

Abstract
Viral myocarditis is common and can progress to cardiac failure. Cardiac cell pro-inflammatory responses are critical for viral clearance, however sustained inflammatory responses contribute to cardiac damage. The transcription factor NF-κB regulates expression of many proinflammatory cytokines, but basal and induced activation of NF-κB in different cardiac cell types have not been compared. Here, we used primary cultures of cardiac myocytes and cardiac fibroblasts to identify cardiac cell type-specific events. We show that while viral infection readily stimulates activation of NF-κB in cardiac fibroblasts, cardiac myocytes are largely recalcitrant to activation of NF-κB. Moreover, we show that cardiac myocyte subpopulations differ in their NF-κB subcellular localization and identify the cis-Golgi as a cardiac myocyte-specific host compartment. Together, results indicate that NF-κB-dependent signaling in the heart is cardiac cell type-specific, likely reflecting mechanisms that have evolved to balance responses that can be either protective or damaging to the heart.
Keywords: NF-κB, myocarditis, cardiac myocyte, cardiac fibroblast, cardiomyocyte, Golgi, reovirus, heart
Introduction
Viral myocarditis is a leading cause of sudden death in young adults and occurs in approximately 10% of patients with unexpected heart failure (1-4). Although most cases of myocarditis are asymptomatic and resolve spontaneously through regulated pro-inflammatory responses, incomplete viral clearance and/or autoimmune responses against cardiac autoantigens can result in chronic inflammation, dilated cardiomyopathy (DCM) and cardiac failure (2-4). Thus while cytokines expressed in the heart help maintain normal cardiac function and repair, their over-production can result in heart disease (5-7).
The expression of many cytokines is regulated by the family of transcription factors, NF-κB, which differ depending on their subunit composition (8). NF-κB is held inactive in a cytoplasmic complex with its inhibitor, IκB. Cell stimulation phosphorylates and inactivates IκB, inducing NF-κB translocation to the nucleus (9). There, NF-κB induces the expression of hundreds of genes that regulate processes including cell survival, apoptosis, inflammation, and antiviral responses (9-11). The consequences of NF-κB activation are cell type-specific and depend on the type, magnitude, and duration of the stimulus (12, 13).
The role of the NF-κB family of transcription factors in cardiac health and disease has been well-studied, particularly during acute ischemia/reperfusion injury (8, 12). In primary cultures of rat cardiac myocytes, the extent of activation of NF-κB upon pro-inflammatory stimulation is dependent on both the developmental stage of the mice used to generate the cultures, as well as the age of the cultures (14). These results likely reflect a developmental impairment of the IκB kinase β (IKKβ) in cardiac myocytes after birth that dampens NF-κB-dependent expression of pro-inflammatory genes (14). Accordingly, conditional or constitutive expression of recombinant IKKβ in cardiac myocytes is highly damaging and can result in their apoptosis, cardiac inflammation, DCM, and heart failure (15, 16). Cardiac damage is also induced by over-expression of TRAF3 interacting protein 2 (TRAFIP2), another activator of NF-κB (17). Thus, activation of NF-κB can be highly deleterious to the heart. However, activation of NF-κB can also be critical to maintain cardiac health. For example, one important NF-κB-regulated cytokine is IFN-β (18, 19), and the increased virus damage induced in mice lacking NF-κB can be ameliorated by treatment with IFN-β (20). The balance between positive and negative impacts of NF-κB therefore must be tightly regulated.
The heart achieves balance for many functions by segregating tasks between its two predominant cell types: cardiac myocytes and cardiac fibroblasts. Cardiac myocytes are highly specialized muscle cells that are poorly replenished, while cardiac fibroblasts, found directly adjacent to cardiac myocytes, maintain cardiac structure and are readily replenished after injury (21). We found that these two cardiac cell types differ dramatically in both their basal and virus-induced IFN-β responses, and together form an integrated network to protect the heart from viral damage (22-26). Importantly, activation of NF-κB in cardiac myocytes has never been compared to any other cardiac cell type, leaving the relative consequences in cardiac myocytes and cardiac fibroblasts entirely unexplored.
Here, we compared basal and stimulated activation of NF-κB between primary cultures of cardiac myocytes and cardiac fibroblasts. Results identified cardiac myocytes as recalcitrant to virus- or cytokine-induced activation of NF-κB relative to cardiac fibroblasts, and identified an unusual localization of NF-κB to the cis-Golgi in cardiac myocytes. It is likely that these distinct cell type-specific phenotypes aid the heart in balancing the positive and negative impacts of NF-κB activation.
Results
TNF-α-induced activation of NF-κB is cardiac cell type-specific
To compare the kinetics of activation of NF-κB between cardiac cell types, primary cultures of murine cardiac fibroblasts and cardiac myocytes were stimulated with TNF-α. Lysates were probed in immunoblots to detect degradation of IκBα and phosphorylation of the NF-κB subunit p65 (also known as RelA), two critical steps required for activation of NF-κB (27) (Figure 1A). In cardiac fibroblasts, TNF-α induced rapid and transient proteolytic degradation of IκBα with concomitant IKKβ-dependent phosphorylation of p65. In cardiac myocytes, however, neither IκBα degradation nor p65 phosphorylation was detected at any time point. Results suggest that TNF-α readily induces activation of NF-κB in cardiac fibroblasts as it does in most cell types, but does not do so in cardiac myocytes. As expected, TNF-α stimulation did not alter levels of IKKβ or its regulatory subunit IKKγ/NEMO in either cell type (Figure 1B). To determine downstream consequences, qRT-PCR was used to quantitate expression of several genes known to be regulated by NF-κB following TNF-α stimulation (28). Following TNF-α stimulation, induction of NF-κB-responsive genes (IL-1β, CCL2/MCP-1, CXCL1, IκBα, and A20) was 2- to 3-fold higher in cardiac fibroblasts than in cardiac myocytes (Figure 1C). Notably, GM-CSF, a leukocyte growth factor whose induction by TNF-α is NF-κB-independent (29), was induced similarly in the two cell types. Finally, several biosynthetic enzymes whose expression is induced at a later phase post-TNF-α stimulation (PGES and COX-2), were induced minimally and similarly in the two cell types. Together, results indicate that the kinetics and magnitude of NF-κB activation is cardiac cell type-specific, with cardiac myocytes displaying an attenuated response to TNF-α stimulation.
Figure 1. The magnitude of NF-κB activation after TNF-α stimulation is cardiac cell type-specific.
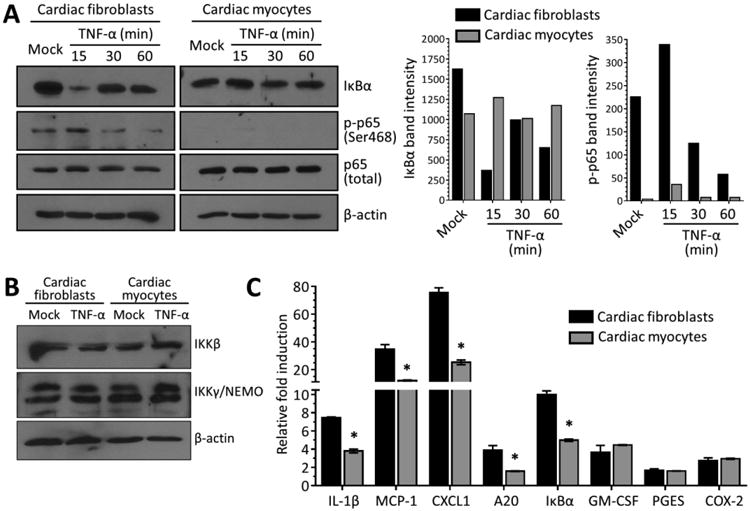
(A) Primary cardiac myocyte or cardiac fibroblast were stimulated with 50 ng/ml TNF-α or media (‘mock’) and whole-cell protein lysates were harvested at the indicated times post-stimulation. Protein lysates were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted using the indicated antibodies. (B) Protein lysates from primary cultures stimulated for 1 h with media or TNF-α were resolved and transferred as in (A) and immunoblotted using the indicated antibodies. (C) Primary cultures were stimulated with TNF-α as in panel A for 60 min prior to mRNA harvest. Levels of mRNA expression for the indicated genes was analyzed by qRT-PCR and normalized to GAPDH expression. Fold induction by TNF-α is expressed relative to ‘mock’ for each culture (mean ± SD) for a representative of at least two independent experiments. *, Significantly different from cardiac fibroblasts (P < 0.05).
