

Abstract
Autophagy is a cellular degradation process integral for promoting cellular adaptation during metabolic stress while also functioning as a cellular homeostatic mechanism. Mounting evidence also demonstrates that autophagy is induced upon loss of integrin-mediated cell attachments to the surrounding extracellular matrix (ECM). Analogous to its established cytoprotective role during nutrient starvation, autophagy protects cells from detachment-induced cell death, termed anoikis. Here, we review the significance of autophagy as an anoikis resistance pathway, focusing on the intracellular signals associated with integrins that modulate the autophagy response and dictate the balance between cell death and survival following loss of cell-matrix contact. In addition, we highlight recent studies demonstrating that autophagy functions in the upstream regulation of integrin-mediated cell adhesion via the control of focal adhesion remodeling, and discuss how these emerging interconnections between integrin-mediated adhesion pathways and autophagy influence cancer progression and metastasis.
Keywords: Autophagy, anoikis, integrins, focal adhesion, metastasis
Graphical abstract
Introduction
Autophagy is a highly conserved cellular catabolic pathway whose activity is finely tuned to promote homeostasis and adaptation to stress. During autophagy, cytoplasmic cargo is sequestered via a double membrane vesicle termed the autophagosome, that later fuses with lysosomes to deposit cargo for degradation. In response to nutrient starvation, the induction of this lysosomal degradation pathway facilitates the bulk degradation and recycling of proteins and organelles to sustain metabolic needs and promote cell survival. In addition, autophagy also occurs at basal levels to selectively degrade specific cytoplasmic components and damaged organelles, and in this way functions as a quality control mechanism to maintain homeostasis [1, 2].
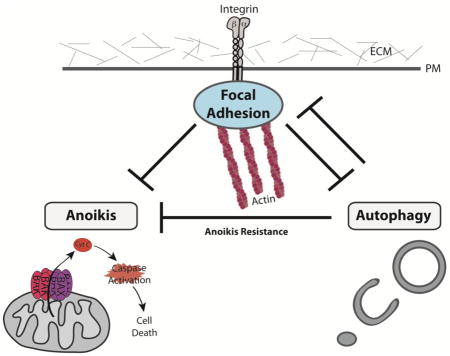
Autophagy is induced by a diverse array of insults in addition to nutrient deprivation, which includes ER stress, hypoxia, and remarkably, the loss of cell attachment to the extracellular matrix (ECM) [3]. Cellular interaction with the ECM via integrins, a large family of heterodimeric transmembrane receptors crucial for cell-matrix attachment, is imperative for cell survival and tissue maintenance and also plays a key role in mediating important cellular biological processes including differentiation and migration. Specifically, prolonged loss of integrin mediated cellular-ECM attachments leads to apoptotic cell death, which has been termed anoikis (Figure 1) [4, 5]. Furthermore, integrin receptors at cellular-ECM attachment sites provide the structural root for the formation of intracellular focal adhesions (FA), containing a number of structural and signaling protein complexes that link integrin mediated ECM connections to the dynamic cytoskeleton [6, 7]. In this way, integrins and their associated focal adhesions translate extracellular changes to intracellular responses and are thus tightly linked to several stress responses, including autophagy. Studies examining hypo-osmotic swelling in liver cells were among the first to demonstrate an inverse functional relationship between integrins and autophagy [8, 9]. Over the past decade, additional studies have more directly corroborated that autophagy is induced in response to loss of integrin-mediated attachment and started to delineate the diverse signaling mechanisms controlling autophagy downstream of integrin signaling. In this review, we discuss these pathways and how they intersect to regulate cell fate and survival.
Figure 1. The balance between autophagy and anoikis following ECM detachment.

Loss of integrin-mediated cell attachment to the extracellular matrix (ECM) promotes autophagy while also leading to cell death by anoikis. Healthy cells initiate an acute autophagy response following detachment, which functions as a cell survival mechanism that represses anoikis, affording cells with time to reattach. However, prolonged ECM detachment tips the balance towards anoikis. In contrast, cancer cells exhibit a durable autophagy response, which protects them from anoikis. Furthermore, by promoting anoikis resistance, autophagy may facilitate the ability of metastatic cancer cells to migrate away from the native ECM environment, intravasate into the systemic circulation and ultimately, to extravasate, reattach and proliferate at the metastatic site.
Furthermore, while integrin-mediated ECM attachments inhibit autophagy, new evidence indicates that autophagy in turn inhibits the dissolution of integrin-mediated attachments. Indeed, recent reports demonstrate that the autophagy pathway targets β1 integrin during nutrient starvation [10], and that selective autophagy specifically degrades focal adhesion proteins and promotes their turnover [11, 12]. Accordingly, we highlight these emerging interrelationships between autophagy, integrin receptors and focal adhesions as well as speculate how these fundamental insights into autophagy in cell adhesion receptor biology may influence cancer progression and metastasis.
Detachment-induced autophagy and anoikis resistance
Cell adhesion to the extracellular matrix (ECM) through integrin-mediated adhesions is critical for parenchymal cell survival and for sustaining proper cellular shape and function. The loss of matrix attachment leads to detachment-induced apoptosis, termed anoikis (Figure 1) [4, 5, 13]. Anoikis has been proposed to play a pivotal role during organ development through the clearance of luminal tissues, to maintain tissue homeostasis as well as safeguard against dysplastic cellular growth [14, 15]. Nevertheless, even in normal tissues, ECM detachment triggers anti-apoptotic signals, presumably as a failsafe mechanism to delay the onset of anoikis and allow cells to survive if they are able to reestablish ECM contact in a timely manner (Figure 1). One such signaling pathway is autophagy, which was initially observed in electron microscopic studies of ECM-detached cells during lumen formation in glandular epithelial structures (acini) grown in three dimensional organotypic cultures [16]. Since those initial observations, studies have uncovered that the induction of autophagy is specifically due to the lack of integrin-mediated cell adhesion [3, 17]. At the molecular level, integrins translate extrinsic environmental changes to the intracellular environment and regulate the signals that promote autophagy induction [3, 18]. Accordingly, the inhibition of ITGB1/integrin β1 via antibody-mediated function blockade is sufficient to induce autophagy in attached cells, while reconstituting a laminin-rich ECM in detached cells abrogates autophagy, corroborating that integrin-binding to ECM proteins negatively regulate autophagy [3].
Increased autophagy promotes epithelial cell survival and fitness in the absence of cell-matrix contact [3]. Upon detachment of non-transformed mammary epithelial cells autophagy is rapidly induced, which protects cells from anoikis over short term periods; accordingly, RNAi-mediated knockdown of key ATG proteins leads to increased caspase-3 activation and results in decreased clonogenic survival following reattachment; taken together, these results indicate that autophagy restricts cell death due to anoikis (Figure 1) [3]. Similar to normal cells, the loss of integrin-mediated ECM cellular attachments robustly induces autophagy in oncogene-transformed cells deprived of ECM contact. Furthermore, inhibiting autophagy in multiple human cancer cell lines either harboring oncogenic Ras or phosphatidylinositide 3-kinase (PI3K) blocks their ability to resist anoikis and proliferate in anchorage independent conditions [19, 20]. Thus, in both transformed and non-transformed cells, the induction of autophagy sufficiently promotes cellular adaptation and survival following the loss of integrin-mediated ECM contact. Furthermore, anoikis resistance in oncogene-expressing cells has been proposed as an important contributor to metastasis in vivo. In order to escape from the primary tumor environment and colonize at distant organ sites, cancerous cells must detach from the native extracellular matrix (ECM) and acquire the pathological ability to evade anoikis in the absence of proper contact to the ECM (Figure 1) [21–23].
