

Abstract
Many clinical treatment studies have reported remarkable interindividual variability in the response to pharmaceutical drugs, and uncovered the existence of inadequate treatment response, non-response, and even adverse drug reactions. Pharmacogenetics addresses the impact of genetic variants on treatment outcome including side-effects. In recent years, it has also entered the field of clinical diabetes research. In modern type 2 diabetes therapy, metformin is established as first-line drug. The latest pharmaceutical developments, including incretin mimetics, dipeptidyl peptidase 4 inhibitors (gliptins), and sodium/glucose cotransporter 2 inhibitors (gliflozins), are currently experiencing a marked increase in clinical use, while the prescriptions of α-glucosidase inhibitors, sulfonylureas, meglitinides (glinides), and thiazolidinediones (glitazones) are declining, predominantly because of reported side-effects. This review summarizes the current knowledge about gene-drug interactions observed in therapy studies with the above drugs. We report drug interactions with candidate genes involved in the pharmacokinetics (e.g., drug transporters) and pharmacodynamics (drug targets and downstream signaling steps) of the drugs, with known type 2 diabetes risk genes and previously unknown genes derived from hypothesis-free approaches such as genome-wide association studies. Moreover, some new and promising candidate genes for future pharmacogenetic assessment are highlighted. Finally, we critically appraise the current state of type 2 diabetes pharmacogenetics in the light of its impact on therapeutic decisions, and we refer to major problems, and make suggestions for future efforts in this field to help improve the clinical relevance of the results, and to establish genetically determined treatment failure.
Keywords: type 2 diabetes, SNP, GWAS, heritability, drug absorption, drug target, trough steady-state
Abbreviations: AMPK - AMP-activated protein kinase; AP - activator protein; ATM - ataxia telangiectasia mutated; DPP4 - dipeptidyl peptidase 4; ENT4 - equilibrative nucleoside transporter 4; GIP - gastric inhibitory peptide; GLP1 - glucagon-like peptide 1; GWAS - genome-wide association study; HbA1c - hemoglobin A1c; HNF - hepatocyte nuclear factor; LKB1 - liver kinase B1; MATE - multidrug and toxin extrusion transporters; MEF - myocyte-specific enhancer factor; MZF1 - myeloid zinc finger 1; Nor1 - neuron-derived orphan receptor 1; OCT - organic cation transporter; OGTT - oral glucose tolerance test; PMAT - plasma membrane monoamine transporter; PPARα - peroxisome proliferator-activated receptor α; SGLT2 - sodium/glucose cotransporter 2; SLC - solute carrier; SNP - single nucleotide polymorphism; STK11 - serine/threonine kinase 11; TCF7L2 - T-cell-specific transcription factor 7-like 2
1. Genetics of type 2 diabetes
With a global prevalence of 9% among adult persons, diabetes is the 7th leading non-comunicable disease world-wide, causing more than 1,500,000 deaths per year (WHO fact sheet N°312, http://www.who.int/mediacentre/factsheets/ fs312/en). Type 2 diabetes accounts for 90% of all diabetes cases. Hallmarks of the disease are a pronounced insulin resistance of peripheral tissues, including skeletal muscle, liver, and adipose tissue, and a failure of pancreatic β-cells to compensate for this metabolic derangement by enhanced insulin secretion. It is mainly caused by lifestyle (sedentary behavior) and environmental (high-caloric nutrition) factors favoring overweight and obesity, but has also a clear genetic component, as has been shown by family and twin studies [1].
Over the last 20 years, innovative biotechnological methods have fostered the development of high-density oligonucleotide microarrays, depicting on small glass chips up to five million single nucleotide polymorphisms (SNPs) with minor allele frequencies that provide near-complete coverage of the common genetic variation present in the human genome. The recent affordability of these chips revolutionized the characterization of the genetic architectures of common non-communicable diseases, such as type 2 diabetes. Huge case-control and prospective study cohorts, including tens of thousands to hundreds of thousands of subjects collected via world-wide consortia, have been genotyped by this methodology, and analyzed for genotype-phenotype associations. This approach, termed genome-wide association study (GWAS), has revealed more than 70 SNPs to be associated with type 2 diabetes on a statistical significance level corrected for multiple testing (genome-wide p-value <5*10-8) [2]. For a closer look at these hits and their nearby genes, see [2] and accompanying articles in this Special Edition of The Review of Diabetic Studies.
Even though we learned from GWAS and other studies assessing the association of diabetes SNPs with metabolic traits that these variants predominantly affect β-cell function and not insulin sensitivity, their functionality and biology is far from being understood [3]. Moreover, their value for risk prediction appears negligible, and all SNPs together explain only about 10% of diabetes heritability [4]. There may be many reasons for this so-called 'missing heritability', including limited value of common heritability estimates (e.g., due to bias by shared environment) or a hitherto underestimated role of low-frequency and rare variants or epigenetic mechanisms in diabetes inheritance. However, the concept of missing heritability also opens the intriguing possibility of gene-gene, gene-lifestyle, gene-environment, and gene-drug interactions that are of major relevance for disease inheritance, prediction, and progression, as well as for the individual responsiveness to preventive and therapeutic measures. In this context, it is noteworthy that lifestyle intervention studies for prevention of type 2 diabetes revealed SNPs in genes very different from those found in GWAS; these genes predicted the magnitude of the individual’s response to lifestyle factors such as diet and exercise [5]. Thus, different and possibly non-overlapping sets of genes may be relevant for gene-gene, gene-lifestyle, gene-environment, and gene-drug interactions.
Gene-drug interactions are the central subject of the new and very promising clinical research discipline termed pharmacogenetics. Despite some success at the research level, the translation of pharmacogenetic results into clinical practice is currently limited, and faces challenges in regulation, infrastructures (e.g., lack of electronical medical recording), and health economics. Apart from the genetic information, the complexity of drug response requires a better understanding of additional information from epigenomics, transcriptomics, proteomics, metabolomics, and microbiomics [6]. Moreover, the patient’s phenotype needs to be comprehensively assessed using novel approaches such as high-throughput, IT-based methods to collect clinical data directly from the patient (e.g., computer-based, direct interview) [7]. In this context, the intelligent, valid, and time- and cost-saving integration of 'big data' needs to be addressed. Thus, the implementation of the concept of precision medicine in clinical practice, with the ultimate goal of establishing treatment algorithms and pharmacogenetic information based on multiscale systems pharmacology approaches [8], may be more challenging than expected.
2. Modern diabetes therapy
The options for drug therapy of type 2 diabetes currently encompass the following 13 drug classes:
α-glucosidase inhibitors
Biguanides
Sulfonylureas
Meglitinides (aka glinides)
Thiazolidinediones (aka glitazones)
Dipeptidyl peptidase 4 (DPP4) inhibitors (aka gliptins)
Incretin mimetics
Sodium/glucose cotransporter 2 (SGLT2) inhibitors (aka gliflozins)
Amylin mimetics
Bile acid sequestrants
Dopamine agonists
Human insulin
Insulin analogues
The drug classes, individual drugs, and their main metabolic effects are listed in Table 1.
Table 1. Anti-diabetic drugs and their major metabolic effects.
