

Abstract
The intermediate filament protein keratin 8 (K8) interacts with the nucleotide‐binding domain 1 (NBD1) of the cystic fibrosis (CF) transmembrane regulator (CFTR) with phenylalanine 508 deletion (ΔF508), and this interaction hampers the biogenesis of functional ΔF508‐CFTR and its insertion into the plasma membrane. Interruption of this interaction may constitute a new therapeutic target for CF patients bearing the ΔF508 mutation. Here, we aimed to determine the binding surface between these two proteins, to facilitate the design of the interaction inhibitors. To identify the NBD1 fragments perturbed by the ΔF508 mutation, we used hydrogen–deuterium exchange coupled with mass spectrometry (HDX‐MS) on recombinant wild‐type (wt) NBD1 and ΔF508‐NBD1 of CFTR. We then performed the same analysis in the presence of a peptide from the K8 head domain, and extended this investigation using bioinformatics procedures and surface plasmon resonance, which revealed regions affected by the peptide binding in both wt‐NBD1 and ΔF508‐NBD1. Finally, we performed HDX‐MS analysis of the NBD1 molecules and full‐length K8, revealing hydrogen‐bonding network changes accompanying complex formation. In conclusion, we have localized a region in the head segment of K8 that participates in its binding to NBD1. Our data also confirm the stronger binding of K8 to ΔF508‐NBD1, which is supported by an additional binding site located in the vicinity of the ΔF508 mutation in NBD1.
Keywords: cystic fibrosis, CFTR, keratin 8, NBD1, hydrogen‐deuterium exchange mass spectrometry, protein structure
Introduction
Cystic fibrosis (CF) is the most common lethal autosomal recessive disease. It is caused by mutations in the gene encoding the CF transmembrane regulator (CFTR), and 90% of CF patients harbor the phenylalanine 508 deletion (ΔF508) at least in one CFTR allele (http://www.genet.sickkids.on.ca/cftr). Although CFTR belongs to the ATP‐binding cassette (ABC) transporter family, it acts as a cAMP‐dependent passive anion channel that is regulated by ATP binding and hydrolysis, rather than as an active transporter of large molecules.1 CFTR is a multidomain single‐chain glycoprotein comprising two cytosolic nucleotide‐binding domains (NBD1 and NBD2), and two membrane‐spanning domains (MSD1 and MSD2) that each include six transmembrane α‐helices linked by intracellular loops (ICL1‐4). CFTR also contains the intrinsically disordered regulatory domain R, which is a primary site of protein kinase‐mediated anion channel regulation.2
As in all members of the ABC transporter family, NBD1 of CFTR comprises three main subdomains: (i) ABCβ, an N‐terminal β‐sheet subdomain containing the ATP‐binding site; (ii) ABCα, an α‐helical subdomain containing F508; and (iii) a central α/β core, analogous to the F1‐type ATPase, that contains a six‐stranded largely parallel β‐sheet.3 The NBD1 of CFTR also contains several unique regions, including a 32‐amino‐acid insert between β‐strands 1 and 2 of the ABCβ subdomain, called the regulatory insertion (RI; residues 402–438), and a 32‐amino‐acid extension at the C‐terminus called the regulatory extension (RE; residues 638–676) (Fig. 1). These unique segments each contain several sites susceptible to PKA phosphorylation, and are involved in CFTR trafficking and in the regulation of its Cl− channel function.3, 4, 5, 6 Both the RI and RE show secondary structure preferences in crystallographic structures of human NBD1 deposited in the protein data bank (PDB IDs: 2BBO, 1XMJ, 2BBS, and 2BBT); however, these fragments exhibit a high B factor, suggesting that they are highly flexible. The ABCα subdomain contains the so‐called structurally diverse region (SDR; L526–T547) that exhibits different conformations for each protein structure found in protein data bank. The secondary structural elements suggested in this fragment by crystallography most likely appear only due to artificial contacts that occur during crystal formation. Prior studies have provided only limited information about CFTR and its domains. We particularly lack knowledge of the complexes formed in solution where the protein retains its native dynamic character in selected regions that are not in agreement with the kinetic snapshots obtained by X‐ray crystallography.6
Figure 1.
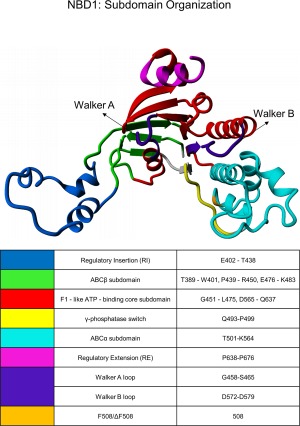
Subdomain organization of the NBD1 domain in its ATP‐bound state, as previously defined by Lewis et al.6
F508 deletion does not dramatically affect NBD1 structure but rather interferes with CFTR maturation and delivery to the plasma membrane.7 This mutation decreases the intrinsically low folding efficiency of CFTR from 20–40% to ∼0.4%. The residual amount of mutated CFTR that reaches the plasma membrane exhibits gating defects but can still exert its function.2
Keratins 8/18 are intermediate filament (IF) proteins characteristic of simple epithelial cells, serving as scaffolding elements and mechanical stress absorbers.8 Keratin IFs occur in nature as obligate heterodimers of two distinct sequence‐related proteins distinguished as type I (keratin 18) and type II (keratin 8, K8) keratins.9 Like all other IF proteins, K8 shares a tripartite organization of non‐α‐helical amino‐terminal head and carboxy‐terminal tail domains of variable length flanking a central α‐helical coil domain. While the head and the tail domains are known to vary considerably in size and composition, the structural organization of the rod domain is highly conserved and is ∼310 amino acids in length, exhibiting a seven‐residue (heptad), and eleven‐residue (hendecad) repeat pattern.10 The K8 rod domain is further subdivided into four regions, coil 1A, coil 1B, coil 2A, and coil 2B, by short unstructured linker regions (designated L1, L12, and L2, respectively).11, 12, 13 Under in vitro conditions, K8 exists as a dimer in the absence of its partner, K18.14, 15
The IFs appear to play a role in the trafficking of both wild‐type (wt)‐CFTR and ΔF508‐CFTR. The IFs: K8 and keratin 18 (K18) prevent the delivery of functional ΔF508‐CFTR to the plasma membrane.16, 17, 18 K18 reportedly binds to the CFTR C‐terminus, regulating its function.19 We recently demonstrated that K8, but not K18, physically binds to the NBD1 of both wt and ΔF508 CFTR,20 with a greater tendency to target the unfolded destabilized conformation caused by F508 deletion. K8 colocalized with ΔF508‐CFTR, but not with wt‐CFTR, in primary cultured human respiratory cells from CF patients bearing the ΔF508 mutation, but not from healthy individuals. Importantly, disruption of this interaction restored CFTR‐dependent chloride transport in cell lines expressing ΔF508‐CFTR and in ΔF508/ΔF508 mice.20
This study aimed to attain further insight into the molecular structure involved in the interaction of K8 with wt‐NBD1/ΔF508‐NBD1. To this end, we used hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS) and surface plasmon resonance (SPR) to investigate the interface between these proteins. The HDX‐MS technique is based on the fact that the rate of exchange of amide hydrogens for deuterons in the solvent reflects the level of structural flexibility or the protein fragment's exposure to solvent. This technique also allows examination of the site of interaction between biomolecules. Due to changes in hydrogen bond stabilities or sequestration from the solvent, amide hydrogens in the complex interface may undergo retarded exchange compared to the separated components. We used SPR to map which K8 fragments bound to either wt‐NBD1 or ΔF508‐NBD1. Our results enabled the identification of a region of K8 that binds to both wt‐NBD1 and ΔF508‐NBD1, and defined a secondary binding region in NBD1 that supports the stronger binding of ΔF508‐NBD1 to K8.