Given the importance of anoikis in both normal tissue homeostasis and cancer progression, the initial discoveries demonstrating the induction of autophagy as an adaptive response to ECM detachment has motivated immense interest in the signals relayed from disengaged integrin receptors to the autophagic machinery. In the following sections, we detail several of the key pathways regulating detachment-induced autophagy identified over recent years. Interestingly, studies also indicate that select microRNAs may play an integral role in promoting anoikis following detachment by negatively regulating autophagy. For example, the loss of ECM cellular contacts leads to increased expression of the non-coding micro RNA (miRNA) miR-181a-5p, which is critical for promoting anoikis in mammary epithelial cell lines. Importantly, increased expression of miR-181a-5p has been found to promote anoikis by attenuating detachment-induced autophagosome formation and turnover in non-transformed MCF10A mammary epithelial cells [24]. This is consistent with previous findings demonstrating that miR-181a-5p negatively regulates starvation-induced autophagy in MCF7 breast cancer cells via targeting of ATG5 [25]. Although, the detailed mechanisms governing how this and other non-coding RNAs regulate detachment-induced autophagy remain to be elucidated, these studies reinforce that increased autophagy following loss of integrin-mediated ECM attachment promotes anoikis resistance.
BH3-only Bcl-2 family proteins in anoikis and autophagy
Anoikis resistance is largely attributed to the down-regulation of several pro-apoptotic BH3-only proteins of the Bcl-2 family that trigger the intrinsic cell death cascade following detachment (Figure 2, top) [13, 14, 26]. Bid and Bim are two members of the BH3-only family that become activated following loss of integrin mediated ECM attachments [27, 28]. When activated, Bid and Bim translocate to the mitochondria and directly promote the pro-apoptotic assembly of Bax-Bak within the outer mitochondrial membrane, which leads to cytochrome c release and promotes anoikis (Figure 2, bottom) [29]. Loss of cell attachments to the ECM promote the dissociation of Bim from the microtubule associated dynein light chain 1 complex where it is normally sequestered away from mitochondria and inactivated [30]. Furthermore, integrin and EGF receptor (EGFR) signaling has been shown to prevent anoikis by negatively regulating Bim expression, as detachment induced expression of Bim requires a lack of β1-integrin engagement and down-regulation of EGFR (Figure 2) [27]. As a result, Bim depletion in mammary epithelial cell lines results in protection from anoikis following detachment [27]. Further studies revealed that increased phosphorylation of Bim by MAPK/ERK kinase signaling, which is often deregulated in cancer, promotes proteasomal degradation of Bim, and leads to increased resistance to anoikis [31–33]. Remarkably, Bim was recently shown to inhibit starvation-induced autophagy by sequestering Beclin-1 to microtubules via interactions with dynein light chain and inhibiting its pro-autophagy function, an event also mediated by ERK/MAPK phosphorylation of Bim (Figure 2) [34]. Because anoikis evasion in inflammatory breast cancer cells is at least partly mediated by Bim sequestration to microtubules [35, 36], these studies suggest that dynamic changes in Bim localization to microtubules and interaction with autophagy regulators such as Beclin-1 may provide a rapid molecular switch that allows cells to tune their fate following loss of integrin-mediated ECM attachments.
Figure 2. BH3-only Bcl-2 family proteins in anoikis and autophagy.

Top: The BH3-only Bcl-2 proteins Bim and Bmf mediate anoikis by activating the intrinsic mitochondrial apoptosis pathway. In attached cells, intact integrin-mediated adhesions and active EGFR signaling repress these apoptotic pathway activators by multiple mechanisms. First, ERK1/2 signaling downstream of EGFR inhibits anoikis by downregulating the transcription of Bim and Bmf. Second, ERK phosphorylation promotes the proteasomal degradation of Bim, as well as promotes the sequestration of phosphorylated-Bim at microtubules. Remarkably, Bim binding to microtubules and the localization of Bmf to actin both function in the upstream regulation of autophagy via inhibiting the critical autophagy regulator, Beclin-1 (ATG6), hence suppressing autophagosome formation. Bottom: The loss of integrin-mediated adhesion attenuates EGFR signaling and destabilizes cytoskeletal-focal adhesion associations. As a result, the mRNA expression of Bim and Bmf is increased and the associations of Bim and Bmf with the cytoskeleton are lost. The subsequent recruitment of Bim and Bmf to mitochondria triggers the intrinsic apoptotic cascade, including BAX/BAK mediated cytochrome c release and caspase activation, leading to anoikis. At the same time, upon disassociation from Bim and Bmf, free Beclin1 promotes autophagy following ECM detachment. These opposing effects of Bim and Bmf on both autophagy and apoptosis likely dictate cell fate in ECM detached cells.
In addition to Bid and Bim, the BH3-only protein Bmf (Bcl2 modifying factor) functions as a sensitizer of cellular anoikis by indirectly promoting the interaction of Bid and Bim with mitochondrial Bax (Figure 2) [38–40]. Bmf is localized to actin filaments via interactions with dynein light chain 2 (DLC2) of the MYO5/myosin V motor complex [41]. Loss of integrin-mediated cellular attachments perturbs actin dynamics leading to the release of BMF from the cytoskeleton. Subsequent translocation of Bmf to the mitochondria enables Bmf to interact with and sequester the anti-apoptotic protein BCL-2, which promotes anoikis [38, 41, 42]. Importantly, recent work also suggests BMF as a potential regulator of autophagy during anoikis [43]. In attached cells, actin-associated Bmf interacts with Beclin1 and stabilizes its inhibitory interaction with Bcl-2, linking this autophagy complex to myosin V and inhibiting autophagosome formation (Figure 2, top) [43–45]. Furthermore, IFN-γ mediated suppression of Bmf promotes autophagy by reducing the interaction between Beclin-1 and Bcl-2 [45]. These recent findings motivate the hypothesis that detachment-induced release of Bmf from actin cables leads to the destabilization of the autophagy inhibitory Bcl-2/Beclin1 complex, resulting in increased autophagy (Figure 2, bottom) [43] [45, 46]. Furthermore, in addition to posttranslational regulation, Bmf is regulated transcriptionally following loss of integrin-mediated cell adhesion. As a result, transcriptional down-regulation of Bmf via diverse oncogenes or in response to hypoxia protects cells from anoikis [26, 47]. An important unanswered question is whether autophagy contributes to anoikis resistance downstream of these emerging pathways that suppress BMF transcription. Overall, these current findings broach previously unappreciated roles for Bim and Bmf, two BH3-only Bcl-2 family proteins that have been classically associated with the intrinsic apoptotic pathway, in mediating the switch between anoikis and autophagy following detachment. In addition to Bim and Bmf, autophagy has been also demonstrated to tune the kinetics and extent of mitochondrial apoptosis by regulating the expression of another BH3-domain containing protein, PUMA [48]. During death receptor-induced apoptosis, autophagy downregulates PUMA levels via an indirect mechanism that attenuates the transcription of PUMA mRNA [48]. As a result, autophagy impedes apoptosis by slowing the kinetics and efficiency of mitochondrial outer membrane permeabilization (MOMP), a cellular event widely considered to be the point of no return that marks commitment to apoptotic cell death. However, it remains to be determined whether this autophagy-dependent regulation of PUMA similarly impacts MOMP during anoikis. Further elucidation of molecular events controlling the transcriptional and spatio-temporal regulation of pro-apoptotic factors Bim, Bmf, and PUMA, as well as how such events modulate autophagy downstream of ECM detachment remain important topics for future study.