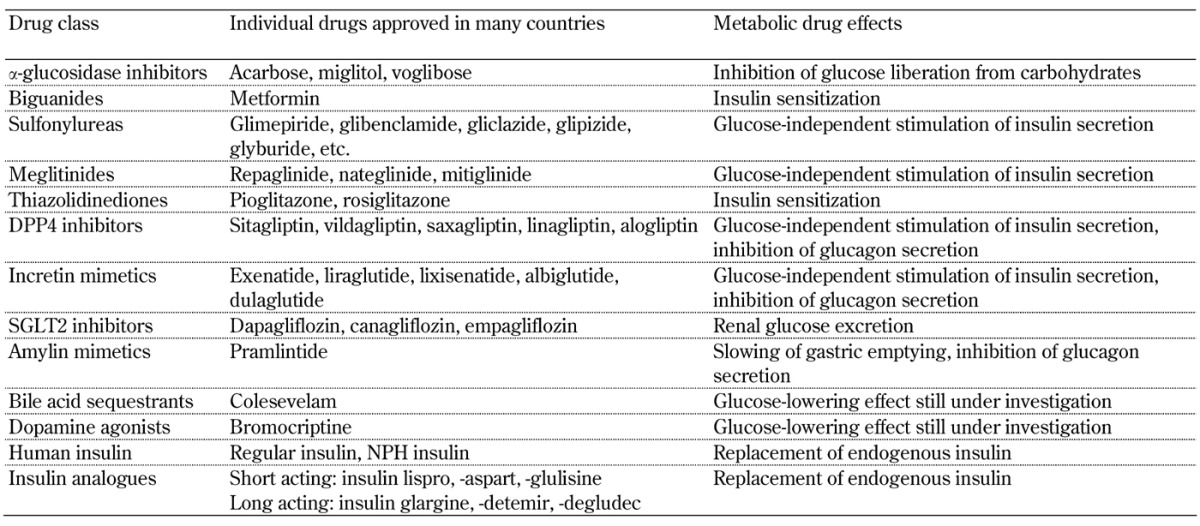
Legend: DPP4 – dipeptidyl peptidase 4; NPH – neutral protamine Hagedorn; SGLT2 – sodium/glucose cotransporter 2.
It is normal practice that any country has its own specific guidelines for diabetes therapy developed by their national expert associations. However, there is world-wide consent that, when lifestyle intervention fails to achieve the individual target hemoglobin A1c (HbA1c), the biguanide metformin is the first-line drug of choice (http://www.idf.org/sites/default/files/IDF-Guideline-for-Type-2-Diabetes.pdf, [9]). If metformin is not well tolerated or contraindicated, an alternative drug from the other drug classes can be prescribed. If metformin is insufficient to reach the individual target HbA1c, it is usually combined with one or more drugs from the other drug classes. Human insulin and insulin analogues are used for intensified and combination therapy when pancreatic β-cell function markedly declines. In fact, the spectrum of drugs predominantly prescribed in modern type 2 diabetes therapies is more limited since α-glucosidase inhibitors are frequently accompanied by adverse gastrointestinal side-effects such as flatulence and diarrhea [10], and thiazolidinediones are no longer marketed in some countries because of reports of the following increased risks: myocardial infarction (rosiglitazone), distal fractures of long bones and bladder cancer (pioglitazone), and heart failure (both glitazones) [11].
Sulfonylureas and meglitinides are β-cell stressors that are suspected to accelerate the exhaustion of endogenous insulin secretion [12]. Also, both classes of insulin secretagogues promote weight gain, and are associated with an increased risk of hypoglycemia [13]. Finally, amylin mimetics, bile acid sequestrants, dopamine agonists, insulin, and insulin analogues have not yet been subjected to pharmacogenetic investigation. Therefore, this review primarily focuses on available reported information about the pharmacogenetic interactions potentially relevant for treatment regimens based on metformin, drugs enhancing the incretin axis, and SGLT2 inhibitors.
3. Pharmacogenetics and drug targets
Many clinical studies reported high biological variance in the response to anti-diabetic drugs. For instance, the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study detected ethnic differences in response to metformin treatment, which seems to be more effective in Hispanics and non-Hispanic Whites than in Afro-Americans [14]. This argues for a role of genetics in drug responses. Among the gene sets that conceivably interact with drugs are:
Genes whose products affect pharmacokinetics, including drug absorption, distribution, metabolism, and elimination (termed ADME genes).
Genes whose products affect pharmacodynamics, including defined drug targets and their upstream and downstream interaction partners.
Known diabetes risk genes.
Finally, GWAS have the potential to identify new drug-interacting genes in a hypothesis-free manner.
3.1 Gene variants affecting the response to metformin
Metformin is the first-line anti-diabetic drug in modern type 2 diabetes therapy, and currently the only available biguanide (Figure 1). Former drugs in this class, i.e., phenformin and buformin, were withdrawn from the market because of an increased risk of lactic acidosis related to these drugs. An increased risk for lactic acidosis caused by metformin is seen in patients with renal insufficiency only [15]. Therefore, metformin is contraindicated in parts of this subgroup of diabetic patients. Metformin displays considerable interindividual variation in efficacy: in the U.K. Diabetes Audit and Research in Tayside Scotland (DARTS) study, some patients experienced extreme drops in HbA1c levels down to 4%, while other patients' HbA1c did not change or even increased under metformin therapy [16]. Christensen et al. reported an 80-fold variability in trough steady-state (i.e. the lowest permanent drug concentration in a patient when intake and elimination of the drug is in dynamic equilibrium) metformin plasma concentration in a randomized, placebo-controlled multicenter study (the South Danish Diabetes Study, SDDS) which included 159 patients on metformin therapy for 24 months [17]. Furthermore, even though considered as safe in general, metformin provokes gastrointestinal side-effects (nausea, vomiting, and diarrhea) in about 25% of patients; these effects are usually transient, but they persist and lead to discontinuation of therapy in about 5% of patients [18]. All these aspects, and in particular the identification of non-responders and even adverse responders, initiated a series of pharmacogenetic investigations with respect to metformin therapy [19].
Figure 1. Transport pathways contributing to the disposition of metformin.

Metformin is not metabolized. It is absorbed, distributed, and eliminated via different members of the solute carrier superfamily. Abbreviations: ENT4 – equilibrative nucleoside transporter 4; OCT1-3 – organic cation transporter 1-3; MATE1/2-K – multidrug and toxin extrusion transporters 1/2-K.
The orally administered anti-diabetic drug metformin acts as an insulin sensitizer, enhancing the sensitivity of insulin-responsive tissues, including liver, skeletal muscle, and adipose tissue, to endogenous and pharmacologically administered insulin [20]. Even though metformin has been in clinical use since 1959, its molecular targets are still insufficiently described. A very compelling hypothesis for its mode of action is that metformin reduces mitochondrial ATP synthesis, thereby increasing intracellular AMP concentrations and activating AMP-activated protein kinase (AMPK) (Figure 2) [21]. AMPK is a central switch between anabolic and catabolic states, favoring catabolic ATP-generating reactions, and concomitantly inhibiting anabolic ATP-consuming pathways such as gluconeogenesis. In fact, inhibition of hepatic glucose production appears to be the primary mechanism of metformin’s anti-hyperglycemic action [22]. Metformin also has the following properties:
Figure 2. Metformin actions in metabolically relevant cell types.

Metformin's primary target in hepatocytes, adipocytes, and myocytes is the mitochondrion. In the liver, metformin inhibits respiratory chain complex I and mGPDH, both located in the inner mitochondrial membrane. Inhibition of complex I increases the AMP/ATP ratio, thereby activating AMPK, a central regulator of anabolic and catabolic pathways and of mitochondrial biogenesis. Inhibition of mGPDH causes elevated NADH concentrations that shift the equilibrium of the lactate dehydrogenase reaction to lactate, resulting in elevated lactate production and reduced gluconeogenic substrate pyruvate levels. In adipocytes and myocytes, AMPK activation leads to inactivation of TBC1D1/D4 proteins. This promotes insulin-independent translocation of GLUT4-containing vesicles from intracellular depots to the plasma membrane, thereby increasing GLUT4 concentration in the plasma membrane. Abbreviations: ACC – acetyl-coenzyme A carboxylase; AMPK – AMP-activated protein kinase; GLUT4 – glucose transporter 4; mGPDH – mitochondrial glycerophosphate dehydrogenase; PFKFP – 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase; PGC1α – peroxisome proliferator-activated receptor γ coactivator 1α; SREBP1c – sterol-regulatory element-binding protein 1c; STK11 – serine/threonine kinase 11; TBC1D1/D4 – TBC1 domain family members D1 and D4.