Results
HDX‐MS reveals regions of stability in NBDs
In the preliminary experiments, we obtained HDX exchange patterns for the human recombinant wt‐NBD1 and ΔF508‐NBD1, which both contained an additional single solubilization mutation (1S) in the linker between the ABCβ and ABCα subdomains (F494N). This 1S mutation did not stabilize the NBD1 structure and only marginally reverted the ΔF508‐CFTR structural defect, justifying its use for structural studies.21 Figure 1 depicts the subdomain organization of the CFTR‐NBD1 as previously defined [see Fig. 1(b) in the study of Lewis et al.6]. Figure 2 shows the HDX patterns for wt‐NBD1 and ΔF508‐NBD1 for 10 s (black) and 1 minute (orange) in D2O. Only these shorter incubation times are shown because ΔF508‐NBD1 showed limited stability/stronger aggregation tendencies over longer incubation periods at room temperature. Additionally, after 20 min of incubation, peptides of the NBDs showed >50% exchange along almost the entire length (data not shown), indicating a high overall level of dynamics in both NBD1 variants.
Figure 2.
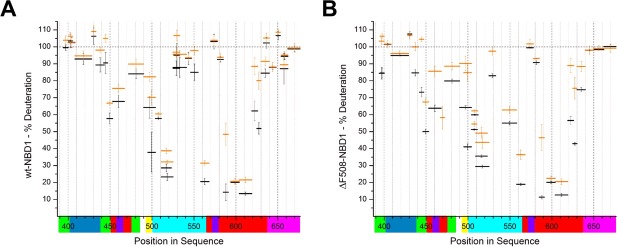
Relative deuterium uptake (% deuteration) in peptic peptides from wt‐NBD1 (A) and ΔF508‐NBD1 (B). Data are shown for the selected shortest peptides that are most representative of the entire protein sequence. Each peptide's position in the sequence is shown on the X‐axis, represented by a horizontal bar with length proportional to the peptide's length. The bar's position on the vertical axis marks the fraction exchanged after 10 s (black) and 1 min (orange) of incubation. Values close to 100% indicate fully unprotected regions, that is, regions of high flexibility. Values close to 0% indicate protected regions, that is, more stable structures. Y‐Axis error bars represent standard deviations calculated from three independent experiments. The colored bars below the X‐axis represent the subdomain organization as defined in Figure 1.
Comparison of the exchange patterns for wt‐NBD1 and ΔF508‐NBD1 revealed high similarity of fast‐ and slow‐exchanging regions. Figure 1 shows the correspondence between the position in the sequence and the structural elements, which are color‐coded on the horizontal axis. The fastest levels of exchange were registered for the regulatory subdomains of the NBDs: RI (E402–T438; Fig. 1, blue) and regulatory extension (RE; P638–P676; Fig. 1, magenta), which are positioned at the N‐ and C‐termini of NBD1. The N‐terminus itself (residues 389–401) was also readily exchangeable, although this region forms a β‐sheet constituting the ABCβ subdomain in the crystal structure. Fast exchange was also exhibited by three other regions within the central part of this domain, which were intertwined with those of greater stability. These fast‐exchanging regions included the ABCβ subdomain (389–401, 470–490; Fig. 1, green), the central region of the ABCα subdomain that corresponds to the structurally diverse region (SDR; L526–T547), and the Walker B loop region (D572–D579; Fig. 1, violet).
The highest stability was observed for the following regions: the N‐termini (residues 495–525) and C‐termini (559–564) of the ABCα subdomain (Fig. 1, cyan), and residues 585–630 from the F1‐like ATP‐binding core subdomain (Fig. 1, red). Moderate levels of exchange were noted for other regions, including part of the F1‐like ATP‐binding core subdomain (G451–L475), which also encompasses the Walker A loop. Previous studies suggest that the Walker A loop physically binds to ATP molecules and the moderate protection within this region reflects the flexibility of the ATP‐binding loop. Figure 3 shows the levels of exchange from Figure 2 (corresponding to different NBD1 subdomains) overlaid on the NBD1 model derived from Mornon et al. (before molecular dynamics simulation, CFTRInitial.pdb).22 Color codes are used to depict different exchange levels at 10 s, from the most protected (violet, slowest exchange) to the least protected (red, fastest exchange). Figure 3 clearly illustrates that regions of higher stability form a single common protein core, with a relatively stable region encompassing the F508 mutation site.
Figure 3.
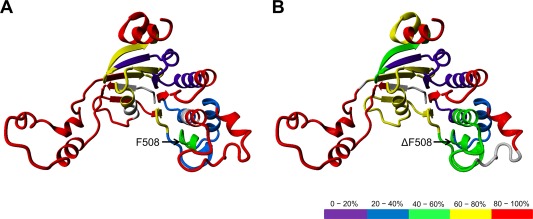
Overlay of the level of exchange after 10 s of incubation (as indicated in black in Fig. 2) on the NBD1 model22 for wt‐NBD1 (A) and ΔF508‐NBD1 (B). Red: >80% of exchange after 10 s (the least stable regions), Yellow: 60–80%; Green: 40–60%; Blue: 20–40%; Violet: <20% (the most stable regions). The regions with no sequence coverage are represented in gray.