ER stress and mTOR signaling during detachment-induced autophagy
Significant interest in the mechanisms that drive anoikis resistance has led to a growing body of evidence indicating that epithelial cells activate ER stress signaling for survival following detachment [50, 51]. In healthy epithelial cells, surviving anoikis may allow for the possibility for reattachment, whereas in cancer, ER-stress signaling is often hijacked to promote long-term survival following loss of integrin-mediated attachments [50–52]. Specifically, recent evidence demonstrates that ECM-detached mammary epithelial cells robustly activate the ER stress kinase PERK, which has been shown to promote survival in a variety of non-transformed cell lines. In addition, PERK promotes anoikis resistance in diverse cancer cells, including ductal carcinoma breast cells, colorectal adenocarcinoma cells, and highly metastatic human fibrosarcoma cells [50, 53].
Following the accumulation of misfolded proteins in the ER, the unfolded protein response (UPR) kinase PERK becomes active and has classically been implicated as a negative regulator of protein translation through direct phosphorylation and inhibition of the translation initiation complex subunit eIF2α (Figure 3) [54, 55]. At the same time, PERK phosphorylation of eIF2α serves a key activator of the integrated stress response, resulting in the upregulation of the transcription factor ATF4, which coordinately promotes a number of cell survival pathways during stress [55, 56]. In particular, loss of ECM-attachment is correlated with a rapid decrease in ATP levels and an increase in ROS production [57, 58]. Detachment-induced PERK activation alleviates these stresses via ATF4-dependent expression of genes that promote ATP production, and the expression of a key antioxidant enzyme, heme oxygenase 1 (HO-1), which promotes the cellular oxidative response [53].
Figure 3. ER stress signaling during detachment-induced autophagy.

ECM detachment activates the ER stress kinase, PERK, which promotes anoikis resistance by activating detachment-induced autophagy. PERK is an EIF2α kinase that mediates the integrated stress response, marked by a global decrease in protein translation and de-repression of the ATF4 transcription factor, which leads to the coordinate induction of numerous autophagy regulatory proteins. PERK also promotes detachment-induced autophagy by activating AMPK, which leads to enhanced autophagy via the negative regulation of mTORC1. Furthermore, PERK phosphorylation of the transcription factor NRF2 promotes its activation and translocation to the nucleus; this results in increased transcription of p62/SQSTM1, which may amplify the autophagic response. Finally, reduced or aberrant integrin signaling has been linked to the activation of MYC in cancer cells, which induces the unfolded protein response (UPR) and promotes PERK activation.
Importantly, PERK-ATF4 signaling supports cell survival following detachment via the activation of autophagy [50, 58, 59]. Studies in diverse cell types demonstrate that during cell stress, activated ATF4 directly binds to and enhances the expression of a growing number of key autophagy related genes, including ATG3, ATG5, ATG7, ATG12, ATG16, Ulk1, Beclin1, MAP1LC3B, as well as the selective autophagy cargo adaptor proteins NBR1 and p62/SQSTM1 (Figure 3) [50, 53, 58–62]. Importantly, a recent study investigating the role of ATF4 in regulating anoikis resistance in highly metastatic human fibrosarcoma cells demonstrated that knockdown of ATF4 led to a significant increase in apoptosis that correlated with diminished autophagosome formation in suspended cells [53]. Furthermore, inhibiting the ATF4 dependent transcriptional upregulation of the key autophagy mediators ATG5, ATG7, or Ulk1 in these cells abrogated the ability of these cells to resist anoikis. Thus ATF4 mediated induction of autophagy in response to matrix detachment acts as a cytoprotective mechanism against anoikis (Figure 3).
However, because ectopic expression of ATF4 in PERK deficient cells fails to completely reverse the anoikis-sensitive phenotype, PERK may promote anoikis resistance via additional signaling mechanisms [53]. In this regard, PERK has been demonstrated to alternatively phosphorylate and activate the transcription factor NRF2, which is also implicated in PERK-mediated cell survival and cancer progression [63, 64]. Activated NRF2 has been demonstrated to enhance the transcription of p62/SQSTM1, which may amplify the autophagy response [65]. Importantly, targeted deletion of NRF2 results in increased cell death following ER-stress, intimating that NRF2, in addition to ATF4, may tune the autophagic response downstream of PERK and promote anoikis resistance [63]. NRF2 has gained significant attention in cancer biology, as it is often up regulated in a number of cancer cells of the prostate, breast, esophagus, and lung [64]. Furthermore, a high level of NRF2 signaling is associated with chemo-resistance to cytotoxic chemotherapeutics including doxorubicin and cisplatin [66, 67]. Future studies elucidating additional transcriptional targets by which non-canonical PERK-NRF2 signaling may regulate autophagy following loss of ECM attachment may reveal novel mechanisms utilized by cancer cells to induce autophagy and promote anoikis resistance and metastasis.
Upstream of PERK, a recent study in spontaneous lymphomas demonstrates a link between Myc-dependent transformation and ER-stress induced autophagy, which may have implications during ECM detachment (Figure 3) [52]. The activation of c-Myc leads to an unchecked increase in protein translation, which induces the UPR and activates PERK [52]. Genetic deletion of PERK in cells with increased UPR signaling due to oncogenic c-Myc leads to an increase in cell death and reduced tumor formation via the inhibition of cytoprotective autophagy. Thus, PERK functions as an enhancer of c-Myc-induced transformation by promoting autophagy. Notably, decreased α6β4 integrin signaling correlates with enhanced activity of the oncogenic transcription factor c-Myc, which has been proposed to promote cancer cell proliferation [68]. Given the importance of Myc in tumor progression and metabolic adaptation [69, 70], an important unanswered question is whether Myc-induced UPR signals contribute to detachment-induced autophagy and anoikis resistance. Furthermore, the mechanisms that induce autophagy downstream of the UPR during Myc transformation remains unclear; thus, further studies of ATF4 or NRF2 may reveal common pathways regulating autophagy following PERK activation downstream of Myc activation.
In addition to the transcriptional upregulation of autophagy genes, PERK has been recently implicated in promoting autophagy during ECM detachment via the inhibition of mTORC1 [51]. Mechanistic TOR (mTOR) kinase functions in two highly conserved and distinct multiprotein complexes, mTORC1 and mTORC2, that collectively regulate discrete aspects of cell growth and metabolism [71]. In particular, mTORC1 is an archetypal negative regulator of autophagy, as it directly phosphorylates and inhibits the autophagy initiating Ulk1-Atg13 complex [72, 73]. In mammary epithelial cells, the disruption of cell adhesion via a β1 integrin-blocking antibody leads to robust activation of the TORC1 inhibitor, AMPK, and subsequent inhibition of mTOR signaling as evidenced by decreased phosphorylation of the mTORC1 effector p70S6K (Figure 3) [51]. Importantly, the genetic deletion of PERK prevents detachment-induced AMPK activation and leads to sustained mTORC1 activity in these cells. The regulation of AMPK by PERK appears to be strictly dependent on ECM detachment rather than by classical ER stress inducers, such as thapsigargin [51]. PERK was found to activate AMPK and inhibit mTORC1 via a mechanism that required the AMPK kinase, LKB1, and subsequent activation of TSC2, an indirect inhibitor of mTORC1 activity. By decreasing mTORC1 activity, activation of PERK following loss of integrin-mediated attachments promoted the rapid induction of autophagy, which may be further tuned by the transcriptional mechanisms governing PERK-mediated autophagy to promote anoikis resistance. Further insight into the direct mechanisms by which integrin-mediated PERK signaling regulates LKB1 is integral to uncovering the distinct role that integrin-mediated adhesions play in autophagy activation. Furthermore, it remains to be determined whether PERK activation directs other AMPK targets to promote autophagy and cell survival following ECM detachment.