NF-κB localizes to perinuclear compartments in a subset of cardiac myocytes
The differences in activation of NF-κB between the two cardiac cell types (Figure 1) suggested that subcellular localization might also differ. Accordingly, the NF-κB subunit p65 was immunostained in primary cultures of cardiac myocytes, cardiac fibroblasts, and undifferentiated and differentiated skeletal muscle cells as additional muscle cell types for comparison (Figure 2A). Cultures were also immunostained for the intermediate filament protein vimentin as a marker that is expressed well in cardiac fibroblasts but minimally in cardiac myocytes (30). As expected, p65 localized to the cytoplasm in unstimulated cardiac fibroblasts and readily translocated to the nucleus upon treatment with TNF-α. However, cardiac myocytes displayed two different phenotypes: some displayed a classic cytoplasmic p65 localization while others also displayed a dramatic perinuclear p65 localization. Perinuclear p65 localization was specific to cardiac myocytes and was never observed in cardiac fibroblasts or skeletal muscle cells.
Figure 2. NF-κB localizes to perinuclear compartments in cardiac myocytes.
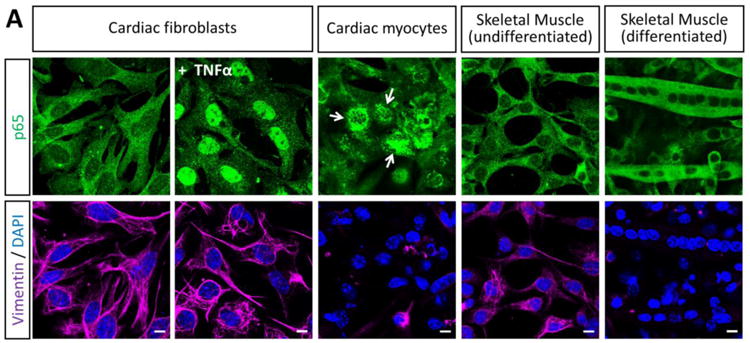
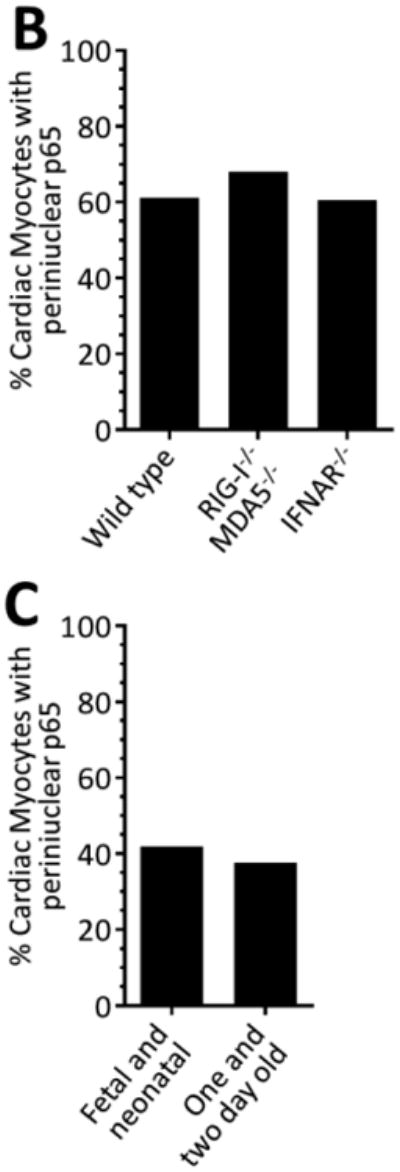
(A) Primary cardiac myocyte, cardiac fibroblast, or skeletal muscle cultures were fixed for immunofluorescent microscopy using antibodies against p65 and vimentin. Nuclei were counterstained with DAPI. Scale bar = 20 μm. (B) Primary cardiac myocyte cultures generated from the indicated mouse strain were immunostained as in panel A and the percentage of cardiac myocytes (defined as ‘vimentin negative’) were scored according to the localization of p65 in each cardiac myocyte (n = 104 – 214 per case) for a representative experiment. (C) Cardiac myocyte cultures were generated from either ‘fetal and neonatal’ or ‘one- and two-day-old’ wild-type mice and scored as in panel B.
Cardiac myocytes express high basal levels of IFN-β and a subset of IFN-stimulated genes (ISGs) compared to cardiac fibroblasts that pre-arm them against viral infection (23-25). While activation of NF-κB is typically required for IFN-β expression, it can also be activated by the ISG PKR (31), thereby providing a positive amplification loop for activation of NF-κB. We hypothesized that cardiac myocytes might sequester NF-κB in perinuclear compartments to dampen IFN-β-induced activation of NF-κB. However, NF-κB remained perinuclear in primary cardiac myocyte cultures generated from mice lacking the IFN-α/β receptor (Figure 2B) or the upstream cytosolic viral RNA sensors RIG-I and MDA5, indicating that basal IFN signaling is not responsible for the unusual localization of NF-κB in cardiac myocytes.
Given the precedent for post-natal attenuation of NF-κB signaling in rat cardiac myocytes (14), we compared NF-κB localization in cardiac myocytes generated from fetal and neonatal mice to those from one- and two-day-old mice (Figure 2C). Results showed no difference in NF-κB distribution in the two populations of cardiac myocytes. Similarly, while prolonged culture conditions can also attenuate NF-κB signaling in cardiac myocytes (14), we found no difference in the localization of NF-κB when comparing two-day-old cultures to four-day-old cardiac myocyte cultures (data not shown). Together, results indicate that the unusual NF-κB localization in cardiac myocytes is not dependent on basal IFN signaling or post-natal changes in the heart.
Perinuclear NF-κB in cardiac myocytes co-localizes with cis-Golgi membranes and is present in the adult murine heart
To identity the subcellular compartments containing NF-κB, we immunostained cardiac myocyte cultures with antibodies against p65 and markers for subcellular organelles. Perinuclear NF-κB structures corresponded to the cis-Golgi, based on p65 co-localization with GM130 (Figure 3A) but not with peroxisomal or mitochondrial markers (data not shown). To further confirm the compartments as cis-Golgi, cultures were treated with the pharmacological agent Brefeldin A (BFA), which reversibly redistributes most Golgi-resident proteins to intermediate compartments (32). Indeed, BFA treatment induced the expected redistribution of GM130 but, remarkably, also eliminated perinuclear localization of NF-κB in cardiac myocytes (Figure 3B), confirming association of NF-κB with the cis-Golgi in these cells. Moreover, this association was evident in a subset of cardiac myocytes in cardiac sections of adult mice (Figure 4), indicating maintenance of NF-κB in the cis-Golgi in situ throughout adulthood.
Figure 3. NF-κB perinuclear compartments are associated with the cis-Golgi.
