctUTP‐B subcomplexes. First, we reproduced the results obtained from the Y2H screen [based on a mating procedure, Fig. 3(A)] by co‐transformation of all combinations of Y2H plasmids coding for members of the ctUTP‐A or the ctUTP‐B complex, respectively [Fig. 3(B)]. This approach largely confirmed all the interactions within the ctUTP‐A and ctUTP‐B complex revealed by the screen. However the co‐transformation procedure revealed additional interactions within the ctUTP‐A complex (ctUtp5–ctUtp10 and ctUtp15–ctUtp17) and the ctUTP‐B complex (ctUtp13–ctUtp12), but missed the interaction between ctUtp21–ctUtp18. These minor differences might be due to ineffective mating or differences in the relative expression levels in diploid and haploid yeast cells. Taken together, the co‐transformation method essentially matched the results from our large‐scale approach.
Figure 3.
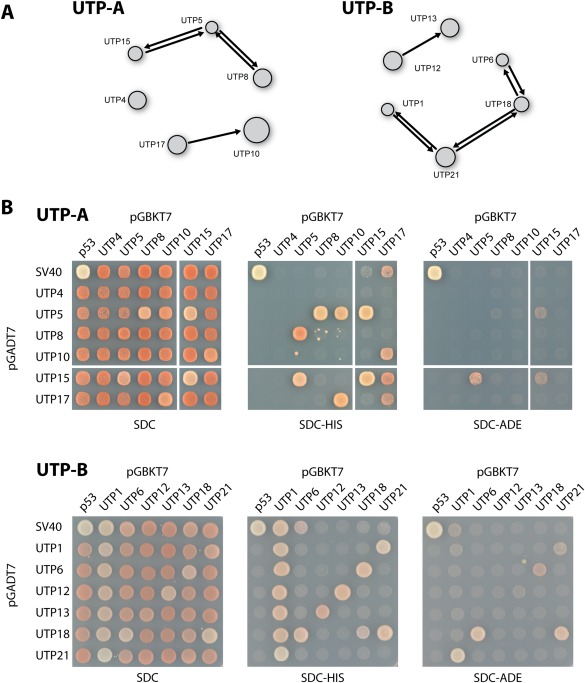
Y2H interaction within the ctUTP‐A and ctUTP‐B complex. (A) The interaction network as derived from the large‐scale screen is depicted. An arrow from the prey protein (AD) towards the bait protein (BD) shows a positive interaction between the connected proteins. The displayed network is derived from the interactive software tool that contains the complete dataset that can be accessed at http://y2h.embl.de. (B) The Y2H interactions from the screen were confirmed by co‐transformation into yeast strain PJ69‐4 (see “Materials and Methods”). The transformants were analyzed for growth on SDC‐Trp‐Leu‐His (SDC‐HIS, weak interactions) and SDC‐Trp‐Leu‐Ade (SDC‐ADE, strong interactions) at 30°C. The growth phenotype after 2 days is shown. The upper panels show the results of the ctUTP‐A complex; the lower panels show the data for ctUTP‐B.
To biochemically verify the observed Y2H interactions, we expressed the proteins in E. coli or S. cerevisiae and used these thermophilic recombinant proteins to test for a direct protein–protein interactions (see “Materials and Methods”). First, we focussed on the binary interactions within the ctUTP‐A complex [Fig. 4(A)]. Accordingly, ctUtp5–ctUtp8 and ctUtp4–ctUtp8 were shown to form stoichiometric complexes that remained stable during size exclusion chromatography (data not shown). Moreover, we could reconstitute the ctUtp5–ctUtp15 dimer and the ctUtp10–ctUtp17–ctUtp5 heterotrimer [Fig. 4(A)]. In a similar way, we biochemically reconstituted, based on our Y2H data, the interactions between the members of the ctUTP‐B complex, which included binary interactions between ctUtp21–ctUtp1, ctUtp12–ctUtp13, ctUtp18–ctUtp6, and ctUtp21–ctUtp18 [Fig. 4(B)]. Thus, all the identified interactions within the ctUTP‐A and ctUTP‐B complex could be biochemically reconstituted.
Figure 4.
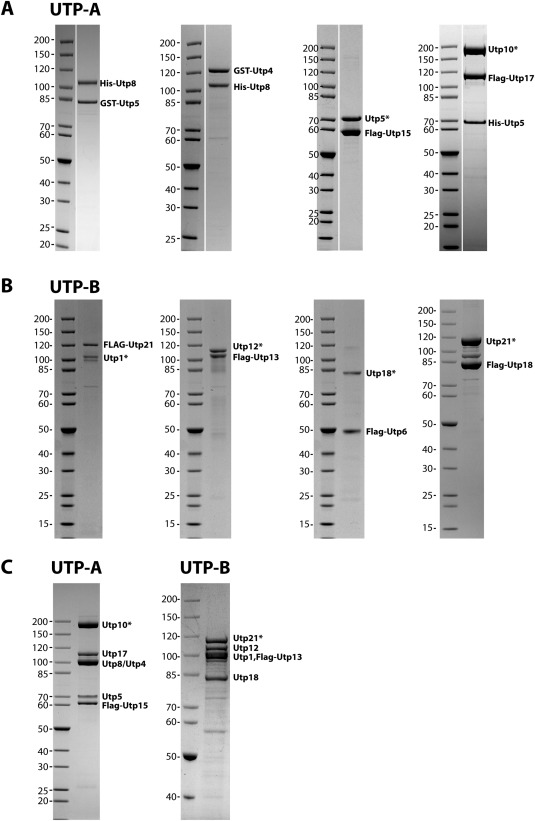
Biochemical reconstitution of the ctUTP‐A and ctUTP‐B complex. Thermophilic proteins were recombinantly co‐expressed in E. coli or S. cerevisiae and co‐purified (for details, see “Materials and Methods”). The dimeric and trimeric reconstituted complexes of ctUTP‐A (A) and ctUTP‐B (B) were analyzed by SDS‐PAGE and subsequent Coomassie blue staining. The label at the right indicates the position of the protein with its tag. A protein A tagged protein, which gets cleaved from its tag during purification is marked with an asterisk. (C) For the reconstitution of the complete ctUTP‐A (left panel) and ctUTP‐B (right panel) complex, all members were recombinantly co‐expressed in yeast and isolated using a split tag approach.