Figure 4 shows the differences in the HDX between wt‐NBD1 and ΔF508‐NBD1 with incubation times of 10 seconds (black) and 1 minute (orange). The exchange pattern was largely similar between wt‐NBD1 and ΔF508‐NBD1 [Fig. 4(A)], although some regions showed slight differences in exchange level between the two variants. Differences revealed at 10 s of incubation indicate changes in dynamic regions, while differences revealed after 1 min of incubation reveal changes in more structured regions. Figure 4(B) shows an overlay of the differences of >10% in the NBD1 model. A region preceding the ΔF508 mutation site (496–504) was more stable in the wt‐NBD1 [Fig. 4(B), red; Supporting Information Fig. S1]. The peptides around the ΔF508 mutation site (505–525) showed a different pattern of pepsin proteolytic cleavage in ΔF508‐NBD1, indicating structural differences in the deletion region (Supporting Information Table S1). The first two segments of the ABCβ subdomain (389–401 and 439–450), and region 615–640 of the F1‐like core subdomain, showed slightly increased stability in ΔF508‐NBD1 [Fig. 4(B), blue].
Figure 4.
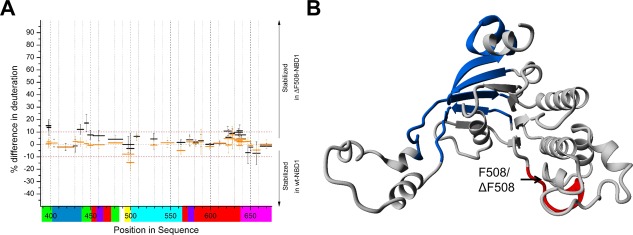
A: Difference in the fraction of exchange (% deuteration) between wt‐NBD1 and ΔF508‐NBD1, after 10 s (black) and 1 min (orange) of incubation. Peptides with differences of >10% are considered more stable in ΔF508‐NBD1, while those with differences below −10% are considered more stable in wt‐NBD1. The colored bars below the X‐axis represent the subdomain organization as defined in Figure 1. B: Overlay of the differences in the NBD1 model. Blue: regions more stable in ΔF508‐NBD1; Red: regions more stable in wt‐NBD1. The ΔF508 mutation caused a shift in protease cleavage pattern between regions 505 and 525. Therefore, we could not compare the peptides from this region between wt‐NBD1 and ΔF508‐NBD1 (see Supporting Information Table S1).
Identification of K8 fragments that interact with NBD1
To identify the NBD1–K8 binding region, we first conducted bioinformatic and literature searches for possible interaction regions. Candidate K8 peptides potentially responsible for interaction with NBD1 were selected according to the following rationale. First, the majority of proteins that interact with IFs target the head domain (desmoplakin,23, 24, 25 plectin,26 fimbrin,27 and S100α/β28). Second, antibodies directed against the N‐terminal fragment of K8 (but not against its C‐terminus) introduced into ΔF508‐CFTR‐expressing HeLa cells reportedly lead to the functional expression of mutated CFTR.29 Third, the dynamic properties of K8 displayed several fragments with high flexibility, with the head region among the regions of highest flexibility.15 Based on these observations, three peptides from the head region and a short N‐terminal fragment of coil 1A of K8 13 were selected for further interaction studies and synthesized:
Peptide 1: Ac‐N83IQAVRTQEKEQIKTLNNKFA SF105‐NH2
Peptide 2: Ac‐T67VNQSLLSPLVLEVDPNIQ85‐NH2
Peptide 3: Ac‐S27GPGSRISSSSFSRVGSSNFRGG LGGG53‐NH2
Peptide 2 was not soluble (50 mM Tris‐HCl, pH 8.4; 150 mM NaCl) and was not investigated further. Peptides 1 and 3 were mixed with NBD1 variants, and the changes in HDX pattern were assessed. A region covering positions 429–433 [Fig. 5(A,B); Supporting Information Fig. S2(A), S2(B)], corresponding to part of the RI subdomain, was commonly stabilized by Peptide 1 in both wt‐NBD1 and ΔF508‐NBD1. The effect was small but reproducible, and was slightly stronger in ΔF508‐NBD1 [Fig. 5(B); Supporting Information Fig. S2(A), panel v] than in wt‐NBD1 [Fig. 5(A); Supporting Information Fig. S2(A), panel iii]. In the ΔF508‐NBD1–Peptide 1 complex, we observed additional stabilization in the regions 400–408 from RI, and in three other peptides between positions 550–650 of the F1‐like core subdomain. This effect was absent in wt‐NBD1. However, wt‐NBD1 alone showed slight stabilization of the region 660–680. In contrast to Peptide 1, Peptide 3 did not lead to any substantial changes when incubated with either NBD1 variant [Fig. 5(C,D)]. These data suggest that Peptide 1 overlays with the K8 region that interacts with NBD1.
Figure 5.
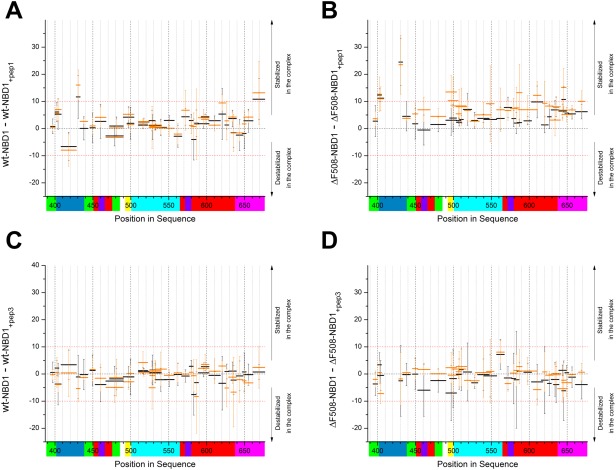
The difference in the fraction of exchange (% deuteration) in wt‐NBD1 (A, C) or ΔF508‐NBD1 (B, D) before and after interaction with K8 Peptide 1 (A, B) or K8 Peptide 3 (C, D) after 10 s (black) and 1 min (orange) of incubation. Peptides with a difference in exchange fraction of >10% were stabilized in the NBD1‐peptide complex, and those with a difference of <−10% were destabilized in this complex. The colored bars below the X‐axis represent the subdomain organization as defined in Figure 1.