It increases muscular glucose uptake by augmenting glucose transporter 4 translocation to the plasma membrane [20].
It exerts anti-steatotic actions by inhibiting lipogenesis and stimulating β-oxidation [23].
It may enhance intestinal glucose uptake and utilization [25].
How AMPK activation increases insulin sensitivity molecularly is still a matter of debate. Recently, selective inhibition of the mitochondrial isoform of glycerophosphate dehydrogenase, which catalyzes the conversion of glycerophosphate to dihydroxyacetone phosphate without changing cellular energy charge or gluconeogenic gene expression in liver, has been reported as a novel mechanistic action of metformin (Figure 2) [26].
Metformin's pharmacokinetics is simplified by the fact that it does not undergo biotransformation in the liver [27]. Therefore, genetic variants of metabolizing enzymes probably do not affect metformin's anti-diabetic action. As a positively charged hydrophilic molecule, metformin has to use transport proteins to cross bio-membranes, and these include the organic cation transporter (OCT) family members OCT1-3 (aka solute carrier (SLC) family members 22A1-3), the equilibrative nucleoside transporter 4 (ENT4, aka SLC29A4), and the multidrug and toxin extrusion transporters 1 and 2-K (MATE1 and MATE2-K, aka SLC47A1 and SLC47A2) [19]. As depicted in Figure 1, metformin is absorbed by enterocytes via ENT4 and OCT3, and released into the bloodstream via OCT1, followed by distribution throughout the body. Hepatocytes take up metformin via OCT1 and OCT3, and excrete it into the bile via MATE1. However, this is not the only way of metformin elimination. Renal tubular cells take up metformin from the circulation via OCT2, and excrete it into the urine via MATE1, MATE2-K, and OCT1. Genetic variants in all these transport proteins are therefore suggested to impact on metformin absorption, distribution, and elimination, thereby altering its half-life in the circulation and its spectrum of activity.
The human OCT1 gene is highly polymorphic, and 23 naturally occurring coding variants of OCT1 have been described [28-30], with seven of them displaying 40-100% reduced metformin transport function in vitro [29]. Among these, Arg61Cys was found to affect OCT1 protein expression in liver specimens from 150 Caucasian donors, with Cys-allele carriers displaying significantly reduced expression [28]. This may result in attenuated metformin uptake in hepatocytes and reduced metformin clearance via the bile. Twenty healthy volunteers carrying at least one of the four variant alleles Arg61Cys, Gly401Ser, Met420del, and Gly465Arg revealed elevated plasma glucose levels during an oral glucose tolerance test (OGTT) after short-term metformin application, pointing to a reduced response to metformin in vivo [29, 31]. In the SDDS, carriers of the Arg61Cys, Ser189Leu, Gly401Ser, Met420del, and Gly465Arg variants showed allele-dose effects; trough steady-state plasma metformin levels and HbA1c reductions decreased with increasing number of variant alleles carried compared with non-carriers [17]. This suggests that a variant-allele-attributable reduction in metformin release from the enterocyte into the bloodstream may be superior compared with the attenuation of hepatocytic metformin uptake. Jablonski et al. tested 47 OCT1 SNPs in 2,994 participants in a randomized, placebo-controlled multicenter diabetes prevention study, the Diabetes Prevention Program (DPP), for effects on diabetes incidence [5]. The authors described a nominally significant interaction of Leu160Phe with response to metformin; the minor Phe-allele impaired the diabetes-protective effect of metformin. In a large intervention study with DNA available for genetic investigations, the Genetics of DARTS (GoDARTS) study, comprising 1,531 diabetic patients on metformin therapy, the two OCT1 variants assessed, i.e., Arg61Cys and Met420del, affected neither the initial reduction in HbA1c, nor the chance of achieving the treatment target (HbA1c < 7 %), nor the average HbA1c level on monotherapy for up to 42 months, nor the hazard of monotherapy failure [32]. However, carriers of two reduced-function alleles were significantly more likely to be metformin-intolerant than carriers of one or none allele [33]. Finally, using a tagging SNP approach, Becker et al. screened the OCT1 locus for representative SNPs, covering all the common genetic variation in the locus (minor allele frequency >5 %), including non-coding variants, and assessed these SNPs' effects on response to metformin in 102 incident metformin users from the Rotterdam Study [34]. They identified a SNP in intron 8 (rs622342 A>C), which interfered with metformin action with each minor C-allele, reducing the metformin-induced drop in HbA1c by 0.28%. It did not interact with metformin action in the DPP [5].
OCT2 accounts for about 80% of total renal clearance of metformin [35, 36]. Although less polymorphic (eight coding variants identified to date [37]), the OCT2 locus harbors a coding variant, i.e., Ala270 Ser, featuring interesting functionality: the minor Ser-allele increased metformin transport in vitro, and was associated with enhanced renal metformin clearance in 23 healthy Caucasian and African American volunteers [38]. This may be due to differences in the haplotype structure between ethnic groups. The same allele was associated with higher plasma metformin levels and reduced renal metformin clearance in healthy Asian volunteers [39, 40]. However, this variant did not affect trough steady-state metformin concentration or metformin-induced reductions in HbA1c in the SDDS [17]. Likewise, Ala270Ser did not affect metformin's effect on diabetes prevention in the DPP [5]. Interestingly, Kashi et al. reported that carriers of the minor Met-allele of the OCT2 variant, Thr201Met, have higher HbA1c concentrations, fasting glucose levels, insulin resistance, and compensatory insulin secretion during metformin therapy than patients with common alleles [41].
Even though genetic variants of OCT3 have been described to associate with reduced OCT3 mRNA expression [28], and thus appear to be functional, pharmacogenetic studies on the role of these variants in metformin transport and action are still lacking.
Chen et al. identified six coding variants of MATE1 with five of them showing 25-100% loss of metformin transport function in vitro [42]. However, these variants have not yet been followed up in clinical studies. Choi et al. described five non-coding variants in the basal promoter region of MATE1, with the minor C-allele of the common variant rs2252281 T>C revealing:
Reduced recruitment of the transcription factor activator protein (AP) 1.
Concomitantly enhanced binding of the transcriptional repressor AP2rep to the MATE1 promoter.
Consequently, diminished reporter gene expression [43].
In line with this finding, the authors also reported reduced MATE1 expression in 38 human kidney samples, suggesting that the minor C-allele is associated with reduced metformin excretion. After short-term application of metformin in 57 healthy volunteers, Stocker et al. demonstrated that homozygous rs2252281 C-allele carriers experience stronger reductions in plasma glucose during OGTT than heterozygous and homozygous carriers of the major T-allele, suggesting an enhanced metformin treatment response in C-allele carriers, possibly due to attenuated metformin elimination [44]. Even though the authors could not detect any effect of the variant on metformin pharmacokinetics, they described a stronger HbA1c-reducing effect of metformin in 249 homozygous C-allele carriers on metformin monotherapy than in the reference group. In the Rotterdam study, Becker et al. performed a tagging SNP approach, and identified only one SNP in the MATE1 gene, i.e., rs2289669 G>A in intron 10, which was associated with the response to metformin in 116 incident users of the drug; each minor A-allele enhanced the Hb1Ac reduction by 0.30% [45]. This was recently confirmed in a study including 148 drug-naïve patients with type 2 diabetes [46], where patients homozygous for the A-allele displayed twofold stronger reductions in HbA1c than patients carrying the major G-allele. A concordant effect was seen in 220 Chinese type 2 diabetic patients who were treated with metformin for one year. Pharmacokinetic parameters indicated that homozygous A-allele carriers had higher plasma concentrations and lower renal clearance of metformin than the reference group [47]. Of note, rs8065082 C>T in intron 12 of the MATE1 gene, which is in 80% linkage with rs2289669, also showed nominally significant association with metformin action in the DPP [5]. In the SDDS however, neither rs2289669 nor the promoter variant rs2252281 affected trough steady-state metformin concentrations or metformin-induced reductions in HbA1c [17].