Recent findings also suggest that impaired autophagy may in turn lead to elevated ER stress. For example, a study examining autophagy and ER stress in aging adipose tissue-derived stromal vascular fraction cells (SVF) demonstrates that aged SVF cells exhibit decreased autophagy and increased ER-stress [74]. Furthermore, the loss of autophagy in mouse colorectal tumors via genetic inhibition of ATG5 or treatment with the lysosomal inhibitor chloroquine lead to elevated ER stress as well as increased CHOP and BiP expression [75]. Notably, this study also uncovers that the inhibition of autophagy and subsequent increase in ER stress results in increased apoptosis. Altogether, these recent findings support that the communication between autophagy and ER stress is bidirectional.
Integrin mediated NFKB-IKK signaling and detachment-induced autophagy
Recent studies further defining the intracellular signals that regulate detachment-induced autophagy demonstrate a link between integrin function and the activation of the IκB kinase (IKK) complex [17]. IKK functions as a central regulator of the nuclear factor kappa B (NFKB) signaling pathway, which has been implicated in autophagy regulation as well as anoikis resistance [76, 77]. NFKB has been shown to induce autophagy by transcriptionally activating the expression autophagy promoting protein Beclin-1 [78]. In attached and non-stressed cells, NFKB is sequestered and inactivated in the cytoplasm via the inhibitory molecule IκB. Activation of IKK family kinases leads to the phosphorylation and proteasomal degradation of IκB, enabling free NFKB dimers to translocate to the nucleus and promote the expression of pro survival genes to prevent apoptosis and enhance cell proliferation [79].
In addition to mediating NFKB activity, IKK phosphorylates additional cellular proteins and promotes a number of NFKB independent pathways, indicating a complex role in cell survival signaling [80]. Importantly, constitutively active IKK in human cells and murine models has been shown to stimulate starvation-induced autophagy via the activation of AMPK, resulting in a subsequent decrease in mTORC1 activity, as well as by activation of the kinase JNK1, which promotes autophagy by phosphorylating and inhibiting Bcl-2 to release Beclin-1 [77]. Importantly, because inhibition of NFKB does not suppress IKK-induced autophagy, these studies indicate that IKK promotes autophagy in an NFKB independent manner. Such results are consistent with reports demonstrating that IKK, but not NFKB, is critical for basal and starvation-induced expression of autophagy genes [81]. Furthermore, a study in mammary epithelial cells demonstrates that the inhibition of IKK or known activators of IKK, such as MAP3K7, attenuates the autophagy response following loss of α3β1 integrin mediated ECM attachments [17]. Importantly, this study demonstrates that IKK mediated activation of detachment-induced autophagy occurs independently of both mTORC1 signaling and NFKB, once again corroborating that an alternative signaling mechanism underlies IKK-mediated regulation of autophagy following matrix detachment. Consistent with a role in promoting autophagy-dependent cell survival, pharmacological IKK inhibition enhanced anoikis and accelerated apoptosis during acinar morphogenesis in 3D mammary epithelial culture [17]. It remains to be determined whether JNK1 signaling plays a role in mediating autophagy in detached mammary epithelial cells downstream of IKK. Further insight into the cellular contexts that influence the IKK signaling cascade to regulate autophagy is necessary given that IKK mediated autophagy regulation is tightly linked to anoikis resistance and cancer progression [80, 82].
Overall, given the complexity of the signaling pathways mediating ECM detachment-induced autophagy, it is likely that both autophagy regulation and anoikis resistance relies on the integration of multiple signaling pathways. Further understanding how these diverse signals cooperate in the temporal regulation of autophagy and anoikis following ECM detachment will provide new perspectives into the highly context-dependent functions of the autophagy pathway in cancer progression in vivo. Finally, though autophagy is traditionally viewed as an autodigestive process, it is also important to recognize that non-canonical autophagy pathways may influence integrin-mediated signaling as well as anoikis resistance. Remarkably, a recent study found that detachment results in the co-internalization of the receptor tyrosine kinase c-Met and β1-integrin from the cell surface; both are subsequently recruited to LC3B-positive autophagy-related endomembranes (ARE) [37]. In detached cells, this internalized pool of unliganded β1-integrin promotes c-Met-dependent phosphorylation and activation of ERK1/2, which supports anchorage-independent cell survival, tumorigenesis, invasion and lung colonization. Importantly, this signaling is dependent on ATG5 and Beclin1, but not on ATG13, thus implicating a non-canonical autophagy pathway in mediating this process [37].
Autophagy dependent regulation of focal adhesion dynamics
As the foundation of cellular-ECM contact sites, integrin mediated focal adhesions not only anchor the cell to the substratum but also play a pivotal role in transducing both mechanical and biochemical signaling events within the cell. As such, focal adhesions comprise both scaffolding and signaling proteins organized into functional layers that link cytoplasmic portions of integrins to signaling and adaptor molecules and facilitate the transmission of forces via interactions with cytoskeletal elements (Figure 4) [6, 7]. Focal adhesions are highly dynamic and the balance of assembly to disassembly from the ECM is often altered in metastatic cancer cells that must detach, undergo migration, and re-reattach at the metastatic site [83].
Figure 4. Autophagy dependent regulation of focal adhesion dynamics.

Focal adhesion turnover is mediated by autophagy. During cell migration, nascent focal adhesions (NA) mature into larger focal adhesions (FA) that are connected to cytoskeletal elements, which transmit the forces necessary to promote forward motility. Subsequently, these FAs must disassemble to allow the cell body to productively move forward, and selective autophagy is crucial for this process. Autophagy promotes focal adhesion disassembly via multiple pathways involving LC3 and autophagy cargo receptors that selectively target FA components. First, NBR1 promotes focal adhesion remodeling by recruiting LC3-containing autophagosomes to FAs. NBR1 binds to focal adhesion proteins, such as vincullin, FAK, paxillin, and zyxin, and promotes their sequestration within autophagosomes. Second, Src-mediated phosphorylation of paxillin targets phosphorylated paxillin to autophagosomes via the direct association with LC3. Third, active Src serves a direct target for the autophagy cargo adaptor, Cbl, which recruits autophagosomes to degrade Src. Finally, autophagic targeting of RhoA via p62/SQSTM1 perturbs FA actin dynamics.
Tension via connections to the cytoskeleton is a key mediator of focal adhesion dynamics. Increased tension induces changes in focal adhesion protein conformations and alters protein phosphorylation states to promote adhesion stabilization and growth [84–87]. One such event includes the phosphorylation of the focal adhesion adaptor protein, paxillin (PXN), by the focal adhesion kinase (FAK) and Src complex [88–90]. Studies on the spatio-temporal regulation of focal adhesions suggest that FAK-Src localizes to nascent focal adhesions prior to PXN and tyrosine phosphorylation by this complex promotes disassembly [86, 90, 91]. In contrast, establishment of tension at nascent focal adhesions promotes the accumulation of de-phosphorylated PXN at focal adhesions and loss of FAK association, which encourages strengthening of focal adhesions and further recruitment of downstream signaling partners (Figure 2) [86, 90, 91].