Analysis of the interaction of peptide 1 with NBD1 by SPR
We also performed SPR experiments to demonstrate the direct binding interaction between Peptide 1 and wt‐NBD1/ΔF508‐NBD1 of CFTR. NBD1 (wt or mutant) was covalently attached to the dextran matrix of the CM5‐sensor chip, and we examined the binding interaction with the K8 peptide (Fig. 6). Kinetic parameters were calculated by quantitative SPR analysis for both complexes [Fig. 6(A,B)]. For the Peptide 1–wtNBD1 complex, values of k a = 73 M−1s−1, k d = 2.27 × 10−3 s−1, and = 31 μM were determined. For the Peptide 1–ΔF508‐NBD1 complex, k a = 113 M−1s−1, k d = 5.18 × 10−4 s−1 and = 4.6 μM. Thus, the K8 Peptide 1 displays a higher affinity for mutated NBD1 than for wt‐NBD1. Moreover, the dissociation rate constant (k d) for the mutated NBD1 complex was 4–fold lower than for wtNBD1, indicating that the Peptide 1–ΔF508‐NBD1 complex is more stable. The randomly scrambled K8 peptide did not interact with immobilized NBD1 [Fig. 6(C)], supporting the specificity of this interaction, and demonstrating that this binding was dependent on the peptide sequence and not the amino‐acid composition.
Figure 6.
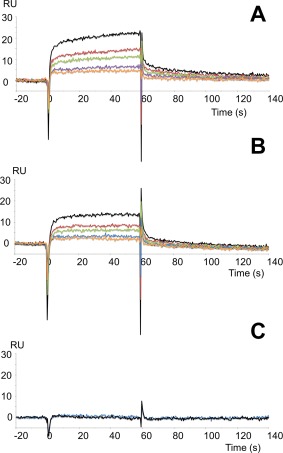
SPR analyses showed the interactions of K8 Peptide 1 with wt‐NBD1 or ΔF508‐NBD1 of CFTR that were covalently immobilized on the Biacore sensor chip. Peptide 1 was injected for 60 s for the association phase, followed by injection of running buffer alone at the same flow rate to support the dissociation phase. The response in resonance units (RUs) is plotted as a function of time (in seconds). Peptide 1 at concentrations of 370 μM (black), 277 μM (red), 185 μM (green), 138 μM (purple), and 69 μM (orange) was injected on a flow‐cell with immobilized wt‐NBD1 (A) or immobilized ΔF508‐NBD1 (B). C: Randomly scrambled K8 peptide at a concentration of 280 μM (blue) was injected on a flow‐cell with immobilized wt‐NBD1. Injection of running buffer alone is shown in black.
Binding with K8 causes allosteric changes in ΔF508‐NBD1
When we added K8 itself (rather than the head peptides) to the NBD1 preparations, we observed a more widespread pattern of changes. Figure 7 summarizes these results for wt‐NBD1 and ΔF508‐NBD1. Binding of full‐length K8 induced changes along the whole sequence of ΔF508‐NBD1, indicating either an alternate binding mode with Peptide 1 region in the context of the full‐length protein, or the existence of alternative binding regions. For the wt‐NBD1–K8 complex, we observed only a small shift in stability in the 429–433 peptide, smaller than observed in complex with Peptide 1 [panel (iii) in Supporting Information Fig. S2(A), S2(B)]. Interestingly, certain short regions in the wt‐NBD1–K8 complex underwent destabilization [Fig. 7(C), red]—namely, positions 496–504 (i.e., the region preceding F508) and the positions 559–568 and 585–591, which flank the Walker B motif and contain the diacidic motifs D565 and D567. These changes extended up to position 620.
Figure 7.
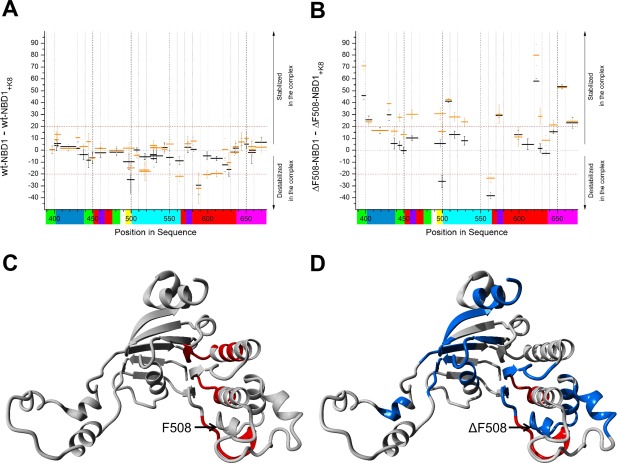
Differences in the fraction of exchange (% deuteration) in wt‐NBD1 (A) and ΔF508‐NBD1 (B) before and after interaction with K8 after 10 s (black) and 1 min (orange) of incubation. Peptides with a difference in exchange fraction of >10% were stabilized upon complex formation, while those with a difference of <−10% were more stable in the unbound state. The colored bars below the X‐axis represent the subdomain organization as defined in Figure 1. C, D: Overlay of differences >20% on the NBD1 model for wt‐NBD1 (C) and ΔF508‐NBD1 (D), indicating the regions that were strongly stabilized (blue) or destabilized (red) upon interaction with K8.
The ΔF508‐NBD1–K8 complex exhibited stronger aggregational tendencies, which might have masked the HDX patterns. It is possible that the observed overall delay in HDX was caused by aggregation and was overlaid on the region‐specific exchange, making it difficult to precisely interpret these results. Nevertheless, RI regions 400–410 and 429–433 were stabilized in the ΔF508‐NBD1–K8 complex [Fig. 7(B)], like in the ΔF508‐NBD1–Peptide 1 complex [Fig. 5(B)]. The other most prominently stabilized regions included a segment of the ABCβ subdomain (389–401), the peptide containing the ΔF508 mutation (505–512), the Walker B loop containing diacidic motifs and its C‐terminal flanking region (570–579), elements of the F1‐like ATP‐binding core subdomain (peptide 619–625), and the RE (peptide 650–662). The regions stabilized when in complex with K8 were not localized to a single portion of the protein structure [Fig. 7(D)]. Interestingly, the areas of ΔF508‐NBD1 that were destabilized in complex with K8 included the region preceding the mutation site (496–504) and the N‐terminal region flanking the Walker B loop (559–568), similar to in wt‐NBD1 [compare Fig. 7(C,D), Supporting Information Fig. S3]. Upon binding to K8, peptides covering the mutation site (Supporting Information Table S1 and Fig. S4) behave differently in wt‐NBD1 and ΔF508‐NBD1, with this region becoming substantially more protected in ΔF508‐NBD1–K8, while protection remains unchanged in wt‐NBD1–K8. Our analysis revealed that parts of the RI and the region preceding the Walker B loop (559–568) were affected by K8 binding similarly in both the wt and mutant NBD1. However, only the mutant NBD1 was affected at the N‐terminus of the ABCα subdomain (389–401), in elements of the F1‐like core domain (619–625), and at the RE (650–662; Fig. 7).