Next, we tested, whether the thermostable proteins are suitable to reconstitute the complete subcomplexes. Here, we used a plasmid system that allows GAL promotor driven expression of up to six different recombinant C. thermophilum subunits in yeast.36 For this purpose, two C. thermophilum assembly factors were tagged for sequential Protein A and Flag purification, whereas the other components remained untagged (see “Materials and Methods”). The reconstituted and purified ctUTP‐A and ctUTP‐B complex consisted of six and five protein factors, respectively, which were verified by mass spectroscopy [Fig. 4(C)]. However, the ctUTP‐B complex did not incorporate ctUtp6, and shuffling of the tags within the ctUTP‐B complex did not improve the association of ctUtp6 (data not shown). Thus, ctUtp6 may be only weakly associated with the ctUTP‐B complex. Our biochemical reconstitutions are consistent with recent biochemical analyses of the mesophilic UTP‐A and UTP‐B subcomplexes.63, 64 However, the data from C. thermophilum, including ours and recently published work65 expands the protein‐protein interaction network within the UTP subcomplexes, by showing the biochemical reconstitution of ctUtp6–Utp18, ctUtp21–Utp18 and ctUtp8–ctUtp5 heterodimers. These additional direct interactions are consistent with recently published crosslink data using the UTP‐A and UTP‐B complexes from S. cerevisiae.66
Next, we used negative stain electron microscopy (nsEM) to structurally characterize the reconstituted ctUTP‐A and ctUTP‐B complexes. Since initial experiments indicated that the complexes are rather flexible (data not shown), we applied the GraFix method67 in order to stabilize the reconstituted complexes (see “Materials and Methods”). After ultracentrifugation on a glycerol gradient including a fixation reagent, the fraction containing the ctUTP‐A and ctUTP‐B complex was analysed by nsEM (Fig. 5). A 2D class averaging of the ctUTP‐A complex (based on 5942 particles) and the ctUTP‐B complex (based on 5000 particles) revealed slightly elongated structures of approximately 20 nm length. The ctUTP‐A complex was built up of globular and extended subdomains, whereas, the ctUTP‐B complex consisted of several globular subdomains. This is consistent with the secondary structure predictions of the individual components, indicating that the ctUTP‐A complex contains six WD40 domains and the large ctUtp10 (>190 kDa) consisting of helical domains. In contrast, the ctUTP‐B complex without ctUtp6 consists mainly of nine WD40 domains with helical extensions. The arrangement of both complexes differs in the class averages, indicating that they are flexible and may adopt different conformations. A highly similar overall structure and an inherent flexibility has recently been reported for the yeast UTP‐A and UTP‐B complexes.65, 66 In contrast, our recently published cryo‐EM structure of the 90S particle revealed, that in the context of the 90S particle, the UTP‐A and UTP‐B complexes take a rigid conformation.24 Taken together, the data presented here support the idea that the direct protein–protein interactions and structural organisation of the UTP‐A and UTP‐B complexes are highly conserved between C. thermophilum and S. cerevisiae, which strongly supports the significance of the Y2H interaction network presented here (Supporting Information Tables S3 and S4).
Figure 5.

Structural characterization of the reconstituted ctUTP‐A and ctUTP‐B complex. The reconstituted ctUTP‐A and ctUTP‐B complexes [see Fig. 2(C)] were purified by the GraFix approach and subsequently analysed by negative stain electron microscopy. Left panel shows a micrograph, the right panel shows a gallery of the representative class averages. Scale bar indicates 10 nm.
Reconstitution of brix proteins with their associated proteins (BAP)
Another important discovery that came out from our Y2H screen was the identification of interactions of the Brix domain factors. The recent structural characterization of Rpf2 in complex with Rrs1,68, 69, 70 and the high sequence homology within the Brix proteins (Supporting Information Fig. S3) indicated that all Brix proteins require a corresponding Brix domain associated protein (BAP)70 that contribute a long α‐helix and an adjacent β‐hairpin to complete the Brix fold [Fig. 6(A)]. Accordingly, our screen identified strong interactions between ctImp4–ctMpp10, ctRpf1–ctMak16, ctBrx1–ctEbp2, and ctSsf1–ctRrp15 (Supporting Information Tables S3 and S4), which are in agreement with previous Y2H data.50, 71, 72 Previously, a fragment of Mpp10 had been shown to biochemically interact with Imp4.52, 73 In contrast, the remaining Brix–BAP dimers, which are exclusively embedded into the eukaryotic pre‐60S ribosome biogenesis pathway, had not been biochemically reconstituted to date. In order to identify the minimal interacting fragment of the BAP factors, we tested various truncations in a Y2H approach [Fig. 6(B–D)], which revealed that the minimal Brix–BAP complexes are built up by ctRpf1 (156–436 aa) and ctMak16 (134–336 aa), ctBrx1 (30–260 aa) and ctEbp2 (178–268 aa), and ctSsf1 (22–359) and ctRrp15 (194–354aa) [Fig. 6(B–D)]. Next, we performed in vitro binding assays to biochemically reconstitute the Brix–BAP dimers, using minimal fragments expressed recombinantly in E. coli (see “Materials and Methods”). These binding assays demonstrated that the Brix domain of Rpf1, Brx1, or Ssf1 alone is sufficient to biochemically interact with a helical fragment of their corresponding BAP binding partner [Fig. 6(E–G) left panel]. Consistently, the thermophilic Brix–BAP complexes remained stable on size exclusion chromatography [Fig. 6(E–G) right panel, Supporting Information Fig. S4]. Despite the fact that all the interacting BAP fragments are predicted to contain similar structural elements, no conserved amino acid motif could be identified (data not shown), suggesting that complex formation occurs only between the respective partners. Taken together, the combination of the Y2H screen and in vitro reconstitution allowed the identification of the Brix‐associated proteins and allowed for the first time the biochemical verification of the missing Brix–BAP complexes that are active in pre‐60S biogenesis.
Figure 6.