A number of signaling mechanisms also regulate the disassembly of focal adhesions, and recently, autophagy has been implicated as a key mediator of this process by targeting directly focal adhesion proteins for degradation (Figure 4) [83]. Phosphorylation of PAX by Src has been shown to promote its interaction with the autophagosome associated LC3 proteins and subsequent autophagic degradation leading to focal adhesion disassembly [12]. Additionally, in cells with reduced or altered FAK signaling, autophagy has been demonstrated to promote the targeted degradation of activated Src, an event mediated by the LC3 interacting autophagy cargo receptor, Cbl (Figure 4) [92]. Furthermore, the degradation of active Src via autophagy results in the subsequent degradation of Ret, a tyrosine kinase that activates FAK at focal adhesions, hence broaching additional molecular links between autophagy and focal adhesion disassembly [93, 94]. Further studies have revealed that the mTORC1 effector p70S6K is the kinase responsible for targeting Src to autophagosomes, further implicating autophagy signaling pathways in the control of integrin-mediated ECM cellular attachments (Figure 4) [95]. In addition to degrading focal adhesion components, autophagy has been demonstrated to selectively target a key regulator of actinomyosin contractility, the GTPase RhoA, which modulates tension via myosin II activation (Figure 4) [96]. When ubiquitinated, RhoA becomes a substrate for the selective autophagy cargo adaptor protein, p62 [97]. While this study mainly focused on the role of autophagy mediated RhoA degradation in mediating cytokinesis, this relationship is consistent with autophagy supporting focal adhesion turnover as RhoA is known to localize at focal adhesions to promote focal adhesion maturation [98]. Furthermore, when bound to unanchored integrins, RhoA induces a tension-dependent cell death response, and degradation by autophagy may provide a mechanism for anoikis resistance [98]. Indeed, initial studies suggest that autophagy may affect migration by capturing RhoA in autophagic vesicles located near the lamellipodia of migrating cells; accordingly, inhibiting autophagosome formation leads to the release of active RhoA to the plasma membrane, resulting in increased actin polymerization and impaired cell motility [99]. Thus, while non-selective autophagy may mediate cell stress responses and metabolic adaptation following ECM detachment, it is becoming apparent that various forms of selective autophagy serve to fine-tune focal adhesion dynamics and signaling.
This notion is further strengthened by most recent findings demonstrating that the selective autophagy cargo adaptor, NBR1, is a key regulator of focal adhesion turnover and migration [11]. Specifically, NBR1 depletion impairs focal adhesion turnover, leading to the accumulation of focal adhesions, increased cell spreading, and reduced cell migration [11]. NBR1 localizes to the leading edge of focal adhesions, and has been shown to biochemically interact with a number of focal adhesion proteins including, PXN, vinculin, zyxin, and FAK, which may mediate its localization to focal adhesions (Figure 4) [11]. However it remains to be determined whether NBR1 directly mediates the autophagic degradation of its associated focal adhesion proteins, which would be consistent with its role in promoting disassembly. Interestingly, both NBR1 and FAK are directly regulated by the glycogen synthase kinase, GSK3β, which has also been implicated in mediating cell migration and spreading by promoting focal adhesion disassembly, suggesting another regulatory link between NBR1 and focal adhesions [100–102]. However, further studies are required to unveil the spatio-temporal regulation of GSK3β in mediating the interplay between NBR1 and FAK at focal adhesions and the implications this may have on cell adhesion and motility. Furthermore, given the existence of a number of autophagy cargo adaptors and the importance of autophagy in mediating focal adhesion dynamics, further studies on the interrelationships between the various autophagy cargo adaptors and focal adhesion proteins is required to unveil a more in-depth mechanism for selective autophagy in tuning cellular adhesions to the ECM.
The selective degradation of cargo by autophagy is often specified by ubiquitination, as demonstrated by RhoA mediated degradation by p62 [103, 104]. Evidence demonstrates a requirement for the ubiquitin-binding (UBA) domain of NBR1 in focal adhesion disassembly, suggesting that NBR1 substrates at focal adhesions may be regulated by ubiquitin [11]. Furthermore, several E3 ubiquitin ligases have been localized at focal adhesions, where they have been linked to the ubiquitination of focal adhesion proteins, such as FAK and paxillin [11, 105–108]. However, the precise role of ubiquitination in the selective autophagy of focal adhesion proteins remains unknown as autophagic targeting of specific substrates has also been shown to occur via ubiquitin-independent mechanisms [92, 103, 109, 110]. Furthermore, the identity of such E3 ligases mediating selective autophagy in focal adhesion turnover remains elusive. In light of recent findings, one tempting candidate may be a member of the cullin-RING ligases (CRL) recently implicated in mediating both focal adhesions as well as autophagy [111, 112]. CRL3 has recently been localized to focal adhesion structures at the cell leading edge and demonstrated to promote cell migration by ubiquitinating the microtubule interacting protein, EB1 [112]. Highlighting the importance of CRL proteins in mediating focal adhesion dynamics, cells with decreased activity of the CRL3 KLHL21 ligase or non-ubiquitinated EB1 are unable to migrate as they exhibit defects in focal adhesion dynamics and in the formation of lamellipodia [112]. Additionally, CRL proteins have been linked to autophagy via the targeted degradation of a number of autophagy regulators including DAPK, which regulates the dissociation of Beclin-1 from Bcl-2; Atg14L, which is involved in autophagosome initiation; and the ULK1 substrate Ambra1, which associates with other CRL proteins to promote autophagy by suppressing mTORC1 [111, 113–116]. Thus, autophagy and CRL family ligases may antagonize each other at focal adhesions, or they may cooperate to regulate the ubiquitination and potential degradation of a number of focal adhesion proteins; however, these possibilities remain open to investigation. Overall, further understanding the interplay between E3 ligases and selective autophagy may reveal novel interactions important for focal adhesion remodeling in both normal and cancerous cells.
Taken together, these current findings demonstrate that autophagy functions to fine-tune focal adhesion dynamics. Thus, one can speculate that heightened autophagic activity positively influences cancer progression and metastatic migration by degrading focal adhesions of cells to the ECM while also preventing anoikis. In support of this model, reduced autophagy in highly aggressive 4T1 mammary carcinoma cells is associated with reduced metastatic dissemination from the primary tumor using in vivo orthotopic transplantation models [12].
Highlighting the pathological importance of maintaining proper focal adhesion dynamics, PXN overexpression and phosphorylation has been demonstrated to promote tumor progression and is associated with poor overall survival in lung cancer patients [117]. Current findings demonstrate that increased Src mediated PXN phosphorylation leads to increased expression of the anti-apoptotic Bcl-2 protein, and that this contributes to resistance of the chemotherapeutic cisplatin [118]. This is supported clinically by the observation that cisplatin-based chemotherapies are less effective on patients with lung tumors expressing heightened PXN signaling and Bcl-2 expression compared to negative tumors [118]. Transcriptional activation of Bcl-2 downstream of FAK-Src PXN signaling was attributed to increased ERK signaling [118]. Moreover, it is interesting to note that mechanical stress has been shown to promote nuclear localization of FAK, thus broaching the possibility that FAK may serve a more direct role in signaling changes to the nucleus to regulate cell fate following detachment [119]. Additionally, autophagic targeting of focal adhesion proteins has been shown to promote cancer cell survival when FAK signaling is reduced or altered [120]. Thus, it is imperative to uncover a more comprehensive map of autophagy targets at focal adhesions and define their physiological roles in order to further understand the tumor promoting mechanisms resulting from increased autophagy.