Figure 7(C,D) highlights the ΔF508‐NBD1 regions most strongly stabilized (blue) or destabilized (red) upon interaction with K8. No strong stabilization was observed in wt‐NBD1. However, many regions were stabilized in ΔF508‐NBD1, including regions flanking the RI domain (395–400 and 429–433) and the C‐terminal RE domain (650–662), elements of the F1‐like ATP‐binding core subdomain (619–626), and a portion of the ABCα subdomain (505–512). This last region was particularly interesting since it encompasses the ΔF508 mutation site. The areas destabilized upon K8 binding included the region preceding the ΔF508 mutation site (496–504) in both wt‐NBD1 and ΔF508‐NBD1. In the full‐length CFTR protein, this domain is normally involved in binding to the intracellular loop 4 (ICL4),22 which is an extension of transmembrane helices 10 and 11 in the cytoplasm. In contrast to wt‐NBD1, binding of K8 to ΔF508‐NBD1 led to the overall stabilization of a set of external loops and helices, without affecting the protein core [Fig. 7(D), blue]. This effect may be due to allosteric changes arising from the stronger binding of K8 to ΔF508‐NBD1. Such allosteric changes may help to discriminate wt‐NBD1 and ΔF508‐NBD1, and provide a means for selecting the mutated form into the degradation pathway.
NBD1 binding mainly affects the K8 head domain
Using data from the same experiment, we compared the HDX patterns for K8 in the presence and absence of wt‐NBD1 and ΔF508‐NBD1 [Fig. 8(A,B)]. Interestingly, the structural consequences on K8 were generally similar after binding to wt‐NBD1 and ΔF508‐NBD1. The head region underwent the most widespread stabilization, which was more pronounced in ΔF508‐NBD1, confirming that this domain is the region that binds NBD1. Substantial stabilization was observed in the head‐coil 1A peptide E79VDPNIQAVRTQEKEQIKTLNNKFASF105 (Fig. 9) that is nearly identical to Peptide 1, supporting that Peptide 1 is indeed a part of the binding site. We also detected increased stability in part of the flexible L12 linker region, which might arrest the mobility between the two coiled‐coil rods of K8. Binding to both versions of NBD1 also destabilized the termini of coil 1B, coil 2A‐L2, and the N‐terminal coil 2B region—which are involved in the intermolecular interactions of K8 dimers, and in its heterocomplexes with K18.15 Since K8 was expected to be dimeric under our experimental conditions, the noted HDX pattern changes could be caused by an equilibrium shift toward K8 monomers. In this interpretation, interaction between the head domains and the NBD1s would pull apart the interactions at the N‐ and C‐termini of the coiled coils that bind the K8 dimer together.
Figure 8.
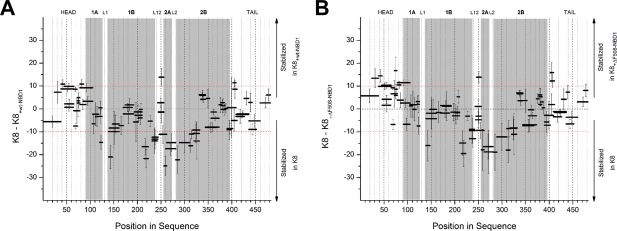
Differences in the fraction of exchange (% deuteration) in K8 peptides before and after interaction with wt‐NBD1 (A) and ΔF508‐NBD1 (B).
Figure 9.
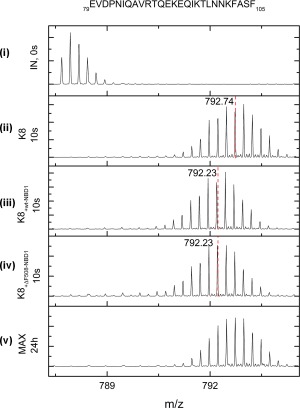
Isotopic envelopes after 10 s of exchange for the peptides, E79VD…ASF105 from the K8 head domain before binding with NBDs (ii), and after binding with wt‐NBD1 (iii) or with ΔF508‐NBD1 (iv). These isotopic envelopes are compared with the minimum possible exchange (IN, 0 s) (i) and the maximum possible exchange (MAX, 24 h) (v). The center of mass (m/z) is marked in red, and the values are flagged.
Discussion
In the present study, we identified the amino‐acid sequence localized in the head region of K8 as the site of interaction with wt‐NBD1 and ΔF508‐NBD1 of CFTR. This region may play a major role in the pathogenic interaction between K8 and ΔF508‐CFTR that has previously been demonstrated in ΔF508‐CFTR‐expressing human cells.20, 30
The global structural dynamic features of NBD1s revealed by HDX‐MS showed good agreement with the crystallographic data. Essentially all protein fragments with an elevated B‐factor also showed a fast HDX rate, with some as high as >80% after 10 s of HDX reaction (Fig. 2).6 This fast exchange was especially observed in relation to the regulatory extension, RI, and SDR. Compared with the HDX‐MS data reported by Lewis et al.,6 we noted elevated HDX rates in the regions of the Walker A and B motifs, which engage in ATP binding. The dilution of the final ATP concentration in our experimental conditions might cause the enhanced flexibility in the ATP‐binding Walker loop regions. Some flexibility in these regions could be expected, as these motifs form flexible loops (PDB: 1XMJ, 2BBO) and are required for docking and hydrolysis of ATP.6 However, the presently observed general HDX rates in wt‐NBD1 and ΔF508‐NBD1 were higher than in the pioneering HDX data reported by Lewis et al.6 One possible explanation for this discrepancy is that the recombinant NBD1s used in our study contained only one solubilization mutation (F494N), while the proteins used by Lewis et al. contained three solubilization mutations. The so‐called, “team mutation,” comprising of 3 mutations more strongly stabilized the NBD1s, almost fully restoring ΔF508‐CFTR to the plasma membrane.21
In consistence with crystal structures, our present results showed that the global HDX rate pattern of wt‐NBD1 was preserved in ΔF508‐NBD1 [Fig. 3(A,B)], indicating that their structures are similar.3, 6, 31, 32, 33 Moreover, the HDX rates of almost all identified peptides of ΔF508‐NBD1 were similar to those obtained for peptides of wt‐NBD1. The exceptions included peptides in direct proximity to the ΔF508 mutation, and the first two segments of the ABCβ subdomain (389–401 and 439–450). The first fragment, which directly precedes the mutation, showed a higher HDX rate in the presence of ΔF508, while the other regions showed slightly increased stability in ΔF508‐NBD1 (Fig. 4).