Choi et al. also screened the MATE2 gene, and identified four coding variants, two of which displayed lower MATE2-K protein expression and reduced metformin transport function upon transfection in HEK293 cells [48]. For MATE2, two splice variants, MATE2-K and MATE2-B, have been identified, with MATE2-K being the only MATE2 protein for which transport activity has been experimentally shown to date [49]. Again, these variants still lack clinical assessment. The authors have described additional variants in the basal promoter region, with carriers of the minor A-allele of the common variant rs12943590 G>A being associated with reduced binding of the transcriptional repressor myeloid zinc finger 1 (MZF1) to the promoter, and consequently increased reporter gene expression. Therefore, it was supposed that A-allele carriers have higher renal MATE2-K expression, and as a result higher metformin excretion. Choi et al. studied 253 diabetic patients on metformin monotherapy, and demonstrated that patients homozygous for the A-allele had a significantly weaker response to metformin treatment with respect to the drop in HbA1c than carriers of the reference G-allele [48]. As expected from these data, Stocker et al. finally reported higher renal metformin clearance and higher glucose levels during OGTT in volunteers carrying the rs12943590 A-allele [44].
As the combined effects of genetic variants in OCT and MATE transporters in an individual patient may result in a more significant alteration of metformin drug disposition and potential consequences on treatment response, as suggested by Christensen et al., future clinical studies are required to clarify comprehensively the gene-gene interactions [50]. In the SDDS, Christensen et al. investigated tagging SNPs of the ENT4 (aka plasma membrane monoamine transporter (PMAT)) gene, and identified five non-coding SNPs, i.e., rs2685753 A>G, rs3889348 C>T, rs4720572 T>C, rs4299914 G>A, and rs6971788 T>A, which were associated with trough steady-state metformin concentration when major allele carriers were compared with heterozygous and homozygous variant allele carriers combined (dominant inheritance model) [17]. Being associated with lower metformin concentrations in blood, the variant alleles are suggested to impair enterocytic metformin uptake, possibly via reduced ENT4 expression. Functional studies are required to confirm this suggestion.
Another level of complexity derives from variation in genes encoding transcription factors that control transporter gene expression. For instance, it is known that:
The transcription factors peroxisome proliferator-activated receptor α (PPARα) and hepatocyte nuclear factor (HNF) 4A control the expression of OCT1 [51-53].
The transcription factor specificity protein 1 (SP1) controls the expression of OCT3 and MATE1 [54, 55].
The transcription factor AP2 controls the expression of MATE1 [43].
Goswami et al. were the first to test SNPs in transcription factor genes for association with metformin treatment response [56]. Among 40 predominantly non-coding SNPs which were associated with treatment HbA1c levels in 440 patients, five SP1 variants and one AP2 variant were concomitantly associated with metformin secretory clearance in 57 healthy volunteers. Seventeen PPARα variants and six HNF4A variants were associated with treatment HbA1c only. Certainly, these very recent findings open up a new field of pharmacogenetic investigation.
Regarding genes related to the current knowledge about the pharmacodynamics of metformin, Jablonski et al. assessed a series of SNPs in or near the genes PRKAA1, PRKAA2, PRKAB1, PRKAB2, PRKAG1, PRKAG2, and PRKAG3 encoding the AMPK subunits α1, α2, β1, β2, γ1, γ2, and γ3, respectively [5]. Among these SNPs, the non-coding variants PRKAA1 rs249429 T>C, PRKAA2 rs9803799 T>G, and PRKAB2 rs6690158 C>T revealed nominally significant interaction with metformin with respect to protection from diabetes progression. In the same study, nominal associations with metformin treatment response were reported for SNPs in the AMPK upstream regulatory kinase serine/threonine kinase 11 (STK11, aka liver kinase B1 (LKB1); rs741765 C>T) (Figure 2), the AMPK downstream transcription factors myocyte-specific enhancer factor (MEF) 2A and MEF2D (rs4424892 A>G and rs6666307 A>T, respectively), and in the type 2 diabetes risk genes HNF1B (rs11868513 G>A), HNF4A (rs11086926 T>G), ABCC8 (rs4148609 G>A), KCNJ11 (rs7124355 G>A), GCK (rs2908289 G>A), and CAPN10 (rs3792269 A>G). Among the aforementioned SNPs, Tkac et al. tested SNPs in PRKAA1, STK11, HNF4A, and CAPN10 for association with treatment success (HbA1c <7 %) and absolute reduction in HbA1c after six months of metformin monotherapy in 148 drug-naïve type 2 diabetes patients [57]. Only the minor G-allele carriers of CAPN10 SNP rs3792269 A>G showed less treatment success and nominally smaller declines in HbA1c than A-allele carriers. Further studies are needed to find out how the cysteine protease calpain 10 (encoded by the CAPN10 gene), which was identified in the pre-GWAS era as a diabetes risk gene affecting insulin sensitivity, fits into the molecular pathways of metformin signaling.
A hypothesis-free, large-scale GWAS for glycemic response to metformin in 1,024 patients of the GoDARTS study, with replication in two cohorts including 1,783 Scottish individuals and 1,113 individuals from the UK Prospective Diabetes Study (UKPDS), revealed 14 SNPs in a 340-kb linkage block, spanning the Ataxia Telangiectasia Mutated (ATM) gene locus, to be associated with treatment HbA1c and the ability to achieve target HbA1c (<7 %) within 18 months of metformin treatment [58]. The SNP with the strongest association in the meta-analysis of the three studies, i.e., rs11212617 A>C, is located 43 kb downstream of the ATM gene (more precisely in an intron of the adjacent open reading frame C11orf65). This SNP increased the treatment success 1.35-fold, and reduced the treatment HbA1c concentration by 0.11% per minor C-allele. Van Leeuwen et al. replicated the effect of this SNP on the ability to reach the target HbA1c (<7%) in a meta-analysis of three independent study populations, the Diabetes Care System (DCS) West-Friesland (n = 929), the Rotterdam Study (n = 182), and the UK Collaborative Atorvastatin Diabetes Study (CARDS, n = 254) [59]. However, the study failed to show an association with treatment HbA1c. Moreover, the SNP was genotyped in the DPP, but no significant interaction effect on diabetes prevention by metformin was observed [60]. In this context, a recent study is of interest, indicating the complexity of pharmacology and drug therapy. In this study, the ATM inhibitor, KU-55933, was shown to inhibit also the OCT1-mediated uptake of metformin. This indicates that KU-55933 inhibits metformin uptake through inhibition of OCT1 rather than ATM [61].
3.2 Gene variants affecting the response to DPP4 inhibitors
As depicted in Figure 3, the ubiquitous membrane protein dipeptidyl peptidase 4 (DPP4) cleaves and inactivates the incretin hormones glucagon-like peptide 1 (GLP1) and gastric inhibitory peptide (GIP), which are released by intestinal L- and K-cells, respectively, in response to diet ingestion, and enhance glucose-stimulated insulin secretion by pancreatic β-cells [62]. To increase circulating levels of the insulinotropic incretins, stable incretin mimetics bypassing DPP4 and low-molecular-weight inhibitors of DPP4 have recently been developed [63]. The orally administered DPP4 inhibitors are generally considered as well tolerated. Interestingly, studies reported considerable biological variance between individuals in the responsiveness to DPP4 inhibitors [63-65]. Even though the reasons underlying good versus poor response are currently unknown, these observations open the possibility of pharmacogenetic interactions.