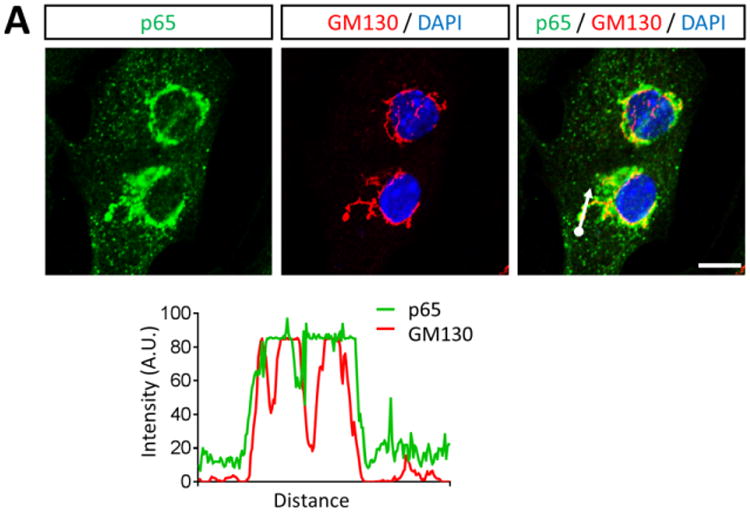
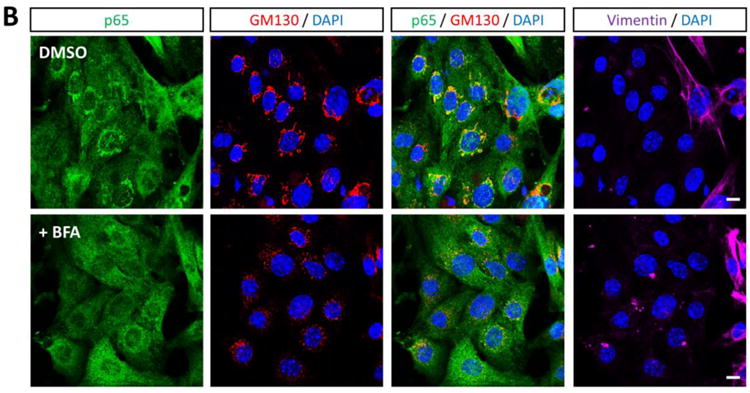
(A) Primary cardiac myocytes were fixed for immunofluorescent microscopy using antibodies against p65 and GM130. Nuclei were counterstained with DAPI. Histograms display measured fluorescence intensity along the drawn line in the overlay panel. (B) Cardiac myocytes were treated with DMSO (vehicle) or 10 μg/ml BFA for 6 h. Cells were then fixed and immunostained as in panel A in addition to vimentin. Scale bar = 10 μm
Figure 4. NF-κB is associated with the cis-Golgi in the adult heart.
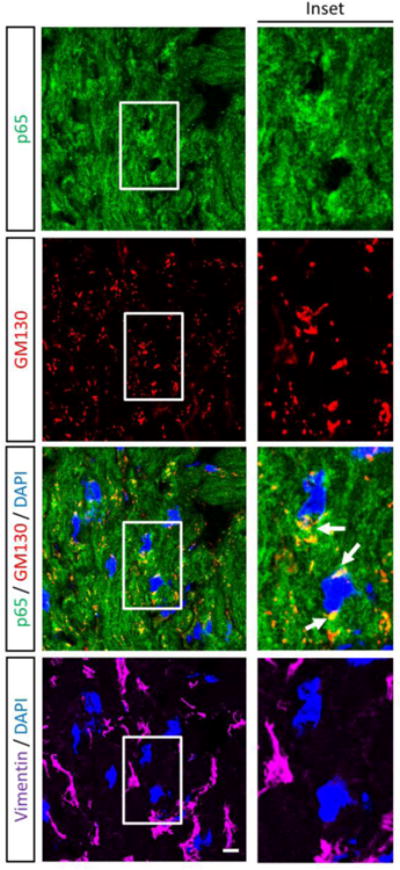
Immunofluorescent microscopy of a cross section of mouse myocardium using antibodies against p65, GM130, and vimentin. Nuclei were counterstained with DAPI. Scale bar = 10 μm
Differential kinetics in basal nucleocytoplasmic shuttling of NF-κB in cardiac cells
Many but not all cardiac myocytes in culture (Figures 2A and 3B) and in situ (Figure 4) displayed perinuclear NF-κB. The existence of two populations of cardiac myocytes distinguished by their ability to support activation of NF-κB has been proposed previously where one subset hosts a functional NF-κB system while the other expresses NF-κB that is resistant to IKKβ-dependent activation and constitutively shuttles between the nucleus and cytoplasm (8). To investigate NF-κB nucleocytoplasmic shuttling, cultures were treated with the irreversible CRM1-specific inhibitor Leptomycin B (LMB) which prevents p65 nuclear export (33) (Figure 5). In cardiac fibroblasts, LMB induced rapid accumulation of p65 in the nucleus as early as 1 h post-treatment, indicating that NF-κB undergoes constant nucleocytoplasmic shuttling in these cells. In contrast, nuclear accumulation of p65 in cardiac myocytes was only minimal and most evident at later times post-LMB treatment. In addition, p65 nuclear intensity was greater in cardiac fibroblasts than in cardiac myocytes at equivalent time points. Overall, results suggest that nucleocytoplasmic shuttling of NF-κB in cardiac myocytes is minimal compared to cardiac fibroblasts and likely present in only a fraction of the cardiac myocyte population.
Figure 5. The magnitude and kinetics of basal NF-κB nucleocytoplasmic shuttling is cardiac cell type-specific.
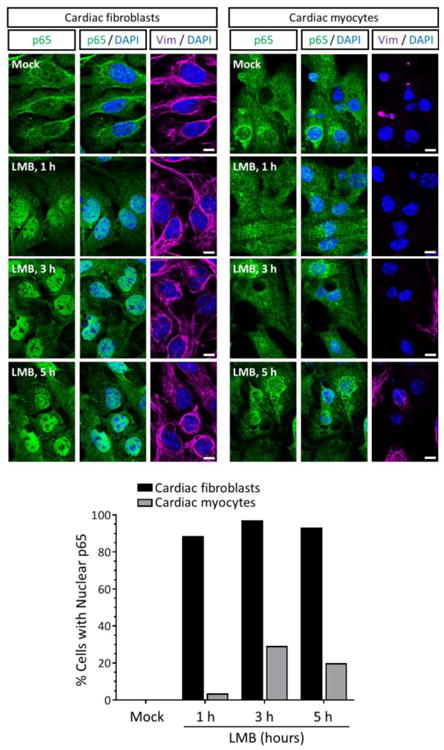
Primary cardiac fibroblast or myocyte cultures were treated with 20 nM LMB or media alone (‘mock’), fixed at the indicated times post-treatment, and immunostained as indicated. Scale bar = 10 μm. The percentage of cardiac fibroblasts (n = 31 – 112 cells per condition) or cardiac myocytes (n = 32 – 61 cells per condition) displaying nuclear p65 is indicated for a representative of two independent experiments.
NF-κB nuclear translocation in cardiac myocytes is largely resistant to the stimulatory effects of TNF-α and Poly(I:C)
The minimal NF-κB nucleocytoplasmic shuttling in cardiac myocytes (Figure 5) suggested they might also be poorly responsive to stimuli. To address this possibility, cultures were stimulated with either TNF-α or Poly(I:C), a synthetic double-stranded RNA that mimics viral infection (Figure 6A, C). In contrast to Figure 5, mock-treated cardiac fibroblast cultures displayed some nuclear NF-κB, likely reflecting the shorter incubation time after media change (one hour vs. five hours) and thus shorter recovery time after transient stress. However as anticipated, stimulation resulted in efficient nuclear translocation of p65 in cardiac fibroblasts. In contrast, nuclear translocation of p65 was almost undetectable in stimulated cardiac myocytes. To assess possible transient translocation, cardiac cultures were stimulated with Poly(I:C) and simultaneously treated with LMB to inhibit nuclear export (Figure 6B, D). As expected, p65 in all fibroblasts was strictly nuclear. Strikingly, nuclear p65 was evident in approximately two-thirds of the LMB-treated, Poly(I:C)-stimulated cardiac myocytes, albeit at a lower intensity than in cardiac fibroblasts. To determine whether LMB could also increase stimulus-induced expression of NF-κB-regulated genes in cardiac myocytes, cultures were treated with LMB, then stimulated with TNF-α, and then harvested for qRT-PCR (Figure 6E). As before (Figure 1C), TNF-α induced expression of NF-κB-regulated genes (MCP-1 and CXCL1) to a greater extent in cardiac fibroblasts than in cardiac myocytes, but induced expression of an NF-κB-independent gene equivalently in the two cell types. Not surprisingly, LMB alone failed to induce expression of any of the genes. However, LMB failed to increase the TNF-α effect in either cardiac fibroblasts or cardiac myocytes, indicating that trapping NF-κB (Figure 6B, D) in the nucleus is insufficient to increase gene expression in either cell type. Results indicate that while most cardiac myocytes support IKKβ-dependent activation of NF-κB, translocation is transient.