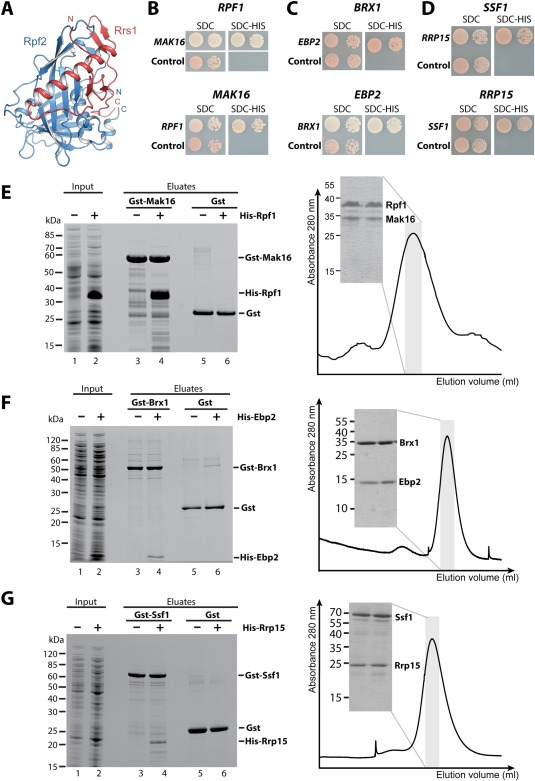
Reconstitution of the Brix–BAP complexes involved in 60S biogenesis. (A) The structure of the Brix domain Rpf2 (blue) in complex with the BAP Rrs1 (red) is shown. The structure is taken from PDB entry 5BY8.69 (B–D) A Y2H analysis of the C. thermophilum Brix proteins with their BAP is shown. The upper panel shows the ctBrix domain fused to the activation domain (pGADT7) without terminal extensions [rpf1 (156–436 aa), brx1 (30–260 aa), and ssf1 (22–359 aa)] tested with the corresponding BAP fused to DNA binding domain (pGBKT7), whereas the lower panel shows the fragment of the ctBAP (pGADT7) (mak16 (134–336 aa), ebp2 (178–268 aa), and rrp15 (194–354 aa) tested with the full‐length Brix protein (pGBKT7). SDC‐Trp‐Leu‐His plates are shown after 3 days incubation at 30°C. (E–G) A binding assay (left panel) and size exclusion chromatography (SEC, right panel) was performed to verify the Y2H interactions. For the binding assay, the indicated GST proteins were immobilized on GSH beads, washed (lanes 3, 5), incubated with E. coli supernatant containing the His6 tagged partner (lane 2), washed and eluted by GSH (lane 4). As a negative control the His6 tagged proteins were incubated with GST alone (lane 6). Right panel shows SEC analysis of the affinity‐purified complexes. SDS‐PAGE shows protein composition of the peak fraction (domain boundaries are identical as in B–D except for SEC analysis of ctBrx1 (34–259 aa)—ctEbp2 (175–282 aa), and ctSsf1 (34–478 aa)—ctRrp15 (173–354 aa) complexes.
Conclusion
The use of thermophilic proteins in a Y2H screen and in biochemical reconstitutions assays enabled us to identify and prove direct protein–protein interactions between ribosome biogenesis factors from C. thermophilum. Moreover, their suitability for structural studies is highlighted by structure determination of numerous proteins and complexes. In order to further exploit the thermophilic properties to study ribosome assembly, or other eukaryotic processes in vivo, additional genetic tools are required. Recently, a transformation protocol for C. thermophilum was established, that allowed the transformation of a plasmid encoding an epitope tagged protein.74 The expression of these tagged proteins also enabled to determine the cellular localization of the tagged proteins. Moreover, in vivo expression of the tagged proteins facilitated the efficient purification of native complexes directly from C. thermophilum. Remarkably, this approach allowed the isolation of higher order subcomplexes, which could not be isolated from mesophilic organisms.74 We expect that such in vivo purifications will allow the identification of novel subcomplexes involved in ribosome biogenesis, and hence will complement the presented Y2H interaction network. Moreover, our collection provides a resource to reconstitute protein complexes from C. thermophilum in vitro. These complementary techniques represent a major step towards establishing C. thermophilum as a model organism for biochemical and structural investigations of ribosome biogenesis.
Materials and Methods
Cultivation of Chaetomium thermophilum, preparation of cDNA and genomic DNA
Cultivation of C. thermophilum was performed as previously described.25, 74 For the preparation of cDNA, approximately 100 mg of mycelium was used to extract total RNA using the “SV total RNA Isolation Kit” from Promega (Z3100). The cDNA was synthesized by the reverse transcriptase “Superscript III” (Invitrogen) according to the manufacturer's instructions. Finally, the cDNA was purified with the PCR Purification Kit (Qiagen). Genomic DNA was isolated as previously described.75
Search, annotation, and cloning of Chaetomium thermophilum genes
The protein sequences of factors involved in ribosome biogenesis from S. cerevisiae were used for a blastp search against the C. thermophilum genome resource (http://ct.bork.embl.de). If no hit was retrieved, homologues from a fungi closely related to C. thermophilum were used (e.g., N. crassa, C. globosum). The predicted protein sequences from C. thermophilum were blasted against the NCBI database, aligned with various species to determine their conservation and confirm the predicted introns (see “Results”, Supporting Information Fig. S1). Subsequently, the ORFs were PCR amplified from cDNA. However, for genes exceeding 2000 bp, fragments were amplified and subsequently fused by PCR to obtain the full ORF. If sequence analysis showed the absence of introns, fragments were amplified using genomic DNA as template. Almost all genes were PCR amplified including terminal NdeI (5′‐end) and BamHI (3′‐end) restriction sites. Internal NdeI or BamHI sites were eliminated by introducing a silent mutation. Finally, the genes were cloned into vectors pGADT7 (AmpR, 2µ, LEU2, PADH1 GAL4‐AD, HA tag, Insert, TADH1), and pGBKT7 (KanR, 2µ, TRP1, PADH1 GAL4‐BD, myc tag, Insert, TADH1) and sequenced (see Supporting Information Table S1). The NdeI/BamHI restriction sites were used for subcloning the ORF's into various S. cerevisiae and E. coli expression plasmids (see below).
Systematic protein expression and crystallization approach
Full‐length targets were cloned in modified pET15b and pET24d vectors with a N or C‐terminal (His)6‐tag (see Supporting Information Table S1), including also a TEV‐cleavage site for some of the constructs. Initially, a miniscale screen was set up for the preparation of 44 targets in parallel. Proteins were immobilized to Ni2+ NTA beads in a buffer containing 250 mM NaCl, 20 mM HEPES, 10 mM KCl, 10 mM MgCl2, 40 mM imidazol, pH 8.0, and eluted with 500 mM imidazol. Protein expression was tested in two different E. coli strains [Rosetta2 (DE3) and BL21 (DE3)] using an auto‐induction protocol.76 Protein solubility analysis was performed by SDS‐PAGE as previously described,77, 78 prior to affinity purification in batch. Additional 35 targets were included in our second screen using only the Rosetta2 (DE3) strain. Large‐scale preparations of promising targets comprised affinity purification followed by size exclusion chromatography (150 mM NaCl, 20 mM HEPES, 10 mM KCl, 10 mM MgCl2, pH 7.5). Crystallization screens were performed at 291 K by the sitting‐drop vapor‐diffusion method using two different protein concentrations (maximum protein concentration close to precipitation and a twofold dilution) across three commercial screens (JCSG+, Classics, and AmSO4). Different strategies were employed for the preparation of the most difficult targets, including construct optimization after limited proteolysis experiments, use of different expression systems including yeast or insect cells, co‐expression with protein partners, and use of carrier‐driven crystallization strategies.79
Crystallization and structure determination of ctYvh1 and ctTif6
The phosphatase domain (PD, aa1–218) of ctYvh1, concentrated to 25 mg/mL was crystallized in 0.2M LiNO3 and 20% PEG3350. ctTif6, was concentrated to 11 mg/mL and crystals were obtained in a condition containing 0.1M Na–citrate pH 5.6, 0.5M ammonium sulfate, and 1M lithium sulfate. For data‐collection single crystals were harvested into reservoir solution supplemented with 20% glycerol and directly transferred into a cold nitrogen stream (Oxford Cryosystems) or liquid nitrogen. Diffraction data was collected at 100 K at ESRF beamline ID23‐280 and processed with XDS81 and AIMLESS82 from the CCP4 package.83 Both structures were solved by molecular replacement with MOLREP.84 For ctTif6 the structure of Saccharomyces cerevisiae Tif6 (PDB‐ID: 1G6285) was used as search model. For ctYvh1‐PD the coordinates of the dual specificity phosphatase domain (PDB‐ID: 1WRM86) of human JSP‐1 was used. Both structures were manually rebuilt with Coot.87 Refinement was carried out with Refmac588 and PHENIX.89 Both structures contain one molecule in the asymmetric unit. Data collection and refinement statistics are summarized in Supporting Information Table S2.