Dynamic changes and allostery in NBD1 upon binding to K8
We used the HDX‐MS technique to investigate the interactions between the NBD1 domain and K8 Peptide 1, K8 Peptide 3, and full‐length K8. No substantial changes were observed after incubation with Peptide 3 [Fig. 5(C,D)]. In contrast, incubation with Peptide 1 caused small but important differences, which were stronger with ΔF508‐NBD1 [Fig. 5(B)] than with wt‐NBD1 [Fig. 5(A)]. In complex with Peptide 1, both wt‐NBD1 and ΔF508‐NBD1 showed decreased HDX at positions 429–433 (the RI region), suggesting that K8 may bind non‐specifically to both NBD1s via this region. However, SPR revealed a higher affinity of Peptide 1 for ΔF508‐NBD1, ( = 4.6 μM) and a 4‐fold lower dissociation rate constant, indicating stronger stabilization with the mutant NBD1 [Fig. 6(A,B)]. In line with this reasoning, the region 496–504 preceding the mutation was strongly destabilized upon binding to both wt‐NBD1 and ΔF508‐NBD1. Moreover, full‐length K8 binds more strongly ( = 29 nM) to ΔF508‐NBD1,20 and our present data indicated that this stronger binding was accompanied by widespread allosteric changes in ΔF508‐NBD1 that were not observed for wt‐NBD1. This further justifies the additional binding sites we observed in the HDX experiments, indicating the higher stability of the ΔF508‐NBD1–K8 complex. These mutant‐specific changes also encompassed the mutation site. Thus, it is possible that the K8 head contains a non‐specific binding site for both NBD1s, along with an additional binding site for ΔF508‐NBD1.
Two types of experiments, analyzing the binding of NBD1s to Peptide 1 and to full‐length K8, suggested that one such ΔF508‐NBD1‐specific binding site may be located near F508. The 505–520 region was stabilized upon K8 binding only in ΔF508‐NBD1. On the other hand, the preceding region 496–504 became strongly destabilized upon binding to both wt‐NBD1 and ΔF508‐NBD1. If K8 binds directly to this region, the binding may involve interactions with side chains, with simultaneous weakening of the H‐bonding networks of amide protons in this region. In ΔF508‐NBD1, this effect may be enhanced by the preferential binding of K8 to the region of the mutation (505–520). The enhanced binding to an alternative binding site led to allosteric stabilization/destabilization of several other subdomains in ΔF508‐NBD1, including the F1‐like ATP‐binding domain, the central strand of the ABCβ subdomain, and the NBD1–NBD2 interface [Fig. 7(C,D)].
These allosteric changes may further alter the availability of potential binding sites for other currently unknown factors, allowing the cell to discriminate ΔF508‐NBD1 from wt‐NBD1 and to direct only the mutated protein into a degradation pathway. For instance, the 3D model shows that the region 491–504 is involved in NBD1 binding to the ICL4 in the case of full‐length CFTR.22 Increased affinity of the mutated NBD1 to K8 may thus lead to competitive shielding of the 491–504 region against binding to the ICL4 loop during CFTR protein folding, which in the case of ΔF508 would preclude escape from degradation pathway and inhibit biosynthesis of the fully matured protein.
Conclusions
In conclusion, we identified a K8 head peptide that participates in the binding to NBD1 and showed preferential binding to ΔF508‐NBD1. Our data suggest that K8 may contain an additional NBD‐binding region that provides an additional mutant‐specific contact site in the vicinity of the ΔF508 mutation. This additional contact increases the stability of the K8‐ΔF508‐NBD1 complex, causing allosteric changes across the entire molecule, and may provide a trigger for its release from the ER and direction to the degradation system.
Materials and Methods
HDX‐MS experiments
The HDX experiments were performed as described previously15 with minor modifications. Briefly, we used stock solutions of human NBD1 (with solubilization mutation F494N, wt‐NBD1, and ΔF508‐NBD1) at a concentration of 4 mg/ml dissolved in reaction buffer and supplemented with 10% glycerol and 2 mM ATP. We diluted 5 µL of NBD1 10× by addition 45 µL of reaction solution with D2O (50 mM Tris‐HCl, pH 8.4; 150 mM NaCl). After 10 s or 1 min, the samples were acidified by addition of 10 µL D2O Stop Buffer (2M glycine buffer, pH 2.5). We then added 2 µL of the K8 peptide solution to obtain a 10‐fold excess of peptide over the protein. To samples without peptide, we added 2 µL of the K8 peptide solvent alone. K8 exists predominantly as a dimer in 5 mM Tris‐HCl (pH 8.4),15 and this buffer condition was chosen as most conducive to our analysis of the protein system because K8 heavily aggregates and precipitates out in high ionic strength buffers. The samples were then acidified by addition of 10 µL H2O stop buffer.
The samples were digested using a 2.1 × 30 mm‐ immobilized pepsin resin column (Porozyme, ABI, Foster City, CA) with 0.07% formic acid in water as the mobile phase and a flow rate of 200 µL/min. The peptides passed directly to the 2.1 × 5 mm2 C18 trapping column (ACQUITY BEH C18 VanGuard precolumn, 1.7‐µm resin; Waters, Milford, MA). Trapped peptides were then eluted onto a reversed‐phase column (Acquity UPLC BEH C18 column, 1.0 × 100 mm, 1.7‐µm resin; Waters, Milford, MA) using a 8–40% gradient of acetonitrile in 0.1% formic acid at 40 µL/min, controlled by the nanoACQUITY Binary Solvent Manager. A single run lasted a total of 13.5 min. All fluidics, valves, and columns were maintained at 0.5°C using the HDX Manager (Waters, Milford, MA)—except the pepsin digestion column, which was maintained at 13°C inside the temperature‐controlled digestion column compartment of the HDX Manager. The C18 column outlet was coupled directly to the ion source of a SYNAPT G2 HDMS mass spectrometer (Waters, Milford, MA) operating in ion mobility mode. Lock mass was activated and performed using leucine‐enkephalin (Sigma). For protein identification, mass spectra were acquired in MSE mode over the m/z range of 50–2000. The spectrometer parameters were as follows: ESI positive mode; capillary voltage, 3 kV; sampling cone voltage, 35 V; extraction cone voltage, 3 V; source temperature, 80°C; desolvation temperature, 175°C; and desolvation gas flow, 800 L/h. The spectrometer was calibrated using standard calibrating solutions.