Figure 3. Pathways contributing to the pharmacodynamics of incretin mimetics and DPP4 inhibitors.

Incretins are released by specialized cells in the intestine (GLP1 by L-cells, GIP by K-cells). They enhance glucose-stimulated insulin secretion via binding to transmembrane receptors in pancreatic β-cells and, at least in part, subsequent activation of transcription factors, such as TCF7L2 and Nor1. Incretins are inactivated by DPP4 which is the target of DPP4 inhibitors. Incretin mimetics are stable incretin analogues bypassing DPP4. Abbreviations: DPP4 – dipeptidyl peptidase 4; GIP(R) – gastric inhibitory peptide (receptor); GLP1(R) – glucagon-like peptide 1 (receptor); GLUT – glucose transporter; Nor1 – neuron-derived orphan receptor 1; TCF7L2 – T-cell-specific transcription factor 7-like 2.
With the exception of saxagliptin, and to a lesser extent sitagliptin as well, DPP4 inhibitors are considered not to be biotransformed in the liver [66]. Saxagliptin is metabolized via the cytochrome P450 isoforms 3A4 and 3A5, and its main metabolite, the primary hydroxylated form, displays twofold less DPP4-inhibitory activity than the parent compound [66]. Several other metabolites have been documented, but most of them are inactive [66]. Regarding the elimination of DPP4 inhibitors, renal excretion is predominant, except for linagliptin which is mainly excreted via the bile [67]. No pharmacogenetic studies with respect to genes important in the pharmacokinetics of DPP4 inhibitors are reported to date.
With respect to the target of the DPP4 inhibitors, we have recently studied SNP rs6741949 G>C in intron 2 of the DPP4 gene in 1,976 participants of the Tübingen Family (TÜF) study for type 2 diabetes. This study showed that carriers of the minor C-allele had markedly reduced glucose-induced GLP1 levels, reduced insulin secretory capacity, and increased fasting and 2-hour glucose concentrations during an OGTT [68]. This may point to higher DPP4 enzyme levels/activity of variant allele carriers as compared to controls, in particular against the background of obesity, where DPP4 as an adipokine is already expressed at a significantly higher level [69]. Even though the SNP's functionality remains to be assessed, this genetic variant is a promising candidate for studying the genetic basis of interindividual differences in the response to DPP4 inhibitors.
Zimdahl et al. reported an interesting interaction between the most important type 2 diabetes gene TCF7L2, encoding T-cell-specific transcription factor 7-like 2, and the DPP4 inhibitor linagliptin [70]: pooled data from four phase-III, placebo-controlled trials (961 patients with type 2 diabetes in total), where subjects received linagliptin for 24 weeks, revealed that homozygous carriers of the diabetes risk allele, the minor T-allele of rs7903146 C>T, have an HbA1c response reduced by 0.26% compared with homozygous C-allele carriers. TCF7L2, a transcription factor downstream of the GLP1 receptor, regulates important β-cells genes, such as insulin, the incretin receptors and the prohormone convertases 1 and 2 (Figure 3) [71-73]. Therefore, the interaction between TCF7L2 and DPP4 inhibitors could indeed represent a pharmacodynamic interaction. Recently, we described another transcription factor in the incretin pathway, i.e., neuron-derived orphan receptor 1 (Nor1), which is also an important regulator of insulin gene expression and insulin secretion [74] (Figure 3). A SNP located in the 3'-flanking region of the Nor1 gene NR4A3, i.e., rs12686676 G>A, which tags a linkage block covering a large part of the second half of the gene, was associated with insulin secretion in the TÜF study and in the Finnish Metabolic Syndrome in Men (METSIM) study, with carriers of the major G-allele showing decreased insulin secretion [75]. Since this SNP interacts with the TCF7L2 SNP [74], compound carriers of the TCF7L2 T-allele and the NR4A3 G-allele could represent a subgroup of individuals displaying a markedly lower response to DPP4 inhibitors or incretin mimetics. This hypothesis is currently under investigation.
A genomic locus not formerly known to interact with the incretin pathway is the CTRB1/CTRB2 locus, harboring the chymotrypsinogen B1 and B2 genes. Using oligonucleotide arrays, depicting roughly 200,000 SNPs of interest for metabolic and atherosclerotic/cardiovascular disease traits (Metabochips), 't Hart et al. identified a non-coding SNP located between CTRB1 and CTRB2, i.e., rs7202877 T>G, that was associated with GLP1-induced insulin secretion during hyperglycemic clamp in 232 non-diabetic subjects [76]. In the DCS West-Friesland and the GoDARTS study, the authors demonstrated that patients carrying the minor G-allele had an HbA1c response to DPP4 inhibitor treatment reduced by 0.5%. Interestingly, there was no interaction with incretin mimetics. Finally, the G-allele was associated with higher chymotrypsinogen B1 and B2 expression in 35 whole pancreata and in 45 human islet samples.
3.3 Gene variants affecting the response to SGLT2 inhibitors
SGLT2 inhibitors represent the latest achievement in the field of type 2 diabetes therapy. Orally administered, they selectively inhibit SGLT2, the transporter responsible for 80-90% of glucose reabsorption in the kidney [77]. Thus, SGLT2 inhibition directly results in glucose excretion into urine and subsequently in reduction of plasma glucose concentrations [78].
Approximately 40% of administered canagliflozin is excreted unmetabolized into feces [79]. A minor part of canagliflozin is metabolized via glucuronidation to two pharmacologically inactive metabolites which are excreted into urine [79]. About seven percent of canagliflozin is metabolized by the cytochrome P450 isoform 3A4 [80]. Dapagliflozin is predominantly metabolized by glucuronidation, and its major metabolite is inactive and eliminated via renal excretion [80]. The pharmacokinetics of empagliflozin is still insufficiently reported. Pharmacogenetic studies investigating the impact of genetic variants in transporters and metabolizing enzymes on the anti-diabetic action of DPP4 inhibitors are currently lacking.
Regarding pharmacodynamics, Enigk et al. used a tagging SNP approach to assess the effect of common genetic variation in the SLC5A2 gene that encodes the drug target SGLT2 on type 2 diabetes and metabolic traits in two populations, Sorbs and participants of the Metabolic Syndrome Berlin Potsdam (MeSyBePo) study [81]. Upon meta-analysis of the data from non-diabetic individuals (n = 2,590), the authors found that carriers of the minor A-allele of the non-coding SNP rs9934336 G>A exhibit nominally lower 2-hour insulin concentrations during OGTT than major G-allele carriers. How this finding is related to SGLT2 function remains to be established; all the more so as the authors did not measure urinary glucose excretion. Nevertheless, SGLT2 is an attractive candidate for pharmacogenetic investigations, and initial studies testing the importance of SGLT2 SNPs for the efficacy of SGLT2 inhibitors are currently underway.
4. Clinical relevance of pharmacogenetic knowledge today
The efforts in genetics over the last two decades have markedly improved our understanding of the complex molecular network underlying the pathophysiology of type 2 diabetes. The clinical relevance of this knowledge is however severely limited by the observation that the sum of all diabetes risk alleles identified so far does not substantially contribute to the prediction of diabetes incidence or explain a relevant proportion of disease inheritance [4]. Therefore, we are currently a long way from a customized SNP array that predicts the individual diabetes risk in a clinically meaningful way.
Pharmacogenetics is a rather novel field in diabetes research, and does not a priori consider the known diabetes risk genes, but rather focuses on genes contributing to the pharmacokinetics and pharmacodynamics of anti-diabetic drugs [82]. Apart from the candidate gene approach, a first hypothesis-free GWAS approach was successful in identifying a previously unknown drug interaction partner, the ATM gene [58]. Both pharmacokinetics and pharmacodynamics will certainly provide novel and interesting information about pharmacogenetic interactions in the years to come. At the current, very early stage of pharmacogenetic investigations however, we experience several limitations:
Functional variants, in particular those leading to non-conservative amino acid exchanges, which are supposed to have the largest effect size, are usually non-frequent or even rare and need to be identified.