Figure 6. NF-κB activation in cardiac myocytes is resistant to the stimulatory effects of TNFα and Poly(I:C).
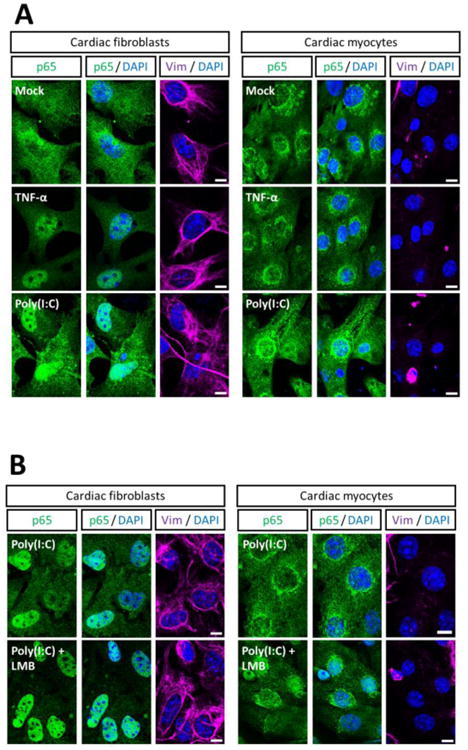
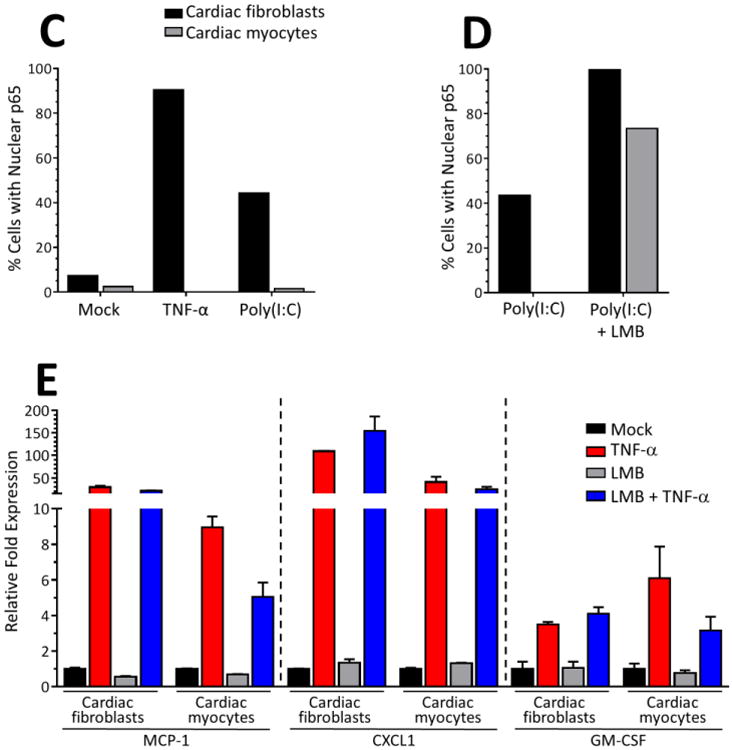
(A, C) Primary cardiac fibroblast or myocyte cultures were stimulated with 50 ng/ml TNF-α for 1 h or transfected with 50 μg/ml Poly(I:C) for 4 h, fixed, and immunostained as indicated. (B, D) Primary cardiac cultures were treated with 20 nM LMB or media and simultaneously transfected with Poly(I:C) as in panel A. Scale bar = 10 μm. The percentage of cardiac fibroblasts (n = 55 – 156 cells per condition) or cardiac myocytes (n = 15 – 131 cells per condition) displaying nuclear p65 is indicated for a representative of two independent experiments. E) Cultures were treated with LMB for 1 h and then stimulated with TNF-α for 1 h, prior to mRNA harvest. Levels of mRNA expression for the indicated genes was analyzed by qRT-PCR and normalized to GAPDH expression. Fold induction by LMB and/or TNF-α is expressed relative to ‘mock’ for each culture (mean ± SD).
Viral infection induces nuclear translocation of NF-κB in cardiac fibroblasts but not in cardiac myocytes
To determine whether cardiac myocytes are relatively recalcitrant to virus-induced NF-κB nuclear translocation, cultures were infected with reovirus type 3 Dearing (T3D), a serotype known to activate NF-κB in different cell types (20, 34-36) (Figure 7). While there were more mock-treated cardiac fibroblasts with nuclear NF-κB than in previous experiments (Figures 5 and 6), likely reflecting the stress induced by two media changes, the number of cells with nuclear NF-κB was still much lower in cardiac fibroblast cultures than in identically-treated cardiac myocyte cultures. Reovirus induced p65 nuclear translocation frequently in infected cardiac fibroblasts but only very rarely in infected cardiac myocytes. Furthermore, while LMB alone or in combination with reovirus infection induced p65 nuclear translocation in all cardiac fibroblasts, nuclear translocation of p65 occurred in many fewer cardiac myocytes. Indeed, nuclear translocation of p65 in LMB-treated cardiac myocytes was not further increased by reovirus infection, suggesting that cardiac myocytes are largely resistant to virus-induced activation of NF-κB.
Figure 7. Virus-induced NF-κB activation is cardiac cell type-specific.