Peptides were identified using ProteinLynx Global Server (PLGS) software (Waters, Milford, MA). We used a randomized database with the following PLGS parameters: minimum fragment ions per peptide, 4; false–positive rate threshold, 4%. The identified peptides—along with peptide m/z, charge, retention time, and ion mobility/drift time—were passed to the DynamX 2.0 hydrogen–deuterium data analysis program (Waters, Milford, MA).
HDX experiments were performed as described for nondeuterated samples, with the reaction buffer prepared using D2O (99.8%; Cambridge Isotope Laboratories), and pH (uncorrected meter reading) adjusted using DCl (Sigma). After mixing 5 µL protein stock with 45 µL D2O reaction buffer, the exchange reactions were conducted at room temperature for varying times, as specified in the text. The exchange was quenched by addition of stop buffer (2M glycine buffer, pH 2.5) and cooling on ice. Immediately after quenching, the sample was manually injected into the nanoACQUITY UPLC system (Waters, Milford, MA). Subsequently, pepsin digestion and liquid chromatography (LC) and MS analyses were performed exactly as described above for non‐deuterated samples.
Two control experiments were performed to account for in‐ and out‐exchange artifacts, as described previously.15 Briefly, to assess minimum exchange (in‐exchange control), D2O reaction buffer was added to stop buffer that had been cooled on ice before protein stock addition, and this mixture was immediately subjected to pepsin digestion and LC‐MS analysis as described above. The deuteration level in the in‐exchange experiment was calculated (as described below) and denoted as 0% exchange (M ex 0). For out‐exchange analysis, 5 µL of protein stock was mixed with 45 µL of D2O reaction buffer, incubated for 24 h, mixed with stop buffer, and analyzed as described above. The deuteration level in an out‐exchange experiment was calculated and denoted as 100% exchange ( ).
The above‐described experiments enabled us to obtain the same set of fragments from the control and HDX experiments. Each experiment was repeated three times, and the results are presented as the mean of these replicates.
HDX data analysis
For each peptide resulting from the exchange, the deuteration level was automatically calculated using DynamX 2.0 software. These calculations were performed using the peptide list obtained from the PLGS program, with further filtering in the DynamX 2.0 program using the following acceptance criteria: minimum intensity threshold, 3000; minimum products per amino acids, 0.3. Post‐exchange isotopic envelopes were analyzed in DynamX 2.0 with the following parameters: RT deviation, ±15 s; m/z deviation, ±12.5 ppm; drift time deviation, ±2‐time bins. The calculated average masses of the peptides in the exchange experiment (M ex) and in the two control experiments (M ex 0 and ) were then verified by visual inspection. Ambiguous or overlapping isotopic envelopes were discarded from further analysis. When a split isotopic envelope was observed, separate M ex values corresponding to each envelope were calculated using the MassLynx program.
Final data were exported to an Excel (Microsoft) spreadsheet for calculation of HDX mass shifts and fractions of exchange. The percentage of relative deuterium uptake (% Deuteration) for a given peptide was calculated by accounting for both control values using the formula:
Exchange fraction (f) error bars were calculated as standard deviations of three independent experiments. The difference in exchange (ΔHDX) between two conditions of interest was determined by subtracting the fraction of exchange measured in these conditions. Errors for ΔHDX values were calculated as the square root of the sum of variances of the subtracted numbers. Final figures were plotted using OriginPro 8.0 (OriginLab) software.
Surface plasmon resonance
To investigate the interaction of Peptide 1 with the wt and ΔF508 NBD1, we used real‐time SPR techniques with the Biacore 2000 system (Biacore AB; GE Healthcare). The wt‐ and ΔF508‐ NBD1 molecules were covalently immobilized via their primary amino groups on a CM5 sensor chip (two independent flow‐cell) as described elsewhere.34 Blank control measurements were performed using one independent flow‐cell on the same sensor chip. Binding experiments were run in TBS buffer (50 mM Tris‐HCl, pH 7.4; 150 mM NaCl; 5 mM MgCl2; 1 mM DTT) containing 0.005% (w/v) surfactant P20. The association between Peptide 1 and immobilized NBD1 was monitored by injecting different concentrations of Peptide 1 (70–370 µM) at 20°C, with a flow rate of 30 µL min−1 over 60 s. Between injections, the sensor chip was regenerated by a single wash with 10 µL of 5 mM NaOH, followed by two washes with 10 µL of 10 mM HCl. To correct curves for nonspecific binding, we subtracted control curves obtained by injection of the different peptide concentrations through the blank flow channel treated with the same immobilization procedure but without NBD1.
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- HDX
hydrogen–deuterium exchange
- IF
intermediate filaments
- K8
keratin 8
- LC
liquid chromatography
- MS
mass spectrometry
- NBD
nucleotide‐binding domain
- SPR
surface plasmon resonance.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Table 1.
Acknowledgments
AK and MFdC were supported by Vaincre la mucoviscidose.
The authors declare that they have no conflicting interests.
Contributor Information
Aleksander Edelman, Email: aleksander.edelman@inserm.fr.
Michał Dadlez, Email: michald@ibb.waw.pl.