Comprehensive Y2H screen
For the analysis of a Y2H interaction, the two proteins of interest were N‐terminally fused to the “DNA‐Binding Domain” (BD) or the “Transcription Activation Domain” (AD) of the Gal4 transcription factor from S. cerevisiae. When two proteins of interest interact, the AD fusion protein will be recruited to the promoter bound BD fusion protein and consequently will activate the transcription of the reporter gene [Fig. 2(A)]. The Y2H yeast reporter strain PJ69‐4 (trp1–901, leu2–3,112, ura3‐52, his3‐200, gal4Δ, gal80Δ, LYS2::GAL1–HIS3, GAL2–ADE2, met2::GAL7‐lacZ 90 has the HIS3, and the ADE2 reporter genes under the control of the GAL1 and GAL2 promotor, respectively, which allows the read out for medium (HIS3 reporter) and strong interactions (ADE2 reporter). For the large‐scale screen S. cerevisiae strain PJ69‐4 MATalpha was transformed with pGBKT7 plasmids. A mix of five transformants was cultivated and stored in 50% glycerol at −80°C. Similarly, the Y2H strain PJ69‐4 MATa (trp1‐901, leu2‐3,112, ura3‐52, his3‐200, gal4Δ, gal80Δ, LYS2::GAL1–HIS3, GAL2–ADE2, met2::GAL7–lacZ 90) was transformed with various pGADT7 plasmids and a mix of five transformants was stored in one distinct slot within a 96 well deep well plate at −80°C. The resulting library consists of two deep well plates (Block I, III) with yeast strain PJ69‐a MATa carrying the different pGADT7 plasmids, including a slot with the control plasmid pTD1‐1 (pGADT7–SV40) and empty slots as controls [Fig. 2(A)]. In addition, a second set of the library (Block II, IV) was prepared analogously with a mix of five different transformants to obtain a second dataset for validation of the interactome. The prey library was used to inoculate a deep well plate containing 96× 1 mL SDC–Leu (30°C), whereas the bait stocks were used to inoculate 20 mL SDC–Trp cultures (30°C). The stationary cultures were mated by mixing 50 µL library, 50 µL bait culture and 200 µL YPD in a sterile deep well plate. After 1 day of incubation at 30°C, the YPD was pipetted off and 1 mL SDC‐Trp‐Leu was added to select for diploid cells containing both plasmids for 2 days at 30°C. After removal of 600 µL of the supernatant medium, the cells were resuspended in the remaining medium and 10 µL were spotted on SDC‐Leu‐Trp, SDC‐Trp‐Leu‐His + 1 mM 3AT, and SDC‐Trp‐Leu‐Ade plates (Onewell plate, Greiner Germany). 3‐Amino‐1,2,4‐triazole (3AT), an inhibitor of the imidazoleglycerol‐phosphate dehydratase, the product of the HIS3 gene, was added to a final concentration of 1 mM to avoid unspecific growth on SDC–His plates. After incubation at 30°C pictures were taken after 4 and 7 days. Pipetting steps were done using a Liquidator with a volume range 5–200 µL (Mettler‐Toledo). For bait proteins that had a low mating efficiency, after 2 days selection of diploid cells, the medium was pipetted off and replaced by fresh SDC‐Trp‐Leu and incubated for additional 4 days at 30°C, before spotting cells on plates. All pictures of the dataset can be accessed under http://y2h.embl.de. Pictures from long selection procedure are marked by a red box.
Biochemical verification of the Y2H interactions
In order to confirm several of the Y2H interactions, ORFs were subcloned into expression plasmids for S. cerevisiae pMT_LEU2_pA‐TEV (PGAL1‐10, 2µ, LEU2) and pMT_TRP1_3xFlag (PGAL1‐10, 2µ, TRP1) using NdeI/BamHI restriction sites (see Supporting Information Table S1). These plasmids [as described in Ref. ( 36)] allow galactose induced expression of N‐terminal tagged proteins in the S. cerevisae strain W303 (MATalpha, ura3‐1, trp1‐1, his3‐11,15, leu2‐3,112, ade2‐1, can1‐100, GAL+). Yeast strain W303, carrying both plasmids was cultured in 30 mL SDC‐Trp‐Leu at 30°C overnight, before inoculating a 1 L YPG culture (o/n, 30°C). After harvesting and cell lysis, the supernatant was used for a split purification procedure via IgG beads (GE Healthcare), and anti‐Flag beads (Sigma) according to the TAP‐protocol described previously.18 For co‐expression of the ctUTP‐A and ctUTP‐B complex, we exploited that the PGAL1‐10 promotor induces transcription in up‐ and downstream directions, thus each plasmid can be used to express two proteins simultaneously. Thus, using a third plasmid pMT_URA (PGAL1‐10, 2µ, URA3), allows the co‐expression of six proteins. Specifically, we used pMT‐TRP1_Flag‐ctUTP15_ctUTP17, pMT_LEU2_pA‐TEV‐ctUTP10_ctUTP5 and pMT_URA3_ctUTP4_ctUTP8 for expression of the ctUTP‐A complex and pMT‐TRP1_Flag‐ctUTP13_ctUTP12, pMT_LEU2_pA‐TEV‐ctUTP21_ctUTP1 and pMT_URA3_ctUTP18_ctUTP6 for expression of the ctUTP‐B. For further information, see Ref. ( 36). Proteins of the ctUTP‐A complex were purified in a buffer containing 150 mM NaCl, 50 mM TRIS pH7.4, 1.5 mM MgCl2, 5% glycerol, and additional 0.1% IGEPAL for the lysis steps. Members of the ctUTP‐B complex were purified in a buffer containing 150 mM NaCl, 50 mM TRIS pH 7.5, 1.5 mM MgCl2, 0.075% IGEPAL, whereas ctUtp18–ctUtp21 was isolated in a buffer containing 200 mM NaCl, 50 mM TRIS pH 7.5, 1.5 mM MgCl2, 0.075% IGEPAL.