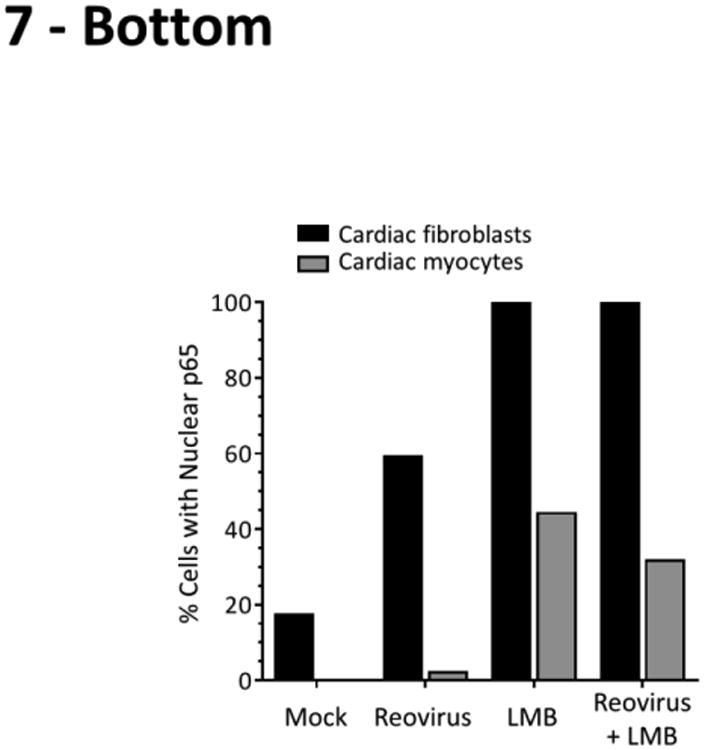
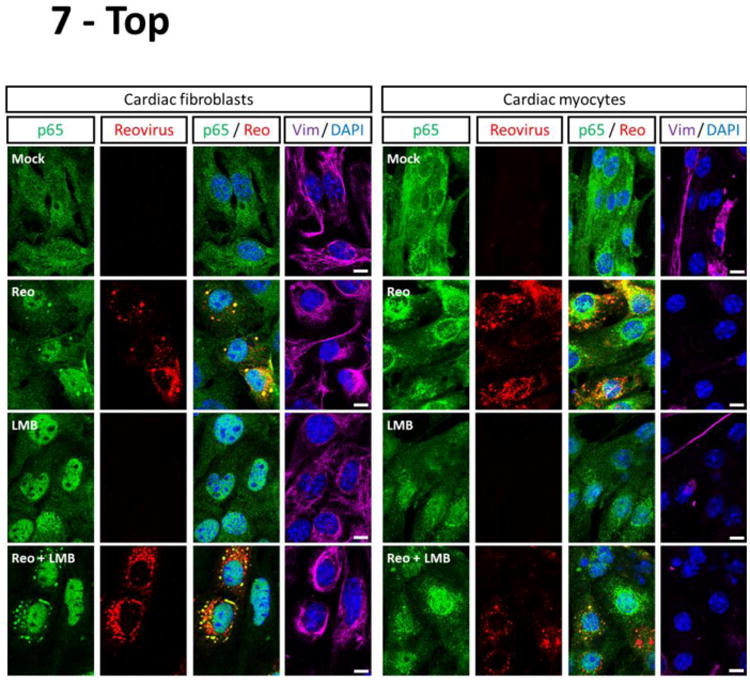
Primary cardiac cultures were infected with reovirus T3D or media alone (‘mock’). After incubation for 2 h, the inoculum was removed and replaced with either media alone or media containing 20 nM LMB. Cells were incubated for another 6 h prior to fixation and immunostained as indicated. The apparent co-localization of RelA within reovirus viral factories is due to cross-reactivity of the p65 antibody with a reovirus protein (data not shown). Scale bar = 10 μm. The percentage of cardiac fibroblasts (n = 8 – 104 cells per condition) or cardiac myocytes (n = 18 – 48 cells per condition) displaying nuclear p65 is indicated for a representative of two independent experiments. For cases that received reovirus inoculum, only cells positive for reovirus antigen were scored.
Pharmacological disruption of the Golgi is insufficient to support virus-induced NF-κB nuclear translocation in cardiac myocytes
To determine whether the cis-Golgi localization of p65 specifically prevents its stimulus-induced nuclear translocation, cultures were treated with BFA to disrupt Golgi structure and challenged with reovirus. (Figure 8A). In cardiac fibroblasts, reovirus induced efficient activation of p65 independent of a functional cis-Golgi (Figure 8B). Notably, BFA activated NF-κB even in uninfected cardiac fibroblasts, likely through the unfolded protein response upon prolonged BFA exposure (37, 38). In contrast, NF-κB was not activated in either uninfected or infected BFA-treated cardiac myocytes despite their loss of Golgi-localized p65 (Figure 8C). Together, results indicate that activation of NF-κB in cardiac myocytes is relatively resistant to sustained endoplasmic reticulum stress and disruption of the Golgi is insufficient to support NF-κB activation upon viral infection.
Figure 8. Pharmacological disruption of the Golgi is insufficient to support virus-induced NF-κB activation in cardiac myocytes.
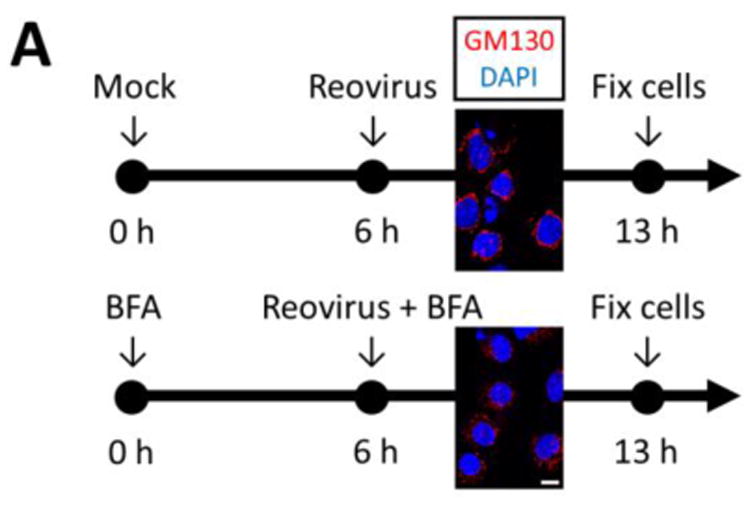
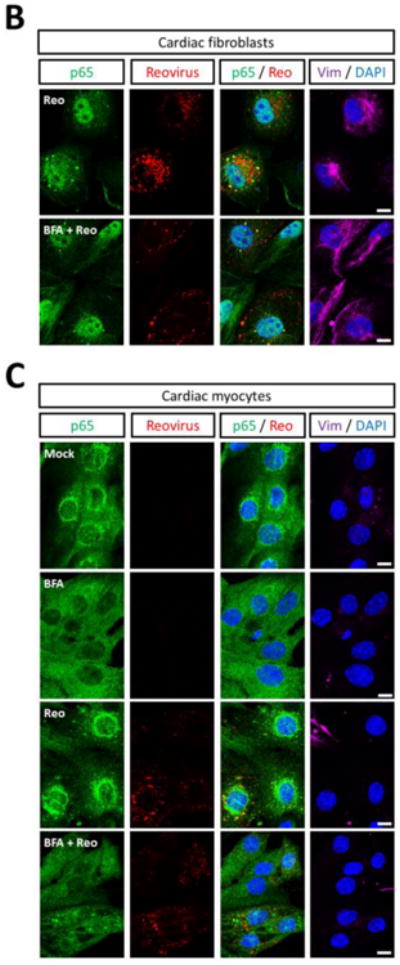
(A) Experimental strategy. Primary cardiac cultures were treated with either control media or media containing 10 μg/ml BFA for 6 h. Disruption of the Golgi at this time was confirmed by GM130 immunostaining. Cells were subsequently inoculated with reovirus in either media alone (‘mock’) or BFA-containing media and fixed 7 h later. Cardiac fibroblast (B) or cardiac myocyte (C) cultures were fixed and immunostained as indicated. Images are representative of at least 20 cells analyzed per condition. Scale bar = 10 μm.
Discussion
Chronic cardiac inflammation is a major issue in cardiovascular health and disease. The role of NF-κB activation in the heart and its critical role in the regulation of cardiac function has been the focus of research for over two decades (8). Cardiac myocytes constitute the majority of cardiac mass and cell number in the heart, however cardiac fibroblasts represent another significant and important population (21, 39). Moreover, while cardiac myocytes have a lifetime turnover estimated to be less than 5% (40-42), cardiac fibroblasts are readily replenished after injury (21). Despite the important role cardiac fibroblasts play in cardiac structure and function and despite the profound differences between the two cell types, most studies of NF-κB activation in the heart have left cardiac fibroblasts unexplored and none have compared them to cardiac myocytes. Here, we show that cardiac fibroblasts are readily stimulated for activation of NF-κB and expression of pro-inflammatory cytokines while in contrast, cardiac myocytes are largely recalcitrant to activation of NF-κB. Moreover, we show that cardiac myocyte subpopulations differ in their NF-κB subcellular localization and identify the cis-Golgi as a cardiac myocyte-specific host compartment. Together, results indicate that NF-κB-dependent signaling in the heart is cardiac cell type-specific, likely reflecting mechanisms that have evolved to balance cardiac responses that can be either protective or damaging to the heart.