References
- 1. Gadsby DC, Vergani P, Csanády L (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- 3. Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S. (2004) Structure of nucleotide‐binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thibodeau PH, Brautigam CA, Machius M, Thomas PJ (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol 12:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, Riordan JR (2010) Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J Mol Biol 401:194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, Lu F, Sauder JM, Molnar KS, Coales SJ, et al. (2010) Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol 396:406–430. [DOI] [PubMed] [Google Scholar]
- 7. Du K, Sharma M, Lukacs GL (2005) The DeltaF508 cystic fibrosis mutation impairs domain‐domain interactions and arrests post‐translational folding of CFTR. Nat Struct Mol Biol 12:17–25. [DOI] [PubMed] [Google Scholar]
- 8. Omary MB, Ku N‐O, Strnad P, Hanada S (2009) Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest 119:1794–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moll R, Divo M, Langbein L (2008) The human keratins: biology and pathology. Histochem Cell Biol 129:705–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H (2004) Crystal structure of the human lamin A coil 2B dimer: implications for the head‐to‐tail association of nuclear lamins. J Mol Biol 343:1067–1080. [DOI] [PubMed] [Google Scholar]
- 11. Chernyatina AA, Guzenko D, Strelkov SV (2015) Intermediate filament structure: the bottom‐up approach. Curr Opin Cell Biol 32:65–72. [DOI] [PubMed] [Google Scholar]
- 12. Herrmann H, Aebi U (2004) Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu Rev Biochem 73:749–789. [DOI] [PubMed] [Google Scholar]
- 13. Herrmann H, Strelkov SV, Burkhard P, Aebi U (2009) Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest 119:1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lichtenstern T, Mücke N, Aebi U, Mauermann M, Herrmann H (2012) Complex formation and kinetics of filament assembly exhibited by the simple epithelial keratins K8 and K18. J Struct Biol 177:54–62. [DOI] [PubMed] [Google Scholar]
- 15. Premchandar A, Kupniewska A, Tarnowski K, Mücke N, Mauermann M, Kaus‐Drobek M, Edelman A, Herrmann H, Dadlez M (2015) Analysis of distinct molecular assembly complexes of keratin K8 and K18 by hydrogen‐deuterium exchange. J Struct Biol 192:426–440. [DOI] [PubMed] [Google Scholar]
- 16. Davezac N, Tondelier D, Lipecka J, Fanen P, Demaugre F, Debski J, Dadlez M, Schrattenholz A, Cahill MA, Edelman A (2004) Global proteomic approach unmasks involvement of keratins 8 and 18 in the delivery of cystic fibrosis transmembrane conductance regulator (CFTR)/deltaF508‐CFTR to the plasma membrane. Proteomics 4:3833–3844. [DOI] [PubMed] [Google Scholar]
- 17. Lipecka J, Norez C, Bensalem N, Baudouin‐legros M, Planelles G, Edelman A (2006) Rescue of DeltaF508‐CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) by Curcumin: Involvement of the Keratin 18 network. J Pharmacol Exp Ther. 317:500–505. [DOI] [PubMed] [Google Scholar]
- 18. Edelman A (2014) Cytoskeleton and CFTR. Int J Biochem Cell Biol 52:68–72. [DOI] [PubMed] [Google Scholar]
- 19. Duan Y, Sun Y, Zhang F, Zhang WK, Wang D, Wang Y, Cao X, Hu W, Xie C, Cuppoletti J, et al. (2012) Keratin K18 increases cystic fibrosis transmembrane conductance regulator (CFTR) surface expression by binding to its C‐terminal hydrophobic patch. J Biol Chem 287:40547–40559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colas J, Faure G, Saussereau E, Trudel S, Rabeh WM, Bitam S, Guerrera IC, Fritsch J, Sermet‐Gaudelus I, Davezac N, et al. (2012) Disruption of cytokeratin‐8 interaction with F508del‐CFTR corrects its functional defect. Hum Mol Genet 21:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, et al. (2012) Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mornon J‐P, Hoffmann B, Jonic S, Lehn P, Callebaut I (2015) Full‐open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell Mol Life Sci 72:1377–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haim M, Trost A, Maier CJ, Achatz G, Feichtner S, Hintner H, Bauer JW, Onder K (2010) Cytokeratin 8 interacts with clumping factor B: a new possible virulence factor target. Microbiology 156:3710–3721. [DOI] [PubMed] [Google Scholar]
- 24. Omary MB, Ku NO, Liao J, Price D (1998) Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell Biochem 31:105–140. [PubMed] [Google Scholar]
- 25. Kouklis PD (1994) Making a connection: direct binding between keratin intermediate filaments and desmosomal proteins. J Cell Biol 127:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sevcík J, Urbániková L, Kost'an J, Janda L, Wiche G (2004) Actin‐binding domain of mouse plectin. Crystal structure and binding to vimentin. Eur J Biochem 271:1873–1884. [DOI] [PubMed] [Google Scholar]
- 27. Correia I, Chu D, Chou YH, Goldman RD, Matsudaira P (1999) Integrating the actin and vimentin cytoskeletons. Adhesion‐dependent formation of fimbrin‐vimentin complexes in macrophages. J Cell Biol 146:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garbuglia M, Verzini M, Donato R (1998) Annexin VI binds S100A1 and S100B and blocks the ability of S100A1 and S100B to inhibit desmin and GFAP assemblies into intermediate filaments. Cell Calcium 24:177–191. [DOI] [PubMed] [Google Scholar]
- 29. Chatin B, Mével M, Edelman A, Pitard B (2013) WS4.4 Restoration of F508Δ‐CFTR trafficking and function by liposome‐mediated delivery of antibodies against cytokeratin 8. J Cyst Fibros 12:S7. [Google Scholar]
- 30. Odolczyk N, Fritsch J, Norez C, Servel N, da Cunha MF, Bitam S, Kupniewska A, Wiszniewski L, Colas J, Tarnowski K, et al. (2013) Discovery of novel potent ΔF508‐CFTR correctors that target the nucleotide binding domain. EMBO Mol Med 5:1484–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, et al. (2005) Impact of the deltaF508 mutation in first nucleotide‐binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem 280:1346–1353. [DOI] [PubMed] [Google Scholar]
- 32. Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, Hendle J, Hunt JF, Lewis HA, Lu F, et al. (2010) Structures of a minimal human CFTR first nucleotide‐binding domain as a monomer, head‐to‐tail homodimer, and pathogenic mutant. Protein Eng Des Sel 23:375–384. [DOI] [PubMed] [Google Scholar]
- 33. Hudson RP, Chong PA, Protasevich II, Vernon R, Noy E, Bihler H, An JL, Kalid O, Sela‐Culang I, Mense M, et al. (2012) Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 287:28480–28494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Faure G, Bakouh N, Lourdel S, Odolczyk N, Premchandar A, Servel N, Hatton A, Ostrowski MK, Xu H, Saul FA, et al. (2016) Rattlesnake phospholipase A2 increases CFTR‐chloride channel current and corrects ΔF508CFTR dysfunction: impact in Cystic Fibrosis. J Mol Biol 428:2898–2915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Table 1.