The recombinant proteins for the reconstitution of ctUtp8–ctUtp5, ctUtp8–ctUtp4, and the Brix–BAF complexes (ctRpf1–ctMak16, ctSsf1–ctRrp15, and ctBrx1–ctEbp2) were expressed in E. coli BL21 (DE3). The respective gene fragments were cloned into plasmid pET24_GST‐TEV (KanR) or pET15b_His6 (AmpR) (see Supporting Information Table S1). Transformants were cultivated in LB medium and expression was induced by adding IPTG (0.2 mM). The ctUtp8–ctUtp5, and ctUtp8–ctUtp4 pairs were coexpressed and purified using GSH beads (Macherey‐Nagel, Germany). For the reconstitution of the dimeric Brix–BAP complexes, the GST tagged protein and a GST control was bound to GSH beads, washed and then incubated for 1 h with the supernatant of the E. coli lysate containing the His6 tagged binding partner. Following a washing step, the proteins were eluted using 20 mM GSH and analyzed by SDS‐PAGE and Coomassie blue staining. In detail, GST‐ctUtp5‐His6‐ctUtp8, and GST‐ctUtp4‐His6‐ctUtp8 were induced at 23°C for 2 h using 0.2 mM IPTG. The purification was performed in a buffer containing 150 mM NaCl, 20 mM HEPES pH 7.5, 5% glycerol, and 0.01% IGEPAL. His6‐ctRpf1 (156–436 aa) and GST‐ctMak16 (134–336 aa) were induced o/n at 16°C and purified using a buffer composed of 100 mM NaCl, 50 mM KCl, 5 mM MgCl2, 30 mM HEPES pH 7.5, 3% glycerol, 0.05% IGEPAL. GST‐ctBrx1 (30–260 aa), His6‐ctEbp2 (178–268 aa) were induced for 4.5 h at 16°C and purified in a buffer containing 650 mM NaCl, 350 mM KCl, 35 mM MgCl2, 30 mM HEPES pH 7.5, 5% glycerol, 0.05% IGEPAL. GST‐ctSsf1 (22–359) and His6‐ctRrp15 (194–354 aa) were induced for 4 h at 18°C and purified in a buffer containing 225 mM NaCl, 75 mM KCl, 15 mM MgCl2, 30 mM HEPES pH 7.5, 5% glycerol, 0.05% IGEPAL.
SEC analysis of the Brix–BAF complexes was performed in the following manner: The (His)6 tagged ctRpf1 (156–436 aa) and GST tagged ctMak16 (134–336 aa) complex was purified by GST affinity chromatography. The GST tag from ctMak16 was removed by the TEV protease and finally, the complex was subjected to SEC on a S200 (16/60) column. The (His)6 tagged ctBrx1 (34–259 aa) and ctEbp2 (175–282 aa) complex was purified by Ni2+–NTA chromatography followed by ion‐exchange chromatography on a S column (GE healthcare) and finally on a S75 (26/60). The ctSsf1 (34–478 aa) and (His)6‐ZZ tagged ctRrp15 (173–354 aa) complex was purified by Ni2+–NTA chromatography followed TEV cleavage to remove ZZ tag from ctRrp15. The ctSsf1–ctRrp15 complex was further purified on a RESOURCE S column (GE Healthcare) and SEC on a S200 (26/60) column.
2D EM sample preparation and image processing
Affinity purified ctUTP‐A and ctUTP‐B complexes were loaded on a glutaraldehyde containing glycerol gradient GraFix, composed of a 200 µL cushion of 7.5% (v/v) glycerol in buffer, followed by a linear 10–30% (v/v) glycerol and 0–0.15% glutaraldehyde gradient.67 Samples were centrifuged (SW 60 Ti; Beckman Coulter) for 16 h at 33,000 rpm and 4°C before 200 µL fractions were collected and analyzed. For negative staining, 5 µL of the complex was placed on a freshly glow‐discharged, carbon‐coated grid, allowed to absorb to the carbon, washed three times with water, stained with uranyl acetate (2% w/v), and dried. Micrographs were recorded using an electron microscope (Tecnai F20; FEI) operating at 200 kV with a bottom‐mounted 4 K, high sensitivity charge‐coupled device camera (Eagle; FEI) at a nominal magnification of 29,000 for the ctUTP‐A complex (calibrated pixel size of 3.81 Å) and 50,000 for the ctUTP‐B complex (calibrated pixel size of 2.2688 Å), respectively. 5942 particles for the ctUTP‐A complex and 5000 particles for the ctUTP‐B complex were processed as described previously.91
Authors Contributions
J.B., I.S. and E.H. designed research, S.G. prepared DNA, J.B., M.K., M.G., M.K., M.T., and E.T. cloned biogenesis factors, Y.L.A, F.R.C, G.S., and S.B. were involved in protein expression, purification and structure determination, J.B., M.K., M.G. and B.B. performed Y2H screen, S.I., N.C., M.K., S.K., and M.K. did biochemical purifications of complexes, D.F. performed EM analysis, J.B., I.S. and E.H. wrote the manuscript
Structure coordinates have been deposited in pdb database (ctTif6 5M3Q, ctYvh1‐PD 5M43)
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Acknowledgments
We appreciate the following people for sharing plasmids: Rizos‐Georgios Manikas, Martin Kos, Clemont Fillias, Adriana Gigova, Miriam Sturm, Lisa Gasse, Benjamin Lau, Sebastien Fereira‐Cerca, Lyudmila Dimitrova, Helge Paternoga, and Stefan Amlacher. Thanks for technical assistance to Beate Jannack. We are grateful to Colin Combe for sharing the xiNET software, and to Hendrik Hofstadt who rewrote the software to display the Y2H dataset. Thanks to Jaime Huerta Cepas and Peer Bork for the integration of our dataset into the “Chaetomium thermophilum genome resource” at http://y2h.embl.de. We thank J. Kopp and C. Siegmann from the BZH/Cluster of Excellence: Cell Networks crystallization platform for protein crystallization, and acknowledge access to the beamlines at the European Synchrotron Radiation Facility (ESRF) in Grenoble and the support of the beamline scientists. This work was supported by the Deutsche Forschungsgemeinschaft DFG [HU363/15‐1, HU363/12‐1 to E.H.]; [BA2316/2‐1 to J.B.]; [SFB 638, Z4 to I.S.]; I.S. and E.H. are investigator of the Cluster of Excellence: CellNetworks and acknowledge support through EcTop1.
Contributor Information
Jochen Baßler, Email: jochen.bassler@bzh.uni-heidelberg.de.
Irmgard Sinning, Email: irmi.sinning@bzh.uni-heidelberg.de.
Ed Hurt, Email: ed.hurt@bzh.uni-heidelberg.de.