Autophagy and cancer metastasis
The lethality of epithelial cancers (carcinomas) is primarily attributed to the unchecked invasion and metastases of tumor cells throughout the body [21, 23]. The loss of integrin-mediated ECM attachments and resistance to anoikis are both intimately linked to the tumorigenesis and metastasis of various cancers and recent evidence suggests that autophagy facilitates the dissemination of tumor cells to metastatic sites [12]. Furthermore, a clinical study focusing on melanoma and breast cancer tumors found strong correlations between high expression levels of the autophagy protein, LC3B, and both increased metastasis and worsened patient outcome [121]. Furthermore, increased expression of Beclin1 protein has been shown to predict poor survival and high occurrence of distant metastasis in nasopharyngeal carcinoma [122]. Heightened Beclin1 activity has also been implicated in the growth and metastasis of colorectal cancer [123]. These findings are corroborated by a recent study of hepatocellular carcinoma (HCC) cell metastasis, which revealed that silencing of autophagy genes BECN1 (Beclin1) or ATG5 significantly decreased the incidence of pulmonary metastases in vivo, attenuated anoikis-resistance via the up regulation of pro-apoptotic factors, and decreased lung colonization of HCC cells [124]. Furthermore, HCC metastases exhibited significantly higher LC3 expression than primary tumors, with the highest levels associating with early metastatic colonies [125]. In addition, the chemical and genetic inhibition of autophagy in experimental metastasis models results in delayed metastasis development, while stimulation of autophagy hastens development [126]. Thus, for several cancer types, the modulation of autophagy is linked to metastatic progression and autophagy-mediated anoikis resistance has been implicated in early stage metastatic disease.
Current studies are only beginning to uncover the how autophagy regulates tumor metastasis. While autophagy does not correlate with invasion and migration of human HCC cells in mouse models, autophagy inhibition decreases the invasive capacity in glioma cells via a mechanism that may involve the autophagy cargo adaptor protein, p62/SQSTM1 [127–129]. However, it remains unclear whether other autophagy cargo adaptors mediate invasion in vivo. Interestingly, during starvation in cell culture, the invasion of human HCC cells is promoted by autophagy-mediated activation of the transforming growth factor beta (TGFβ) signaling pathway, which stimulates the epithelial-to-mesenchymal transition (EMT) [130]. Remarkably, in addition to regulating autophagy, integrins have been shown to modulate TGFβ signaling and EMT progression in mammary epithelial cells [131], broaching the hypothesis that integrin-mediated regulation of autophagy may act as a context dependent promoter of metastasis by driving pro-metastatic EMT signals. However, other studies support an inhibitory role for autophagy during EMT. A study investigating the role of death-effector domain-containing DNA-binding protein (DEDD) in EMT and metastasis revealed that increased DEDD expression inhibits EMT by activating autophagy-dependent degradation of SNAIL and TWIST, two principal transcriptional EMT regulators [132]. Interestingly, DEDD levels promotes autophagy via direct interaction with class III PI-3-kinase (PI3KC3)/Beclin1. However, it remains to be determined whether autophagy functions as an in vivo suppressor of EMT during metastasis [133].
In addition to modulating EMT, integrin signaling promotes metastasis via the formation of integrin β1 adhesion plaques and filopodium-like protrusions in extravasated cancer cells, which allows for proliferation of micro-metastases into the surrounding tissue parenchyma. Because early colonization is a rate-limiting step in metastasis [134], one can speculate that integrin-mediated autophagic signaling may modulate these later stages of adhesion to promote the attachment and proliferation of disseminated metastatic cells. Importantly, the proliferative outgrowth of micrometastases after extravasating into the parenchyma has been shown to be highly dependent on the integrin β1-mediated activation of focal adhesion kinase (FAK) [135]. While autophagy promotes the selective degradation and turnover of integrin-mediated focal adhesions, more direct roles for autophagy in mediating FAK-driven proliferative signaling remains unknown.
In addition to modulating migration and adhesion dynamics, other mechanisms are likely at play with regards to the role of autophagy in metastasis. For example, autophagy promotes oncogenic RAS-driven invasion and metastasis by regulating the secretion of a multiple pro-invasive factors, including the pro-migratory cytokine, interleukin-6 (IL-6) [136]. Nevertheless, given the roles of integrin-mediated signaling and autophagy in anoikis resistance and metastasis, further investigation into the interrelationships of the autophagy pathway with cell-matrix adhesion signaling cascades should provide unique insights into the complex functions of autophagy during cancer metastasis in vivo.
Concluding remarks
Integrin-focal adhesion signaling and autophagy exhibit an inverse relationship, as the loss of ECM-cellular attachments or disengagement of integrin receptors induces a protective autophagy response that negatively regulates anoikis and promotes cell survival. While healthy cells possess the capability of tipping the balance towards cell death following prolonged detachment, cancer cells rely on a durable autophagy response to overcome anoikis and survive in the absence of integrin-mediated cellular adhesions to the ECM. Moreover, the emerging cell biological roles of autophagy in promoting focal adhesion turnover and integrin–mediated intrinsic signaling provide new perspective into the mechanisms through which autophagy influences cancer progression. Further investigating how autophagy controls cell adhesion and how integrin-mediated signaling pathways in turn regulate autophagy will enhance our understanding of how these pathways influence important biological processes such as cellular motility and differentiation. Finally, such research will undoubtedly offer fresh insight into the paradoxical roles of autophagy in cancers, especially in the progression towards metastasis.
Highlights.
Loss of integrin-mediated cell matrix adhesion induces autophagy, which protects cells from detachment-induced cell death (anoikis).
Multiple intracellular signaling pathways regulate detachment-induced autophagy.
Autophagy regulates integrin-mediated cell adhesion by promoting focal adhesion remodeling.