Variants identified by GWAS or tagging SNP approaches are usually more common, but have small effect sizes, and are non-coding with no proven functionality, requiring a subsequent systematic resequencing approach.
Many of the findings reported to date have not yet been replicated, or replication has even failed, possibly because of confounders such as non-genetic factors (e.g., age, body composition, dietary habits, background medication, renal function) and epigenetic alterations (e.g., DNA methylation, miRNAs).
Studies addressing the positive predictive value and the penetrance of the tested variants are still lacking. Therefore, genetic testing has currently not yet been included in national guidelines for the treatment of type 2 diabetes.
How can these problems be overcome if we believe in pharmacogenetics as an important underlying cause of inadequate treatment response, non-response, or even adverse drug reactions? Possibly, novel 'omics' methodologies, such as whole-genome sequencing, epigenomics, and metabolomics, and subsequent systems biology analysis strategies, including computational modeling [83], will help identify true causal variants. Of note, a very recent publication reports on a promising novel, and even cost-effective approach, i.e., methyl-C capture sequencing (MCC-Seq), for sequencing of functional methylomes, while simultaneously providing information about genetic variation [84].
For successful replication, standardized large-scale studies with comprehensive DNA sampling and comprehensive phenotypic characterization of patients, as already performed by some pharmaceutical companies, should become state-of-the-art for pharmacogenetics in type 2 diabetes research. Finally, prediction modeling including all causal variants in genes known to affect the pharmacokinetics and pharmacodynamics of a drug, but also other confounding factors (as excellently proven by the warfarin dosing algorithm, http://www.warfarindosing.org/Source/Home.aspx), will help to improve the clinical relevance of pharmacogenetic findings, and establish genetically determined treatment failure.
Thus, implementation of precision medicine including pharmacogenetic information is still challenging. Independently of all the issues mentioned above, aspects related to the education and training of clinicians need to be considered more appropriately. The correct interpretation of pharmacogenetic test results and subsequent dosing recommendations based on pharmacogenetic information require international standards and international pharmacogenetic networks (e.g., CPIC – Clinical Pharmacogenetics Implementation Consortium; https://www.pharmgkb.org/page/cpic; [85]). In this context, a major initiative in the United States, the Electronic and Medical Records and Genomics (eMERGE) network, aims to promote the implementation of personalized drug therapy by the use of electronic medical records and biobanks as optimal tools for the integration of genome and phenotype data [86]. Thus, major efforts are required in the future.
Acknowledgments
Disclosures
The authors have no conflicts of interest related to this work.
References
- 1.Pal A, McCarthy MI. The genetics of type 2 diabetes and its clinical relevance. Clin Genet. 2013;83(4):297–306. doi: 10.1111/cge.12055. [DOI] [PubMed] [Google Scholar]
- 2.Hara K, Shojima N, Hosoe J, Kadowaki T. Genetic architecture of type 2 diabetes. Biochem Biophys Res Commun. 2014;452(2):213–220. doi: 10.1016/j.bbrc.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Staiger H, Machicao F, Fritsche A, Haring HU. Pathomechanisms of type 2 diabetes genes. Endocr Rev. 2009;30(6):557–585. doi: 10.1210/er.2009-0017. [DOI] [PubMed] [Google Scholar]
- 4.Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci. 2010;1212:59–77. doi: 10.1111/j.1749-6632.2010.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jablonski KA, McAteer JB, de Bakker PI, Franks PW, Pollin TI, Hanson RL, Saxena R, Fowler S, Shuldiner AR, Knowler WC. et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes. 2010;59(10):2672–2681. doi: 10.2337/db10-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer UA, Zanger UM, Schwab M. Omics and drug response. Annu Rev Pharmacol Toxicol. 2013;53:475–502. doi: 10.1146/annurev-pharmtox-010510-100502. [DOI] [PubMed] [Google Scholar]
- 7.Zakim D, Schwab M. Data collection as a barrier to personalized medicine. Trends Pharmacol Sci. 2015;36(2):68–71. doi: 10.1016/j.tips.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Wang RS, Maron BA, Loscalzo J. Systems medicine: evolution of systems biology from bench to bedside. Wiley Interdiscip Rev Syst Biol Med. 2015;7(4):141–161. doi: 10.1002/wsbm.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Metthews DR. Management of hyperglycaemia in type 2 diabetes, 2015: a patient-centred approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2015;58(3):429–442. doi: 10.1007/s00125-014-3460-0. [DOI] [PubMed] [Google Scholar]
- 10.Stein SA, Lamos EM, Davis SN. A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin Drug Saf. 2013;12(2):153–175. doi: 10.1517/14740338.2013.752813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha B, Ghosal S. Pioglitazone - do we really need it to manage type 2 diabetes? Diabetes Metab Syndr. 2013;7(4):243–246. doi: 10.1016/j.dsx.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2):S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 13.McIntosh B, Cameron C, Singh SR, Yu C, Ahuja T, Welton NJ, Dahl M. Second-line therapy in patients with type 2 diabetes inadequately controlled with metformin monotherapy: a systematic review and mixed-treatment comparison meta-analysis. Open Med. 2011;5(1):e35–e48. [PMC free article] [PubMed] [Google Scholar]
- 14.Zeitler P, Hirst K, Pyle L, Linder B, Copeland K, Arslanian S, Cuttler L, Nathan DM, Tollefsen S, Wilfley D. et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N Engl J Med. 2012;366(24):2247–2256. doi: 10.1056/NEJMoa1109333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasquel FJ, Klein R, Adigweme A, Hinedi Z, Coralli R, Pimentel JL, Umpierrez GE. Metformin-associated lactic acidosis. Am J Med Sci. 2015;349(3):263–267. doi: 10.1097/MAJ.0b013e3182a562b7. [DOI] [PubMed] [Google Scholar]
- 16.Rena G, Pearson ER, Sakamoto K. Molecular action and pharmacogenetics of metformin: current understanding of an old drug. Diabetes Management. 2012;2:439–452. [Google Scholar]
- 17.Christensen MM, Brasch-Andersen C, Green H, Nielsen F, Damkier P, Beck-Nielsen H, Brosen K. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–850. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]
- 18.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334(9):574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 19.Pawlyk AC, Giacomini KM, McKeon C, Shuldiner AR, Florez JC. Metformin pharmacogenomics: current status and future directions. Diabetes. 2014;63(8):2590–2599. doi: 10.2337/db13-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29(4 Pt 2):6S28–6S35. doi: 10.1016/s1262-3636(03)72785-2. [DOI] [PubMed] [Google Scholar]
- 21.Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56(9):1898–1906. doi: 10.1007/s00125-013-2991-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pernicova I, Korbonits M. Metformin - mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 23.Anfossi G, Russo I, Bonomo K, Trovati M. The cardiovascular effects of metformin: further reasons to consider an old drug as a cornerstone in the therapy of type 2 diabetes mellitus. Curr Vasc Pharmacol. 2010;8(3):327–337. doi: 10.2174/157016110791112359. [DOI] [PubMed] [Google Scholar]