References
- 1. Woolford JL, Jr. , Baserga SJ (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae . Genetics 195:643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kressler D, Hurt E, Baßler J (2010) Driving ribosome assembly. Biochim Biophys Acta 1803:673–683. [DOI] [PubMed] [Google Scholar]
- 3. Strunk BS, Karbstein K (2009) Powering through ribosome assembly. RNA 15:2083–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues‐Ferrer M, Mougin A, Henry Y (2008) The post‐transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65:2334–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Granneman S, Baserga SJ (2004) Ribosome biogenesis: of knobs and RNA processing. Exp Cell Res 296:43–50. [DOI] [PubMed] [Google Scholar]
- 6. Fromont‐Racine M, Senger B, Saveanu C, Fasiolo F (2003) Ribosome assembly in eukaryotes. Gene 313:17–42. [DOI] [PubMed] [Google Scholar]
- 7. Fatica A, Tollervey D (2002) Making ribosomes. Curr Opin Cell Biol 14:313–318. [DOI] [PubMed] [Google Scholar]
- 8. Brighenti E, Trere D, Derenzini M. 2015. Targeted cancer therapy with ribosome biogenesis inhibitors: A real possibility? Oncotarget. 6:38617–38627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD (2014) Targeting the nucleolus for cancer intervention. Biochim Biophys Acta 1842:802–816. [DOI] [PubMed] [Google Scholar]
- 10. Burger K, Muhl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, Kellner M, Gruber‐Eber A, Kremmer E, Holzel M, Eick D. 2010. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 285:12416–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schäfer T, Kuster B, Tschochner H, Tollervey D, Gavin AC, Hurt E. 2002. 90s pre‐ribosomes include the 35s pre‐rrna, the u3 snornp, and 40s subunit processing factors but predominantly lack 60s synthesis factors. Mol Cell. 10:105–115. [DOI] [PubMed] [Google Scholar]
- 12. Bassler J, Grandi P, Gadal O, Leßmann T, Tollervey D, Lechner J, Hurt EC (2001) Identification of a 60s pre‐ribosomal particle that is closely linked to nuclear export. Mol Cell 8:517–529. [DOI] [PubMed] [Google Scholar]
- 13. Nissan TA, Bassler J, Petfalski E, Tollervey D, Hurt EC (2002) 60s pre‐ribosome formation viewed from assembly in the nucleolus until export to the cytoplasm. EMBO J 21:5539–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fatica A, Cronshaw AD,MD, Tollervey D (2002) Ssf1p prevents premature processing of an early pre‐60s ribosomal particle. Mol Cell 9:341–351. [DOI] [PubMed] [Google Scholar]
- 15. Harnpicharnchai P, Jakovljevic J, Horsey E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, Shabanowitz J, Hunt DF, Woolford JL. 2001. Composition and functional characterization of yeast 66s ribosome assembly intermediates. Mol Cell. 8:505–515. [DOI] [PubMed] [Google Scholar]
- 16. Schäfer T, Strauss D, Petfalski E, Tollervey D, Hurt EC (2003) The path from nucleolar 90s to cytoplasmic 40s pre‐ribosomes. EMBO J 22:1370–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weis F, Giudice E, Churcher M, Jin L, Hilcenko C, Wong CC, Traynor D, Kay RR, Warren AJ (2015) Mechanism of eif6 release from the nascent 60s ribosomal subunit. Nat Struct Mol Biol 22:914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bradatsch B, Leidig C, Granneman S, Gnadig M, Tollervey D, Bottcher B, Beckmann R, Hurt E (2012) Structure of the pre‐60s ribosomal subunit with nuclear export factor arx1 bound at the exit tunnel. Nat Struct Mol Biol 19:1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leidig C, Thoms M, Holdermann I, Bradatsch B, Berninghausen O, Bange G, Sinning I, Hurt E, Beckmann R (2014) 60s ribosome biogenesis requires rotation of the 5s ribonucleoprotein particle. Nat Commun 5:3491. [DOI] [PubMed] [Google Scholar]
- 20. Barrio‐Garcia C, Thoms M, Flemming D, Kater L, Berninghausen O, Bassler J, Beckmann R, Hurt E (2016) Architecture of the rix1‐rea1 checkpoint machinery during pre‐60s‐ribosome remodeling. Nat Struct Mol Biol 23:37–44. [DOI] [PubMed] [Google Scholar]
- 21. Greber BJ, Gerhardy S, Leitner A, Leibundgut M, Salem M, Boehringer D, Leulliot N, Aebersold R, Panse VG, Ban N (2016) Insertion of the biogenesis factor rei1 probes the ribosomal tunnel during 60s maturation. Cell 164:91–102. [DOI] [PubMed] [Google Scholar]
- 22. Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL, 3rd , Karbstein K, Skiniotis G (2011) Ribosome assembly factors prevent premature translation initiation by 40s assembly intermediates. Science 333:1449–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu S, Tutuncuoglu B, Yan K, Brown H, Zhang Y, Tan D, Gamalinda M, Yuan Y, Li Z, Jakovljevic J, Ma C, Lei J, Dong MQ, Woolford JL, Jr. , Gao N. 2016. Diverse roles of assembly factors revealed by structures of late nuclear pre‐60s ribosomes. Nature. 534:133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kornprobst M, Turk M, Kellner N, Cheng J, Flemming D, Kos‐Braun I, Kos M, Thoms M, Berninghausen O, Beckmann R, Hurt E. 2016. Architecture of the 90s pre‐ribosome: A structural view on the birth of the eukaryotic ribosome. Cell. 166:380–393. [DOI] [PubMed] [Google Scholar]
- 25. Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E (2011) Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146:277–289. [DOI] [PubMed] [Google Scholar]
- 26. Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T (2016) Architecture of human mtor complex 1. Science 351:48–52. [DOI] [PubMed] [Google Scholar]
- 27. Hondele M, Stuwe T, Hassler M, Halbach F, Bowman A, Zhang ET, Nijmeijer B, Kotthoff C, Rybin V, Amlacher S, Hurt E, Ladurner AG. 2013. Structural basis of histone h2a‐h2b recognition by the essential chaperone fact. Nature. 499:111–114. [DOI] [PubMed] [Google Scholar]
- 28. Jiao L, Liu X (2015) Structural basis of histone h3k27 trimethylation by an active polycomb repressive complex 2. Science 350:aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin Z, Luo X, Yu H. 2016. Structural basis of cohesin cleavage by separase. Nature. 532:131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bozkurt G, Stjepanovic G, Vilardi F, Amlacher S, Wild K, Bange G, Favaloro V, Rippe K, Hurt E, Dobberstein B, Sinning I. 2009. Structural insights into tail‐anchored protein binding and membrane insertion by get3. Proc Natl Acad Sci U S A. 106:21131–21136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leidig C, Bange G, Kopp J, Amlacher S, Aravind A, Wickles S, Witte G, Hurt E, Beckmann R, Sinning I (2013) Structural characterization of a eukaryotic chaperone—The ribosome‐associated complex. Nat Struct Mol Biol 20:23–28. [DOI] [PubMed] [Google Scholar]