The NF-κB dimer can be comprised of different subunits and in cardiac myocytes the p65 subunit is the predominant activated subunit in health (43) and following myocardial infarction (44). Activation of NF-κB has been demonstrated to promote adverse cardiac remodeling, hypertrophy, endoplasmic reticulum stress, and apoptosis (44-48). Not surprisingly, cardiac myocytes have evolved at least one mechanism to limit NF-κB activation: impaired activation of the IKK complex (14). Accordingly, over-expression of pathway components to constitutively activate NF-κB in cardiac myocytes results in myocarditis and heart defects (15-17). Here, results indicate that only a minority of cardiac myocytes (∼20-30%) permit basal NF-κB nucleocytoplasmic shuttling, and that even in those cells, nuclear NF-κB is minimal (Figure 5). IKKβ-dependent stimulation induces a larger fraction of cardiac myocytes (∼70%) to support NF-κB nuclear translocation but it is remarkably transient (Figure 6). Results are consistent with a previously proposed model for two populations of cardiac myocytes in the heart that differ in their capacities to support activation of NF-κB (8). The identification of a population of cardiac myocytes characterized by Golgi-associated NF-κB (Figures 3-4) suggests one possible underlying mechanism. While pharmacological disruption of the Golgi did not render cardiac myocytes susceptible to virus-induced activation of NF-κB (Figure 8), NF-κB may bind to a Golgi-associated protein that prevents NF-κB nuclear translocation independent of Golgi structure. Future studies will address the role of the Golgi in regulating activation of NF-κB, and determine whether it relates to the Golgi-associated upstream component TRAF3 (49).
One important NF-κB-regulated cytokine is IFN-β (18, 19). We have previously shown that cardiac myocytes express high basal levels of IFN-β relative to cardiac fibroblasts, which serves to pre-arm the cardiac myocytes for protection against viral infection (23-25). Therefore, we anticipated we would find a similar difference in basal activation of NF-κB. Surprisingly, we found the opposite: cardiac myocytes display minimal basal activation of NF-κB (Figure 1A) and minimal basal NF-κB nuclear translocation (Figure 2). One possibility is that NF-κB is indeed activated in cardiac myocytes but that the specific subunit p65 is not involved in basal IFN-β expression. This seems unlikely given that p65 is the primary NF-κB subunit in cardiac myocytes and the myocardium (43, 44). An alternative hypothesis is that basal activation of NF-κB that is undetectable by immunoblot or immunofluorescent microscopy is nonetheless sufficient to induce minimal IFN-β expression, which is then further amplified by the IFN-induced transcription factor IRF7 in an autocrine positive amplification loop (50). This possibility is consistent with the requirement for p65 for basal expression of IRF7 in mouse embryo fibroblasts (19, 33), and with the >400-fold decrease in basal IRF7 expression in cardiac myocytes lacking the IFN-α/β receptor compared to wild type cardiac myocytes (24). Indeed, this would be an elegant mechanism by which cardiac myocytes could capture the protective anti-viral impact of basal NF-κB activation without triggering damaging effects.
Viral infection can activate NF-κB resulting in further induction of IFN-β and cytokines, and either direct or cytokine-induced apoptosis. The IFN-α/β response is the primary determinant of protection against reovirus-induced myocarditis (22, 26, 51) and is similarly critical for protection against other viruses (72). We have previously shown that reovirus induces IFN-β more robustly in cardiac myocytes than in cardiac fibroblasts, likely due to the IRF7-mediated amplification of IFN-β expression as described above (24, 25). Thus reovirus induction of IFN-β in cardiac myocytes would require minimal activation of NF-κB, consistent with our evidence that cardiac myocytes are relatively recalcitrant to NF-κB activation (Figures 1 and 6-8). While IFN-β plays an important antiviral role, pro-inflammatory cytokines recruit cells that clear viral infection. Cardiac fibroblasts are exquisitely responsive to stimulation of NF-κB and secrete a wide range of cytokines to regulate cardiac function (52-56). Indeed, reovirus induction of pro-inflammatory cytokines in cardiac fibroblasts correlates inversely with the capacity to induce myocarditis (57). Thus, activation of NF-κB in cardiac fibroblasts can be protective to the heart. Accordingly, we found that TNF-α activated NF-κB (Figure 1A) and induced NF-κB-dependent cytokine responses (Figure 1C) more readily in cardiac fibroblasts than in cardiac myocytes. The detectable TNF-α induction of cytokines in cardiac myocytes despite the undetectable activation of NF-κB in those cells could reflect effects of the transcription factors ATF-2 and c-Jun, which are known to be activated by TNF-α and to stimulate pro-inflammatory gene expression (58). Finally, reovirus-induced myocarditis is the result of a direct viral cytopathic effect to cardiac myocytes (59), which is largely apoptotic (57). Reovirus-induced apoptosis requires activation of NF-κB in most cell types (20, 34-36, 60-62) but not in cardiac myocytes (20, 36). Thus reovirus-induced myocarditis does not require activation of NF-κB in cardiac myocytes, consistent with evidence here that cardiac myocytes are relatively recalcitrant to NF-κB activation (Figures 1 and 6-8). Instead, the minimal activation of NF-κB in cardiac myocytes is sufficient to stimulate a protective IFN-β response while the robust activation of NF-κB in cardiac fibroblasts stimulates a protective proinflammatory cytokine response.
In sum, both basal and stimulated activation of NF-κB differ remarkably between cardiac myocytes and cardiac fibroblasts, and within subsets of cardiac myocytes themselves. These cell type-specific differences have likely evolved as a balance between the protective and damaging effects of NF-κB activation on the heart.
Materials and Methods
Primary cardiac cultures
C57BL/6, RIG-I-/- MDA5-/- (63), and IFN-α/β-R-/- (64) mice were maintained as an in-house colony in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All animal procedures were approved by the North Carolina State University Institutional Animal Care and Use Committee (IACUC). Primary cardiac myocyte cultures were generated as previously described (65) from 1-day-old neonatal and term fetal mice (unless otherwise indicated) resulting from timed pregnancies. Cardiac myocyte and fibroblast cultures were plated in Dulbecco MEM (DMEM; Gibco #11965-092) supplemented to contain 7% fetal calf serum (FCS; Atlanta Biologicals) and 10 μg/ml gentamycin (Sigma, #G1272) in 24-well or 48-well clusters for RNA or protein harvest, or in 8-well poly-D-lysine-coated chamber slides (Corning) for immunofluorescent microscopy. Cardiac myocyte cultures were also supplemented to contain 0.06% thymidine (to inhibit cardiac fibroblast growth). Cardiac myocytes and cardiac fibroblasts cultures were ≥ 90% and ≥ 95% pure, respectively, as estimated by immunostaining against sarcomeric actin (alpha Sr-1) and vimentin (data not shown). For all experiments, cultures were incubated at 37°C in 5% CO2 for two days post-seeding before use, and were never passaged.
Primary skeletal muscle cultures
Timed-pregnant mice and mouse colonies for timed matings were maintained as above, and all procedures were IACUC-approved. Muscle from the limbs of neonatal mice less than 24 h old was dissected away from other tissues into Hanks' balanced salt solution (HBSS; Corning, #21-023-CV), the HBSS was aspirated, and the muscle was minced with sterile scissors and incubated in freshly prepared 2% Type II collagenase (Worthington Biochemical, #LS004174) in HBSS for 30 min with vortexing every 10 min. Cells were pelleted by centrifugation at 1800 × g for 5 m at room temperature, resuspended in HBSS, and re-pelleted as above. Cells were resuspended in DMEM supplemented to contain 6% FCS, 2 mM L-glutamine (Corning #25-005-Cl), and 10 μg/ml gentamycin, and passed through a 100 μm cell strainer (Falcon, #352360). Cells were resuspended in supplemented DMEM and plated in 8-well poly-D-lysine-coated chamber slides for immunofluorescent microscopy. After incubation at 37°C in 5% CO2 for two days, media was replaced with DMEM supplemented as above (for undifferentiated skeletal muscle cell cultures) or DMEM supplemented as above but at 3% instead of 6% FCS (for differentiated skeletal muscle cell cultures). Cultures were incubated an additional two days before use to allow differentiation, and were never passaged.