- 24.Haffner S, Temprosa M, Crandall J, Fowler S, Goldberg R, Horton E, Marcovina S, Mather K, Orchard T, Ratner R. et al. Intensive lifestyle intervention or metformin on inflammation and coagulation in participants with impaired glucose tolerance. Diabetes. 2005;54(5):1566–1572. doi: 10.2337/diabetes.54.5.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mithieux G, Rajas F, Zitoun C. Glucose utilization is suppressed in the gut of insulin-resistant high fat-fed rats and is restored by metformin. Biochem Pharmacol. 2006;72(12):1757–1762. doi: 10.1016/j.bcp.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ. et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emami RA, Fisel P, Nies AT, Schaeffeler E, Schwab M. Metformin and cancer: from the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol Sci. 2013;34(2):126–135. doi: 10.1016/j.tips.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R, Zanger UM, Keppler D, Schwab M, Schaeffeler E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50(4):1227–1240. doi: 10.1002/hep.23103. [DOI] [PubMed] [Google Scholar]
- 29.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG. et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117(5):1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shikata E, Yamamoto R, Takane H, Shigemasa C, Ikeda T, Otsubo K, Ieiri I. Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J Hum Genet. 2007;52(2):117–122. doi: 10.1007/s10038-006-0087-0. [DOI] [PubMed] [Google Scholar]
- 31.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM. et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou K, Donnelly LA, Kimber CH, Donnan PT, Doney AS, Leese G, Hattersley AT, McCarthy MI, Morris AD, Palmer CN. et al. Reduced-function SLC22A1 polymorphisms encoding organic cation transporter 1 and glycemic response to metformin: a GoDARTS study. Diabetes. 2009;58(6):1434–1439. doi: 10.2337/db08-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: A GoDARTS Study. Diabetes. 2015;64(5):1786–1793. doi: 10.2337/db14-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009;9(4):242–247. doi: 10.1038/tpj.2009.15. [DOI] [PubMed] [Google Scholar]
- 35.Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20(5):379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- 36.Leabman MK, Giacomini KM. Estimating the contribution of genes and environment to variation in renal drug clearance. Pharmacogenetics. 2003;13(9):581–584. doi: 10.1097/00008571-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Leabman MK, Huang CC, Kawamoto M, Johns SJ, Stryke D, Ferrin TE, DeYoung J, Taylor T, Clark AG, Herskowitz I. et al. Polymorphisms in a human kidney xenobiotic transporter, OCT2, exhibit altered function. Pharmacogenetics. 2002;12(5):395–405. doi: 10.1097/00008571-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, Urban TJ, Chen L, Yee SW, Choi JH. et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19(7):497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, Shin JG. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther. 2008;84(5):559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 40.Wang ZJ, Yin OQ, Tomlinson B, Chow MS. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genomics. 2008;18(7):637–645. doi: 10.1097/FPC.0b013e328302cd41. [DOI] [PubMed] [Google Scholar]
- 41.Kashi Z, Masoumi P, Mahrooz A, Hashemi-Soteh MB, Bahar A, Alizadeh A. The variant organic cation transporter 2 (OCT2)-T201M contribute to changes in insulin resistance in patients with type 2 diabetes treated with metformin. Diabetes Res Clin Pract. 2015;108(1):78–83. doi: 10.1016/j.diabres.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Teranishi K, Li S, Yee SW, Hesselson S, Stryke D, Johns SJ, Ferrin TE, Kwok P, Giacomini KM. Genetic variants in multidrug and toxic compound extrusion-1, hMATE1, alter transport function. Pharmacogenomics J. 2009;9(2):127–136. doi: 10.1038/tpj.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ha CJ, Wah YS, Kim MJ, Nguyen L, Ho LJ, Kang JO, Hesselson S, Castro RA, Stryke D, Johns SJ. et al. Identification and characterization of novel polymorphisms in the basal promoter of the human transporter, MATE1. Pharmacogenet Genomics. 2009;19(10):770–780. doi: 10.1097/FPC.0b013e328330eeca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stocker SL, Morrissey KM, Yee SW, Castro RA, Xu L, Dahlin A, Ramirez AH, Roden DM, Wilke RA, McCarty CA. et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2013;93(2):186–194. doi: 10.1038/clpt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes. 2009;58(3):745–749. doi: 10.2337/db08-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tkac I, Klimcakova L, Javorsky M, Fabianova M, Schroner Z, Hermanova H, Babjakova E, Tkacova R. Pharmacogenomic association between a variant in SLC47A1 gene and therapeutic response to metformin in type 2 diabetes. Diabetes Obes Metab. 2013;15(2):189–191. doi: 10.1111/j.1463-1326.2012.01691.x. [DOI] [PubMed] [Google Scholar]
- 47.He R, Zhang D, Lu W, Zheng T, Wan L, Liu F, Jia W. SLC47A1 gene rs2289669 G>A variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res Clin Pract. 2015;109(1):57–63. doi: 10.1016/j.diabres.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Choi JH, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, Mefford JA, Hesselson SE, Schlessinger A, Jenkins G. et al. A common 5'-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90(5):674–684. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisel P, Renner O, Nies AT, Schwab M, Schaeffeler E. Solute carrier transporter and drug-related nephrotoxicity: the impact of proximal tubule cell models for preclinical research. Expert Opin Drug Metab Toxicol. 2014;10(3):395–408. doi: 10.1517/17425255.2014.876990. [DOI] [PubMed] [Google Scholar]
- 50.Christensen MM, Pedersen RS, Stage TB, Brasch-Andersen C, Nielsen F, Damkier P, Beck-Nielsen H, Brosen K. A gene-gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet Genomics. 2013;23(10):526–534. doi: 10.1097/FPC.0b013e328364a57d. [DOI] [PubMed] [Google Scholar]
- 51.Nie W, Sweetser S, Rinella M, Green RM. Transcriptional regulation of murine Slc22a1 (Oct1) by peroxisome proliferator agonist receptor-alpha and -gamma. Am J Physiol Gastrointest Liver Physiol. 2005;288(2):G207–G212. doi: 10.1152/ajpgi.00057.2004. [DOI] [PubMed] [Google Scholar]
- 52.Saborowski M, Kullak-Ublick GA, Eloranta JJ. The human organic cation transporter-1 gene is transactivated by hepatocyte nuclear factor-4alpha. J Pharmacol Exp Ther. 2006;317(2):778–785. doi: 10.1124/jpet.105.099929. [DOI] [PubMed] [Google Scholar]
- 53.Kamiyama Y, Matsubara T, Yoshinari K, Nagata K, Kamimura H, Yamazoe Y. Role of human hepatocyte nuclear factor 4alpha in the expression of drug-metabolizing enzymes and transporters in human hepatocytes assessed by use of small interfering RNA. Drug Metab Pharmacokinet. 2007;22(4):287–298. doi: 10.2133/dmpk.22.287. [DOI] [PubMed] [Google Scholar]
- 54.Kajiwara M, Terada T, Asaka J, Ogasawara K, Katsura T, Ogawa O, Fukatsu A, Doi T, Inui K. Critical roles of Sp1 in gene expression of human and rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol. 2007;293(5):F1564–F1570. doi: 10.1152/ajprenal.00322.2007. [DOI] [PubMed] [Google Scholar]
- 55.Chen L, Hong C, Chen EC, Yee SW, Xu L, Almof EU, Wen C, Fujii K, Johns SJ, Stryke D. et al. Genetic and epigenetic regulation of the organic cation transporter 3, SLC22A3. Pharmacogenomics J. 2013;13(2):110–120. doi: 10.1038/tpj.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goswami S, Yee SW, Stocker S, Mosley JD, Kubo M, Castro R, Mefford JA, Wen C, Liang X, Witte J. et al. Genetic variants in transcription factors are associated with the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2014;96(3):370–379. doi: 10.1038/clpt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tkac I, Javorsky M, Klimcakova L, Zidzik J, Gala I, Babjakova E, Schroner Z, Stolfova M, Hermanova H, Habalova V. A pharmacogenetic association between a variation in calpain 10 (CAPN10) gene and the response to metformin treatment in patients with type 2 diabetes. Eur J Clin Pharmacol. 2015;71(1):59–63. doi: 10.1007/s00228-014-1774-y. [DOI] [PubMed] [Google Scholar]