- 32. Kressler D, Bange G, Ogawa Y, Stjepanovic G, Bradatsch B, Pratte D, Amlacher S, Strauss D, Yoneda Y, Katahira J, Sinning I, Hurt E. 2012. Synchronizing nuclear import of ribosomal proteins with ribosome assembly. Science. 338:666–671. [DOI] [PubMed] [Google Scholar]
- 33. Bock T, Chen WH, Ori A, Malik N, Silva‐Martin N, Huerta‐Cepas J, Powell ST, Kastritis PL, Smyshlyaev G, Vonkova I, Kirkpatrick J, Doerks T, Nesme L, Bassler J, Kos M, Hurt E, Carlomagno T, Gavin AC, Barabas O, Muller CW, van Noort V, Beck M, Bork P. 2014. An integrated approach for genome annotation of the eukaryotic thermophile chaetomium thermophilum. Nucleic Acids Res. 42:13525–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu Y, Hirsch JP (1995) An essential gene pair in Saccharomyces cerevisiae with a potential role in mating. DNA Cell Biol 14:411–418. [DOI] [PubMed] [Google Scholar]
- 35. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. http://www.jalview.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thierbach K, von Appen A, Thoms M, Beck M, Flemming D, Hurt E (2013) Protein interfaces of the conserved nup84 complex from Chaetomium thermophilum shown by crosslinking mass spectrometry and electron microscopy. Structure 21:1672–1682. [DOI] [PubMed] [Google Scholar]
- 37. Fischer J, Teimer R, Amlacher S, Kunze R, Hurt E (2015) Linker nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nat Struct Mol Biol 22:774–781. [DOI] [PubMed] [Google Scholar]
- 38. Bassler J, Paternoga H, Holdermann I, Thoms M, Granneman S, Barrio‐Garcia C, Nyarko A, Stier G, Clark SA, Schraivogel D, Kallas M, Beckmann R, Tollervey D, Barbar E, Sinning I, Hurt E. 2014. A network of assembly factors is involved in remodeling rrna elements during preribosome maturation. J Cell Biol. 207:481–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thoms M, Ahmed YL, Maddi K, Hurt E, Sinning I (2015) Concerted removal of the erb1‐ytm1 complex in ribosome biogenesis relies on an elaborate interface. Nucleic Acids Res. [DOI] [PMC free article] [PubMed]
- 40. Stelter P, Huber FM, Kunze R, Flemming D, Hoelz A, Hurt E (2015) Coordinated ribosomal l4 protein assembly into the pre‐ribosome is regulated by its eukaryote‐specific extension. Mol Cell 58:854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pausch P, Singh U, Ahmed YL, Pillet B, Murat G, Altegoer F, Stier G, Thoms M, Hurt E, Sinning I, Bange G, Kressler D. 2015. Co‐translational capturing of nascent ribosomal proteins by their dedicated chaperones. Nature communications. 6:7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferreira‐Cerca S, Sagar V, Schafer T, Diop M, Wesseling AM, Lu H, Chai E, Hurt E, LaRonde‐LeBlanc N (2012) Atpase‐dependent role of the atypical kinase rio2 on the evolving pre‐40s ribosomal subunit. Nat Struct Mol Biol 19:1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Noort V, Bradatsch B, Arumugam M, Amlacher S, Bange G, Creevey C, Falk S, Mende DR, Sinning I, Hurt E, Bork P. 2013. Consistent mutational paths predict eukaryotic thermostability. BMC Evol Biol. 13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monecke T, Haselbach D, Voss B, Russek A, Neumann P, Thomson E, Hurt E, Zachariae U, Stark H, Grubmuller H, Dickmanns A, Ficner R. 2013. Structural basis for cooperativity of crm1 export complex formation. Proc Natl Acad Sci U S A. 110:960–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aibara S, Valkov E, Lamers MH, Dimitrova L, Hurt E, Stewart M (2015) Structural characterization of the principal mrna‐export factor mex67‐mtr2 from chaetomium thermophilum. Acta Crystallogr F71:876–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. 2002. Structural genomics of the thermotoga maritima proteome implemented in a high‐throughput structure determination pipeline. Proc Natl Acad Sci U S A. 99:11664–11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christendat D, Yee A, Dharamsi A, Kluger Y, Savchenko A, Cort JR, Booth V, Mackereth CD, Saridakis V, Ekiel I, Kozlov G, Maxwell KL, Wu N, McIntosh LP, Gehring K, Kennedy MA, Davidson AR, Pai EF, Gerstein M, Edwards AM, Arrowsmith CH. 2000. Structural proteomics of an archaeon. Nat Struct Biol. 7:903–909. [DOI] [PubMed] [Google Scholar]
- 48. Eswaran J, Knapp S (2010) Insights into protein kinase regulation and inhibition by large scale structural comparison. Biochim Biophys Acta 1804:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess‐Brown NA, Muller S, Knapp S (2009) Large‐scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCann KL, Charette JM, Vincent NG, Baserga SJ (2015) A protein interaction map of the lsu processome. Genes Dev 29:862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Combe CW, Fischer L, Rappsilber J (2015) Xinet: crosslink network maps with residue resolution. Mol Cell Proteom 14:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Granneman S, Gallagher JE, Vogelzangs J, Horstman W, van Venrooij WJ, Baserga SJ, Pruijn GJ (2003) The human imp3 and imp4 proteins form a ternary complex with hmpp10, which only interacts with the u3 snorna in 60–80s ribonucleoprotein complexes. Nucleic Acids Res 31:1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perez‐Fernandez J, Roman A, De Las Rivas J, Bustelo XR, Dosil M (2007) The 90s preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol Cell Biol 27:5414–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wegierski T, Billy E, Nasr F, Filipowicz W (2001) Bms1p, a g‐domain‐containing protein, associates with rcl1p and is required for 18s rRNA biogenesis in yeast. RNA 7:1254–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karkusiewicz I, Rempola B, Gromadka R, Grynberg M, Rytka J (2004) Functional and physical interactions of faf1p, a Saccharomyces cerevisiae nucleolar protein. Biochem Biophys Res Commun 319:349–357. [DOI] [PubMed] [Google Scholar]
- 56. Zheng S, Lan P, Liu X, Ye K (2014) Interaction between ribosome assembly factors krr1 and faf1 is essential for formation of small ribosomal subunit in yeast. J Biol Chem 289:22692–22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li L, Ye K (2006) Crystal structure of an h/aca box ribonucleoprotein particle. Nature 443:302–307. [DOI] [PubMed] [Google Scholar]