Viral infections
CsCl-purified reovirus type 3 Dearing (T3D) was maintained as a low-passage laboratory stock and stored at −80°C. T3D was chosen as a strong inducer of NF-κB activity (20, 34-36, 60, 61). Unless stated otherwise, cardiac cultures plated in 8-well chamber slides were inoculated with reovirus T3D at 50 plaque forming units (PFU) per cell and were fixed at the indicated times post-infection. Mock-infected cultures were treated similarly but received supplemented DMEM as the inoculum.
Stimulants of NF-κB activation and chemical treatments
For stimulation of NF-κB activation, cardiac cultures were treated with 50 ng/ml of mouse TNF-α (Sigma-Aldrich; #T7539) diluted in supplemented DMEM or transfected with 50 μg/ml Poly(I:C) (Invivogen; #tlrl-pic) using Lipofectamine 3000 (Thermo-Fisher; #L3000) according to the manufacturer's instructions and fixed at the indicated times. For disruption of Golgi structure, cells were treated for 6 h with 10 μg/ml Brefeldin A (Sigma-Aldrich; #B7651) maintained as a 5 mg/ml stock in ethanol and diluted in supplemented DMEM. For the irreversible inhibition of Crm1-dependent nuclear export, cells were incubated with 20 nM Leptomycin B (Sigma-Aldrich; #L2913) diluted in supplemented DMEM and fixed at the indicated times.
Quantitative (real-time) reverse transcription-PCR (qRT-PCR)
Total RNA was harvested using an RNeasy kit (Qiagen, Inc.), treated with RNase-free DNase I (Qiagen, Inc.), converted to cDNA by reverse transcription and used for real-time PCR on a LightCycler® 480 fluorescence thermocyler (Roche Life Science). Reactions contained 1× Quantitech SYBR green master mix (Qiagen, Inc.) and 0.3 μM (each) forward and reverse primers. The primers used were: IL-1β (forward: 5′–TCACAGCAGCACATCAACAA–3′; and reverse: 5′–TGTCCTCATCCTGGAAGGTC–3′), CCL2/MCP-1 (forward: 5′–CCCAATGAGTAGGCTGGAGA–3′; and reverse: 5′–TCTGGACCCATTCCTTCTTG–3′), CXCL1 (forward: 5′– TGTTGTGCGAAAAGAAGTGC–3′; and reverse: 5′–TACAAACACAGCCTCCCACA–3′), A20 (forward: 5′–AAAAAGGACCAGCCCAGATT–3′; and reverse: 5′–TCCTTGAGCTCCTCCACTGT–3′), IκBα (forward: 5′– CTCCAGATGCTACCCGAGAG–3′; and reverse: 5′– TAGGGCAGCTCATCCTCTGT–3′), PGES (forward: 5′– GTACACACCGTGGCCTACCT–3′; and reverse: 5′– GCCATGGAGAAACAGGAGAA–3′), COX-2 (forward: 5′– TGCAGAATTGAAAGCCCTCT–3′; and reverse: 5′– CCCCAAAGATAGCATCTGGA–3′), GM-CSF (forward: 5′– GGCAGCAGATGGAAAACCTA–3′; and reverse: 5′– CTGGAAGGCAGAAGTGAAGG–3′), and GAPDH (forward: 5′– GGGTGTGAACCACGAGAAAT–3′; and reverse: 5′– CCTTCCACAATGCCAAAGTT–3′). Relative mRNA abundance for all genes was normalized to the expression of GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and TNF-α-mediated fold-induction of mRNA expression was calculated relative to mock-treated cells for each cell type.
Antibodies
Antibodies used for immunoblotting and their corresponding dilutions were anti-p65/RelA (Santa Cruz Biotech #sc-372; 1:500), anti-phospho-RelA/p65 (Ser468) (Cell Signaling #3039; 1:500), anti-IκBα (Santa Cruz Biotech #sc-371; 1:300), anti-IKKβ (Cell Signaling #2378; 1:800), anti-IKKy (Santa Cruz #sc-8330; 1:500), anti-β-actin (Santa Cruz Biotech #sc-1615-hrp, 1:2,000), goat anti-rabbit IgG-HRP (Millipore #12-348; 1:2,000), and goat anti-mouse IgG-HRP (Millipore #12-349; 1:2,000). Primary antibodies used for immunofluorescence were anti-p65/RelA (Santa Cruz Biotech #sc-372; 1:50), anti-GM130 (BD Biosciences #610822; 1:250), anti-vimentin (Thermo-Fisher #PA1-16759; 1:2,000), and mouse anti-reovirus antisera (generated in the Sherry laboratory; 1:5,000). Secondary antibodies were Alexa 488-, Alexa 594-or Alexa 647-conjugated goat anti-mouse, rabbit or chicken IgG (Thermo-Fisher; 1:1000).
SDS-PAGE and immunoblotting
Whole cell protein extracts were obtained two days post-plating using RIPA lysis buffer (50 mM Tris HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented to contain 1% sodium dodecylsulfate (SDS), a cocktail of protease and phosphatase inhibitors (Sigma; #P8340 and #P2850), and 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma, #P7626). Protein lysates were resolved by SDS-PAGE and transferred to a nitrocellulose membrane (GE Healthcare). Membranes were probed with the indicated antibodies, developed using Amersham enhanced chemiluminiscence (ECL) or ECL Prime kits, exposed to film and scanned using an HP Scanjet G4050. Band intensities were determined using the LI-COR BioSciences Image Studio™ Lite Software (version 5.x).
Indirect immunofluorescence of primary cardiac cultures
Cells in poly-D-Lysine-coated chamber slides (BD Biosciences) were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in phosphate-buffered saline (PBS) and permeabilized with 0.25% Triton X-100 (Sigma, #X100). Slides were blocked with normal goat serum (Sigma; #G9023), incubated in DAPI (4′,6-diamidino-2-phenylindole; Sigma; #D8417), immunostained with the indicated primary and secondary antibodies, and preserved with ProLong Gold (Invitrogen).
Indirect immunofluorescence of heart sections from adult mice
Hearts from adult C57BL/6 mice were excised, snap frozen in liquid nitrogen-cooled isopentane, and stored at −35°C. Transverse cryosections (1 μm) were collected on SuperFrost/Plus slides and stored at −35°C. Sections were thawed and permeabilized in 0.25% Triton X-100 in PBS for 10 min prior to blocking in 20% normal goat serum (Sigma-Aldrich) in PBS for 2 h at room temperature. Primary antibodies were diluted in 0.1% IgG- and protease-free BSA and samples were incubated simultaneously overnight at 4°C in a humidity chamber. Samples were washed with PBS and incubated with a mix of Alexa 488-conjugated goat anti-rabbit IgG, Alexa 594-conjugated goat anti-mouse IgG, and Alexa 647-conjugated goat anti-chicken IgG (1:1,000 each; Thermo-Fisher) diluted in 0.1% IgG- and protease-free BSA for 1 h at room temperature. Samples were washed with PBS and coverslips were mounted with ProLong Gold Antifade (Thermo-Fisher).