Acknowledgments
Grant support includes the NIH (CA126792, CA188404 to JD), the DOD BCRP (W81XWH-11-1-0130 to JD), and the Samuel Waxman Cancer Research Foundation (to JD). The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–72. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 2.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–37. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–6. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- 6.Geiger B, Yamada KM. Molecular Architecture and Function of Matrix Adhesions. Csh Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanchanawong P, Shtengle G, Ramko EB, Davidson MW, Hess HF, Waterman C. Nanoscale Protein Architecture of Focal Adhesions. Biophys J. 2010;98:211a–a. [Google Scholar]
- 8.vom Dahl S, Schliess F, Reissmann R, Gorg B, Weiergraber O, Kocalkova M, et al. Involvement of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver. The Journal of biological chemistry. 2003;278:27088–95. doi: 10.1074/jbc.M210699200. [DOI] [PubMed] [Google Scholar]
- 9.Schliess F, Reissmann R, Reinehr R, vom Dahl S, Haussinger D. Involvement of integrins and Src in insulin signaling toward autophagic proteolysis in rat liver. The Journal of biological chemistry. 2004;279:21294–301. doi: 10.1074/jbc.M313901200. [DOI] [PubMed] [Google Scholar]
- 10.Tuloup-Minguez V, Hamai A, Greffard A, Nicolas V, Codogno P, Botti J. Autophagy modulates cell migration and beta1 integrin membrane recycling. Cell Cycle. 2013;12:3317–28. doi: 10.4161/cc.26298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, et al. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol. 2016;212:577–90. doi: 10.1083/jcb.201503075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, et al. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016;15:1660–72. doi: 10.1016/j.celrep.2016.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 14.Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124:3189–97. doi: 10.1242/jcs.072165. [DOI] [PubMed] [Google Scholar]
- 15.Debnath J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy. 2008;4:351–3. doi: 10.4161/auto.5523. [DOI] [PubMed] [Google Scholar]
- 16.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 17.Chen N, Debnath J. IkappaB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy. 2013;9:1214–27. doi: 10.4161/auto.24870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 19.Chen N, Eritja N, Lock R, Debnath J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3-kinase in three-dimensional culture. Oncogene. 2013;32:2543–54. doi: 10.1038/onc.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5:402–18. doi: 10.1016/j.apsb.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sethi N, Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer. 2011;11:735–48. doi: 10.1038/nrc3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei JL, Li YC, Ma ZL, Jin YX. MiR-181a-5p promotes anoikis by suppressing autophagy during detachment induction in the mammary epithelial cell line MCF10A. Protein Cell. 2016;7:305–9. doi: 10.1007/s13238-016-0255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy. 2013;9:374–85. doi: 10.4161/auto.23117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whelan KA, Caldwell SA, Shahriari KS, Jackson SR, Franchetti LD, Johannes GJ, et al. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell. 2010;21:3829–37. doi: 10.1091/mbc.E10-04-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5:733–40. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- 28.Valentijn AJ, Gilmore AP. Translocation of full-length Bid to mitochondria during anoikis. The Journal of biological chemistry. 2004;279:32848–57. doi: 10.1074/jbc.M313375200. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–7. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 30.Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–96. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- 31.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–93. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 32.Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–25. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao L, Zhang Z, Han L, Du J, Liang X, Liu Y, et al. Mitogen-activated protein kinase pathway is pivotal for anoikis resistance in metastatic hepatoma cells. Mol Med Rep. 2014;9:1121–7. doi: 10.3892/mmr.2014.1952. [DOI] [PubMed] [Google Scholar]
- 34.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–70. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchheit CL, Angarola BL, Steiner A, Weigel KJ, Schafer ZT. Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ. 2015;22:1275–86. doi: 10.1038/cdd.2014.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchheit CL, Schafer ZT. BIM-EL localization: The key to understanding anoikis resistance in inflammatory breast cancer cells. Mol Cell Oncol. 2016;3:e1011474. doi: 10.1080/23723556.2015.1011474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrow-McGee R, Kishi N, Joffre C, Menard L, Hervieu A, Bakhouche BA, et al. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat Commun. 2016;7:11942. doi: 10.1038/ncomms11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hausmann M, Leucht K, Ploner C, Kiessling S, Villunger A, Becker H, et al. BCL-2 modifying factor (BMF) is a central regulator of anoikis in human intestinal epithelial cells. The Journal of biological chemistry. 2011;286:26533–40. doi: 10.1074/jbc.M111.265322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–12. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- 40.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 42.Kutuk O, Letai A. Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ. 2010;17:1624–35. doi: 10.1038/cdd.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgado M, Tesfaigzi Y. Is BMF central for anoikis and autophagy? Autophagy. 2014;10:168–9. doi: 10.4161/auto.26759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delgado M, Tesfaigzi Y. BH3-only proteins, Bmf and Bim, in autophagy. Cell Cycle. 2013;12:3453–4. doi: 10.4161/cc.26696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Contreras AU, Mebratu Y, Delgado M, Montano G, Hu CA, Ryter SW, et al. Deacetylation of p53 induces autophagy by suppressing Bmf expression. J Cell Biol. 2013;201:427–37. doi: 10.1083/jcb.201205064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graef M, Friedman JR, Graham C, Babu M, Nunnari J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell. 2013;24:2918–31. doi: 10.1091/mbc.E13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, et al. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3787–92. doi: 10.1073/pnas.0700115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, et al. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7:45–52. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014;16:47–54. doi: 10.1038/ncb2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616–29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013;32:4932–40. doi: 10.1038/onc.2012.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–34. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest. 2015;125:2592–608. doi: 10.1172/JCI78031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16:5493–501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16:533–44. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singleton DC, Harris AL. Targeting the ATF4 pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:1189–202. doi: 10.1517/14728222.2012.728207. [DOI] [PubMed] [Google Scholar]
- 57.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–13. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF, Aguirre-Ghiso JA. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS One. 2007;2:e615. doi: 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.B'Chir W, Maurin Ac, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8:3838–47. doi: 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- 61.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–35. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 62.Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. The Journal of Clinical Investigation. 120:127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ryoo IG, Lee SH, Kwak MK. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid Med Cell Longev. 2016;2016:2428153. doi: 10.1155/2016/2428153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. The Journal of biological chemistry. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433–8. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dydensborg AB, Teller IC, Groulx JF, Basora N, Pare F, Herring E, et al. Integrin alpha6Bbeta4 inhibits colon cancer cell proliferation and c-Myc activity. BMC Cancer. 2009;9:223. doi: 10.1186/1471-2407-9-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11–21. doi: 10.1016/j.semcdb.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of Cell Science. 2009;122:3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–4. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghosh AK, Mau T, O'Brien M, Garg S, Yung R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging (Albany NY) 2016;8:2525–37. doi: 10.18632/aging.101083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakitani K, Hirata Y, Hikiba Y, Hayakawa Y, Ihara S, Suzuki H, et al. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer. 2015;15:795. doi: 10.1186/s12885-015-1789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yan SR, Joseph RR, Rosen K, Reginato MJ, Jackson A, Allaire N, et al. Activation of NF-kappaB following detachment delays apoptosis in intestinal epithelial cells. Oncogene. 2005;24:6482–91. doi: 10.1038/sj.onc.1208810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29:619–31. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29:2594–608. doi: 10.1128/MCB.01396-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 80.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–13. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 81.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene. 2011;30:1727–32. doi: 10.1038/onc.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yeh PY, Lu YS, Ou DL, Cheng AL. IkappaB kinases increase Myc protein stability and enhance progression of breast cancer cells. Mol Cancer. 2011;10:53. doi: 10.1186/1476-4598-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kenific CM, Wittmann T, Debnath J. Autophagy in adhesion and migration. J Cell Sci. 2016;129:3685–93. doi: 10.1242/jcs.188490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc T. 2004;32:416–20. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- 85.Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nature Cell Biology. 2007;9:858–68. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. Journal of Cell Science. 2007;120:137–48. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