- 58. Hierlmeier T, Merl J, Sauert M, Perez‐Fernandez J, Schultz P, Bruckmann A, Hamperl S, Ohmayer U, Rachel R, Jacob A, Hergert K, Deutzmann R, Griesenbeck J, Hurt E, Milkereit P, Bassler J, Tschochner H. 2013. Rrp5p, noc1p and noc2p form a protein module which is part of early large ribosomal subunit precursors in s. Cerevisiae. Nucleic Acids Res. 41:1191–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Castle CD, Sardana R, Dandekar V, Borgianini V, Johnson AW, Denicourt C (2013) Las1 interacts with grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae . Nucleic Acids Res 41:1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gasse L, Flemming D, Hurt E (2015) Coordinated ribosomal its2 RNA processing by the las1 complex integrating endonuclease, polynucleotide kinase, and exonuclease activities. Mol Cell 60:808–815. [DOI] [PubMed] [Google Scholar]
- 61. Liu Q, Greimann JC, Lima CD (2006) Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127:1223–1237. [DOI] [PubMed] [Google Scholar]
- 62. Thoms M, Thomson E, Baßler J, Gnädig M, Griesel S, Hurt E (2015) The exosome is recruited to RNA substrates through specific adaptor proteins. Cell 162:1029–1038. [DOI] [PubMed] [Google Scholar]
- 63. Poll G, Li S, Ohmayer U, Hierlmeier T, Milkereit P, Perez‐Fernandez J (2014) In vitro reconstitution of yeast tUTP/UTP‐A and UTP-B subcomplexes provides new insights into their modular architecture. PloS One 9:e114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, Luo SK, Ding YH, Wang LH, Hao Z, Xiu LY, Chen S, Ye K, He SM, Dong MQ. 2012. Identification of cross‐linked peptides from complex samples. Nat Methods. 9:904–906. [DOI] [PubMed] [Google Scholar]
- 65. Zhang C, Sun Q, Chen R, Chen X, Lin J, Ye K (2016) Integrative structural analysis of the UTPB complex, an early assembly factor for eukaryotic small ribosomal subunits. Nucleic Acids Res 44:7475–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hunziker M, Barandun J, Petfalski E, Tan D, Delan‐Forino C, Molloy KR, Kim KH, Dunn‐Davies H, Shi Y, Chaker‐Margot M, et al. (2016) UtpA and UtpB chaperone nascent pre‐ribosomal rna and u3 snorna to initiate eukaryotic ribosome assembly. Nat Commun 7:12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, Uchtenhagen H, Urlaub H, Herzog F, Peters JM, Poerschke D, Luhrmann R, Stark H. 2008. Grafix: Sample preparation for single‐particle electron cryomicroscopy. Nat Methods. 5:53–55. [DOI] [PubMed] [Google Scholar]
- 68. Asano N, Kato K, Nakamura A, Komoda K, Tanaka I, Yao M (2015) Structural and functional analysis of the rpf2–rrs1 complex in ribosome biogenesis. Nucleic Acids Res 43:4746–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kharde S, Calvino FR, Gumiero A, Wild K, Sinning I (2015) The structure of rpf2–rrs1 explains its role in ribosome biogenesis. Nucleic Acids Res 43:7083–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Madru C, Lebaron S, Blaud M, Delbos L, Pipoli J, Pasmant E, Rety S, Leulliot N (2015) Chaperoning 5s RNA assembly. Genes Dev 29:1432–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shimoji K, Jakovljevic J, Tsuchihashi K, Umeki Y, Wan K, Kawasaki S, Talkish J, Woolford JL, Jr. , Mizuta K (2012) Ebp2 and brx1 function cooperatively in 60s ribosomal subunit assembly in Saccharomyces cerevisiae . Nucleic Acids Res 40:4574–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane‐Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M. 2008. High‐quality binary protein interaction map of the yeast interactome network. Science. 322:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee SJ, Baserga SJ (1999) Imp3p and imp4p, two specific components of the u3 small nucleolar ribonucleoprotein that are essential for pre‐18s rRNA processing. Mol Cell Biol 19:5441–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kellner N, Schwarz J, Sturm M, Fernandez‐Martinez J, Griesel S, Zhang W, Chait BT, Rout MP, Kuck U, Hurt E (2016) Developing genetic tools to exploit Chaetomium thermophilum for biochemical analyses of eukaryotic macromolecular assemblies. Sci Rep 6:20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Al‐Samarrai TH, Schmid J (2000) A simple method for extraction of fungal genomic DNA. Lett Appl Microbiol 30:53–56. [DOI] [PubMed] [Google Scholar]
- 76. Studier FW (2005) Protein production by auto‐induction in high density shaking cultures. Prot Expr Purif 41:207–234. [DOI] [PubMed] [Google Scholar]
- 77. Berrow NS, Bussow K, Coutard B, Diprose J, Ekberg M, Folkers GE, Levy N, Lieu V, Owens RJ, Peleg Y, Pinaglia C, Quevillon‐Cheruel S, Salim L, Scheich C, Vincentelli R, Busso D. 2006. Recombinant protein expression and solubility screening in escherichia coli: A comparative study. Acta crystallographica Section D, Biological crystallography. 62:1218–1226. [DOI] [PubMed] [Google Scholar]
- 78. Sala E, de Marco A (2010) Screening optimized protein purification protocols by coupling small‐scale expression and mini‐size exclusion chromatography. Prot Expr Purif 74:231–235. [DOI] [PubMed] [Google Scholar]
- 79. Corsini L, Hothorn M, Scheffzek K, Sattler M, Stier G (2008) Thioredoxin as a fusion tag for carrier‐driven crystallization. Protein Sci 17:2070–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Flot D, Mairs T, Giraud T, Guijarro M, Lesourd M, Rey V, van Brussel D, Morawe C, Borel C, Hignette O, Chavanne J, Nurizzo D, McSweeney S, Mitchell E. 2010. The id23‐2 structural biology microfocus beamline at the esrf. J Synchrotron Radiat. 17:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kabsch W (2010) Xds. Acta Crystallogr D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the ccp4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vagin A, Teplyakov A (2010) Molecular replacement with molrep. Acta Crystallogr D66:22–25. [DOI] [PubMed] [Google Scholar]
- 85. Groft CM, Beckmann R, Sali A, Burley SK (2000) Crystal structures of ribosome anti‐association factor if6. Nat Struct Biol 7:1156–1164. [DOI] [PubMed] [Google Scholar]
- 86. Yokota T, Nara Y, Kashima A, Matsubara K, Misawa S, Kato R, Sugio S (2007) Crystal structure of human dual specificity phosphatase, jnk stimulatory phosphatase‐1, at 1.5 a resolution. Proteins 66:272–278. [DOI] [PubMed] [Google Scholar]
- 87. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of coot. Acta Crystallogr D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. Phenix: A comprehensive python‐based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. James P, Halladay J, Craig EA (1996) Genomic libraries and a host strain designed for highly efficient two‐hybrid selection in yeast. Genetics 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dimitrova L, Valkov E, Aibara S, Flemming D, McLaughlin SH, Hurt E, Stewart M (2015) Structural characterization of the Chaetomium thermophilum trex‐2 complex and its interaction with the mrna nuclear export factor mex67:Mtr2. Structure 23:1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.