- 58.Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ. et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43(2):117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Leeuwen N, Nijpels G, Becker ML, Deshmukh H, Zhou K, Stricker BH, Uitterlinden AG, Hofman A, van 't RE, Palmer CN. et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia. 2012;55(7):1971–1977. doi: 10.1007/s00125-012-2537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Florez JC, Jablonski KA, Taylor A, Mather K, Horton E, White NH, Barrett-Connor E, Knowler WC, Shuldiner AR, Pollin TI. The C allele of ATM rs11212617 does not associate with metformin response in the Diabetes Prevention Program. Diabetes Care. 2012;35(9):1864–1867. doi: 10.2337/dc11-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yee SW, Chen L, Giacomini KM. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44(4):359–360. doi: 10.1038/ng.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60(4):470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monami M, Cremasco F, Lamanna C, Marchionni N, Mannucci E. Predictors of response to dipeptidyl peptidase-4 inhibitors: evidence from randomized clinical trials. Diabetes Metab Res Rev. 2011;27(4):362–372. doi: 10.1002/dmrr.1184. [DOI] [PubMed] [Google Scholar]
- 64.Aschner P, Chan J, Owens DR, Picard S, Wang E, Dain MP, Pilorget V, Echtay A, Fonseca V. Insulin glargine versus sitagliptin in insulin-naive patients with type 2 diabetes mellitus uncontrolled on metformin (EASIE): a multicentre, randomised open-label trial. Lancet. 2012;379(9833):2262–2269. doi: 10.1016/S0140-6736(12)60439-5. [DOI] [PubMed] [Google Scholar]
- 65.Aso Y, Ozeki N, Terasawa T, Naruse R, Hara K, Suetsugu M, Takebayashi K, Shibazaki M, Haruki K, Morita K. et al. Serum level of soluble CD26/dipeptidyl peptidase-4 (DPP-4) predicts the response to sitagliptin, a DPP-4 inhibitor, in patients with type 2 diabetes controlled inadequately by metformin and/or sulfonylurea. Transl Res. 2012;159(1):25–31. doi: 10.1016/j.trsl.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 66.Golightly LK, Drayna CC, McDermott MT. Comparative clinical pharmacokinetics of dipeptidyl peptidase-4 inhibitors. Clin Pharmacokinet. 2012;51(8):501–514. doi: 10.1007/BF03261927. [DOI] [PubMed] [Google Scholar]
- 67.Scheen AJ. Pharmacokinetics of dipeptidylpeptidase-4 inhibitors. Diabetes Obes Metab. 2010;12(8):648–658. doi: 10.1111/j.1463-1326.2010.01212.x. [DOI] [PubMed] [Google Scholar]
- 68.Staiger H, Staiger K, Böhm A, Heni M, Linder K, Machicao F, Stefan N, Fritsche A, Häring HU. DPP4 gene variation affects GLP-1 secretion, insulin secretion, and glucose tolerance in humans with high body adiposity. Diabetes. 2013;62(Suppl 1):A433. doi: 10.1371/journal.pone.0181880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, Eckardt K, Kaufman JM, Ryden M, Muller S. et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60(7):1917–1925. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimdahl H, Ittrich C, Graefe-Mody U, Boehm BO, Mark M, Woerle HJ, Dugi KA. Influence of TCF7L2 gene variants on the therapeutic response to the dipeptidylpeptidase-4 inhibitor linagliptin. Diabetologia. 2014;57(9):1869–1875. doi: 10.1007/s00125-014-3276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.da Silva Xavier G, Mondragon A, Sun G, Chen L, McGinty JA, French PM, Rutter GA. Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia. 2012;55(10):2667–2676. doi: 10.1007/s00125-012-2600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet. 2009;18(13):2388–2399. doi: 10.1093/hmg/ddp178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loos RJ, Franks PW, Francis RW, Barroso I, Gribble FM, Savage DB, Ong KK, O'Rahilly S, Wareham NJ. TCF7L2 polymorphisms modulate proinsulin levels and beta-cell function in a British Europid population. Diabetes. 2007;56(7):1943–1947. doi: 10.2337/db07-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ordelheide AM, Gerst F, Rothfuss O, Heni M, Haas C, Thielker I, Herzberg-Schafer S, Bohm A, Machicao F, Ullrich S. et al. Nor-1, a novel incretin-responsive regulator of insulin genes and insulin secretion. Mol Metab. 2013;2(3):243–255. doi: 10.1016/j.molmet.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weyrich P, Staiger H, Stancakova A, Schafer SA, Kirchhoff K, Ullrich S, Ranta F, Gallwitz B, Stefan N, Machicao F. et al. Common polymorphisms within the NR4A3 locus, encoding the orphan nuclear receptor Nor-1, are associated with enhanced beta-cell function in non-diabetic subjects. BMC Med Genet. 2009;10:77. doi: 10.1186/1471-2350-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.'t Hart LM, Fritsche A, Nijpels G, van Leeuwen N, Donnelly LA, Dekker JM, Alssema M, Fadista J, Carlotti F, Gjesing AP. et al. The CTRB1/2 locus affects diabetes susceptibility and treatment via the incretin pathway. Diabetes. 2013;62(9):3275–3281. doi: 10.2337/db13-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91(2):733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 78.Jung CH, Jang JE, Park JY. A novel therapeutic agent for type 2 diabetes mellitus: SGLT2 inhibitor. Diabetes Metab J. 2014;38(4):261–273. doi: 10.4093/dmj.2014.38.4.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haas B, Eckstein N, Pfeifer V, Mayer P, Hass MD. Efficacy, safety and regulatory status of SGLT2 inhibitors: focus on canagliflozin. Nutr Diabetes. 2014;4:e143. doi: 10.1038/nutd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scheen AJ. Drug-drug interactions with sodium-glucose cotransporters type 2 (SGLT2) inhibitors, new oral glucose-lowering agents for the management of type 2 diabetes mellitus. Clin Pharmacokinet. 2014;53(4):295–304. doi: 10.1007/s40262-013-0128-8. [DOI] [PubMed] [Google Scholar]
- 81.Enigk U, Breitfeld J, Schleinitz D, Dietrich K, Halbritter J, Fischer-Rosinsky A, Enigk B, Muller I, Spranger J, Pfeiffer A. et al. Role of genetic variation in the human sodium-glucose cotransporter 2 gene (SGLT2) in glucose homeostasis. Pharmacogenomics. 2011;12(8):1119–1126. doi: 10.2217/pgs.11.69. [DOI] [PubMed] [Google Scholar]
- 82.Emami-Riedmaier A, Schaeffeler E, Nies AT, Morike K, Schwab M. Stratified medicine for the use of antidiabetic medication in treatment of type II diabetes and cancer: where do we go from here? J Intern Med. 2015;277(2):235–247. doi: 10.1111/joim.12330. [DOI] [PubMed] [Google Scholar]
- 83.Claussnitzer M, Dankel SN, Klocke B, Grallert H, Glunk V, Berulava T, Lee H, Oskolkov N, Fadista J, Ehlers K. et al. Leveraging cross-species transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell. 2014;156(1-2):343–358. doi: 10.1016/j.cell.2013.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allum F, Shao X, Guenard F, Simon MM, Busche S, Caron M, Lambourne J, Lessard J, Tandre K, Hedman AK. et al. Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants. Nat Commun. 2015;6:7211. doi: 10.1038/ncomms8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caudle KE, Klein TE, Hoffman JM, Muller DJ, Whirl-Carrillo M, Gong L, McDonagh EM, Sangkuhl K, Thorn CF, Schwab M. et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209–217. doi: 10.2174/1389200215666140130124910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, Kannry J, Zinberg R, Basford MA. et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15(10):761–771. doi: 10.1038/gim.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]