- 87.del Rio A, Perez-Jimenez R, Liu RC, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009;323:638–41. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aponte M, Jiang W, Lakkis M, Li MJ, Edwards D, Albitar L, et al. Activation of platelet-activating factor receptor and pleiotropic effects on tyrosine phospho-EGFR/Src/FAK/paxillin in ovarian cancer. Cancer Res. 2008;68:5839–48. doi: 10.1158/0008-5472.CAN-07-5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sachdev S, Bu Y, Gelman IH. Paxillin-Y118 phosphorylation contributes to the control of Src-induced anchorage-independent growth by FAK and adhesion. Bmc Cancer. 2009:9. doi: 10.1186/1471-2407-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature Cell Biology. 2004;6:154. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 91.Hu YL, Lu SY, Szeto KW, Sun J, Wang YX, Lasheras JC, et al. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci Rep-Uk. 2014:4. doi: 10.1038/srep06024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signallin. Nature Cell Biology. 2012;14:51–U76. doi: 10.1038/ncb2386. [DOI] [PubMed] [Google Scholar]
- 93.Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells (vol 13, pg 733, 2012) Embo Rep. 2012;13:867. doi: 10.1038/embor.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Plaza-Menacho I, Morandi A, Mologni L, Boender P, Gambacorti-Passerini C, Magee AI, et al. Focal Adhesion Kinase (FAK) Binds RET Kinase via Its FERM Domain, Priming a Direct and Reciprocal RET-FAK Transactivation Mechanism. Journal of Biological Chemistry. 2011;286:17292–302. doi: 10.1074/jbc.M110.168500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sandilands E, Schoenherr C, Frame MC. p70S6K is regulated by focal adhesion kinase and is required for Src-selective autophagy. Cell Signal. 2015;27:1816–23. doi: 10.1016/j.cellsig.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. Journal of Cell Biology. 1999;144:1235–44. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, et al. Autophagy Plays a Critical Role in the Degradation of Active RHOA, the Control of Cell Cytokinesis, and Genomic Stability. Cancer Res. 2013;73:4311–22. doi: 10.1158/0008-5472.CAN-12-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma Z, Myers DP, Wu RF, Nwariaku FE, Terada LS. P66(Shc) mediates anoikis through RhoA. Journal of Cell Biology. 2007;179:23–31. doi: 10.1083/jcb.200706097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ, et al. Autophagy and SQSTM1 on the RHOA(d) again: emerging roles of autophagy in the degradation of signaling proteins. Autophagy. 2014;10:201–8. doi: 10.4161/auto.27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, et al. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol Cell Biol. 2006;26:898–911. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bianchi M, De Lucchini S, Marin O, Turner DL, Hanks SK, Villa-Moruzzi E. Regulation of FAK Ser-722 phosphorylation and kinase activity by GSK3 and PP1 during cell spreading and migration. Biochem J. 2005;391:359–70. doi: 10.1042/BJ20050282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nicot AS, Lo Verso F, Ratti F, Pilot-Storck F, Streichenberger N, Sandri M, et al. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy. 2014;10:1036–53. doi: 10.4161/auto.28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nature Cell Biology. 2010;12:836–41. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 104.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang C. Roles of E3 ubiquitin ligases in cell adhesion and migration. Cell Adhes Migr. 2010;4:10–8. doi: 10.4161/cam.4.1.9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schiller HB, Fassler R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. Embo Rep. 2013;14:509–19. doi: 10.1038/embor.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wolfenson H, Lavelin I, Geiger B. Dynamic Regulation of the Structure and Functions of Integrin Adhesions. Dev Cell. 2013;24:447–58. doi: 10.1016/j.devcel.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deng SS, Huang C. E3 ubiquitin ligases in regulating stress fiber, lamellipodium, and focal adhesion dynamics. Cell Adhes Migr. 2014;8:49–54. doi: 10.4161/cam.27480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- 110.Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24:787–95. doi: 10.1038/cr.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cui D, Xiong X, Zhao Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016;11:8. doi: 10.1186/s13008-016-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Courtheoux T, Enchev RI, Lampert F, Gerez J, Beck J, Picotti P, et al. Cortical dynamics during cell motility are regulated by CRL3(KLHL21) E3 ubiquitin ligase. Nat Commun. 2016;7:12810. doi: 10.1038/ncomms12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang T, Dong K, Liang W, Xu D, Xia H, Geng J, et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. Elife. 2015;4:e06734. doi: 10.7554/eLife.06734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. 2014;31:734–46. doi: 10.1016/j.devcel.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 115.McEwan DG, Dikic I. Cullins keep autophagy under control. Dev Cell. 2014;31:675–6. doi: 10.1016/j.devcel.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 116.Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM, Chen RH. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010;29:1748–61. doi: 10.1038/emboj.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu DW, Cheng YW, Wang J, Chen CY, Lee H. Paxillin predicts survival and relapse in non-small cell lung cancer by microRNA-218 targeting. Cancer Res. 2010;70:10392–401. doi: 10.1158/0008-5472.CAN-10-2341. [DOI] [PubMed] [Google Scholar]
- 118.Wu DW, Wu TC, Wu JY, Cheng YW, Chen YC, Lee MC, et al. Phosphorylation of paxillin confers cisplatin resistance in non-small cell lung cancer via activating ERK-mediated Bcl–2 expression. Oncogene. 2014;33:4385–95. doi: 10.1038/onc.2013.389. [DOI] [PubMed] [Google Scholar]
- 119.Cardoso AC, Pereira AH, Ambrosio AL, Consonni SR, Rocha de Oliveira R, Bajgelman MC, et al. FAK Forms a Complex with MEF2 to Couple Biomechanical Signaling to Transcription in Cardiomyocytes. Structure. 2016;24:1301–10. doi: 10.1016/j.str.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 120.Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14:51–60. doi: 10.1038/ncb2386. [DOI] [PubMed] [Google Scholar]
- 121.Lazova R, Camp Rl, Klump V, Siddiqui Sf, Amaravadi Rk, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. doi: 10.1158/1078-0432.CCR-11-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wan XB, Fan Xj, Chen My, Xiang J, Huang Py, Guo L, Wu X-Y, et al. Elevated Beclin 1 expression is correlated with HIF-1alpha in predicting poor prognosis of nasopharyngeal carcinoma. doi: 10.4161/auto.6.3.11303. [DOI] [PubMed] [Google Scholar]
- 123.Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br J Cancer. 2010;103:1209–14. doi: 10.1038/sj.bjc.6605904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9:2056–68. doi: 10.4161/auto.26398. [DOI] [PubMed] [Google Scholar]
- 125.Peng YF, Shi YH, Shen YH, Ding ZB, Ke AW, Zhou J, et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS One. 2013;8:e74407. doi: 10.1371/journal.pone.0074407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barnard RA, Regan DP, Hansen RJ, Maycotte P, Thorburn A, Gustafson DL. Autophagy Inhibition Delays Early but Not Late-Stage Metastatic Disease. J Pharmacol Exp Ther. 2016;358:282–93. doi: 10.1124/jpet.116.233908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beckner ME, Fellows-Mayle W, Zhang Z, Agostino NR, Kant JA, Day BW, et al. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer. 2010;126:2282–95. doi: 10.1002/ijc.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 2012;11:2022–9. doi: 10.4161/cc.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699–712. doi: 10.1038/onc.2012.111. [DOI] [PubMed] [Google Scholar]
- 130.Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34:1343–51. doi: 10.1093/carcin/bgt063. [DOI] [PubMed] [Google Scholar]
- 131.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. The Journal of biological chemistry. 2001;276:46707–13. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 132.Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, et al. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012;72:3238–50. doi: 10.1158/0008-5472.CAN-11-3832. [DOI] [PubMed] [Google Scholar]
- 133.Ramos JW, Kojima TK, Hughes PE, Fenczik CA, Ginsberg MH. The death effector domain of PEA-15 is involved in its regulation of integrin activation. The Journal of biological chemistry. 1998;273:33897–900. doi: 10.1074/jbc.273.51.33897. [DOI] [PubMed] [Google Scholar]
- 134.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706–21. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A. 2009;106:10290–5. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–79. doi: 10.1158/2159-8290.CD-13-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]