Confocal microscopy and image analysis
A Zeiss LSM 710 confocal microscope equipped with a 40× C-apochromat / 1.1 NA water immersion objective from the Cellular and Molecular Facility (CMIF) at NC State University was used for all experiments. The pinhole diameter was maintained at 1 Airy unit (A.U.) and all images were obtained using multitrack sequential scanning for each fluorophore to prevent bleed-through. The excitation/emission wavelengths during micrograph acquisition were 488 nm/492-554 nm for Alexa Fluor® 488, 561 nm/584-666 nm for Alexa Fluor® 594, 633 nm/650-709 nm for Alexa Fluor® 647, and 405 nm/407-507 nm for DAPI. Images were processed for presentation using Photoshop® CS4, and intensity plot profiles were generated using ImageJ software (66). The number of cells displaying nuclear NF-κB were scored manually and, for each quantitation, cell identity was assessed by vimentin co-immunostaining (i.e. cardiac fibroblasts were identified by their high levels of vimentin, and cardiac myocytes by their lack of vimentin expression) and only the cell type in question was scored for each condition analyzed.
Statistical analysis
A Student's two-sample t test (pooled variance) was applied using Systat software. Results were considered significant if the P value was < 0.05.
Research Highlights.
Insults to the heart induce both protective and damaging NF-κB-regulated responses
Cardiac myocytes are resistant to NF-κB activation relative to cardiac fibroblasts
NF-κB is predominantly in cis-Golgi bodies in cardiac myocytes but not fibroblasts
Even in virus-infected cardiac myocytes, NF-κB is predominantly in cis-Golgi bodies
Results demonstrate that cardiac NF-κB-dependent responses are cell type-specific
Acknowledgments
We thank Shannon Chiera for excellent technical assistance and suggestions; and Tiffany Benzine and Nicole DeAngelis for helpful discussions. We thank Dr. Michael Gale Jr. (Univ. of Washington) for providing the RIG-I-/- MDA5-/- mice. This research was supported by the National Institutes of Health (NIH) grant R01 AI083333 (B.S.), a Research Supplement to Promote Diversity in Health-Related Research through the parental NIH grant (B.S. and E.E.R.S.), and a U.S. Department of Education Graduate Assistance in Areas of National Need (GAANN) Fellowship (E.E.R-.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drory Y, Turetz Y, Hiss Y, Lev B, Fisman EZ, Pines A, Kramer MR. Sudden unexpected death in persons less than 40 years of age. Am J Cardiol. 1991;68:1388–1392. doi: 10.1016/0002-9149(91)90251-f. [DOI] [PubMed] [Google Scholar]
- 2.Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 3.Cooper LT., Jr Myocarditis. N Engl J Med. 2009;360:1526–1538. doi: 10.1056/NEJMra0800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274–288. doi: 10.1016/j.pcad.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corsten MF, Schroen B, Heymans S. Inflammation in viral myocarditis: friend or foe? Trends Mol Med. 2012;18:426–437. doi: 10.1016/j.molmed.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–249. doi: 10.1016/s1071-9164(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 7.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 8.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 9.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 10.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubio D, Xu RH, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, Balachandran S, Alcami A, Norbury CC, Sigal LJ. Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell Host Microbe. 2013;13:701–710. doi: 10.1016/j.chom.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 2010;118:593–605. doi: 10.1042/CS20090557. [DOI] [PubMed] [Google Scholar]
- 13.Dhingra R, Shaw JA, Aviv Y, Kirshenbaum LA. Dichotomous actions of NF-kappaB signaling pathways in heart. J Cardiovasc Transl Res. 2010;3:344–354. doi: 10.1007/s12265-010-9195-5. [DOI] [PubMed] [Google Scholar]
- 14.Goren N, Cuenca J, Martin-Sanz P, Bosca L. Attenuation of NF-kappaB signalling in rat cardiomyocytes at birth restricts the induction of inflammatory genes. Cardiovasc Res. 2004;64:289–297. doi: 10.1016/j.cardiores.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 15.Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, Klingel K, Bottger T, Braun T, Wirth T. Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2012;109:11794–11799. doi: 10.1073/pnas.1116584109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kraut B, Maier HJ, Kokai E, Fiedler K, Boettger T, Illing A, Kostin S, Walther P, Braun T, Wirth T. Cardiac-Specific Activation of IKK2 Leads to Defects in Heart Development and Embryonic Lethality. PLoS One. 2015;10:e0141591. doi: 10.1371/journal.pone.0141591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yariswamy M, Yoshida T, Valente AJ, Kandikattu HK, Sakamuri SS, Siddesha JM, Sukhanov S, Saifudeen Z, Ma L, Siebenlist U, Gardner JD, Chandrasekar B. Cardiac-restricted Overexpression of TRAF3 Interacting Protein 2 (TRAF3IP2) Results in Spontaneous Development of Myocardial Hypertrophy, Fibrosis, and Dysfunction. J Biol Chem. 2016;291:19425–19436. doi: 10.1074/jbc.M116.724138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, Garcia-Sastre A, Balachandran S, Beg AA. NF-κB RelA subunit is crucial for early IFN-β expression and resistance to RNA virus replication. J Immunol. 2010;185:1720–1729. doi: 10.4049/jimmunol.1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Donnell SM, Hansberger MW, Connolly JL, Chappell JD, Watson MJ, Pierce JM, Wetzel JD, Han W, Barton ES, Forrest JC, Valyi-Nagy T, Yull FE, Blackwell TS, Rottman JN, Sherry B, Dermody TS. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irvin SC, Zurney J, Ooms LS, Chappell JD, Dermody TS, Sherry B. A single-amino-acid polymorphism in reovirus protein mu2 determines repression of interferon signaling and modulates myocarditis. J Virol. 2012;86:2302–2311. doi: 10.1128/JVI.06236-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Sherry B. IFN-alpha expression and antiviral effects are subtype and cell type specific in the cardiac response to viral infection. Virology. 2010;396:59–68. doi: 10.1016/j.virol.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart MJ, Smoak K, Blum MA, Sherry B. Basal and reovirus-induced beta interferon (IFN-beta) and IFN-beta-stimulated gene expression are cell type specific in the cardiac protective response. J Virol. 2005;79:2979–2987. doi: 10.1128/JVI.79.5.2979-2987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zurney J, Howard KE, Sherry B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J Virol. 2007;81:13668–13680. doi: 10.1128/JVI.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwabe RF, Sakurai H. IKKbeta phosphorylates p65 at S468 in transactivaton domain 2. Faseb j. 2005;19:1758–1760. doi: 10.1096/fj.05-3736fje. [DOI] [PubMed] [Google Scholar]
- 28.Zhou A, Scoggin S, Gaynor RB, Williams NS. Identification of NF-κB-regulated genes induced by TNFα utilizing expression profiling and RNA interference. Oncogene. 2003;22:2054–2064. doi: 10.1038/sj.onc.1206262. [DOI] [PubMed] [Google Scholar]
- 29.Meja KK, Seldon PM, Nasuhara Y, Ito K, Barnes PJ, Lindsay MA, Giembycz MA. p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NF-kappa B-independent mechanism. Br J Pharmacol. 2000;131:1143–1153. doi: 10.1038/sj.bjp.0703684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, Rice M, Borg TK. Organization of fibroblasts in the heart. Dev Dyn. 2004;230:787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 31.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saraste J, Svensson K. Distribution of the intermediate elements operating in ER to Golgi transport. J Cell Sci. 1991;100(Pt 3):415–430. doi: 10.1242/jcs.100.3.415. [DOI] [PubMed] [Google Scholar]
- 33.Basagoudanavar SH, Thapa RJ, Nogusa S, Wang J, Beg AA, Balachandran S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J Virol. 2011;85:2599–2610. doi: 10.1128/JVI.02213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. J Virol. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holm GH, Zurney J, Tumilasci V, Leveille S, Danthi P, Hiscott J, Sherry B, Dermody TS. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- 36.Stebbing RE, Irvin SC, Rivera-Serrano EE, Boehme KW, Ikizler M, Yoder JA, Dermody TS, Sherry B. An ITAM in a nonenveloped virus regulates activation of NF-kappaB, induction of beta interferon, and viral spread. J Virol. 2014;88:2572–2583. doi: 10.1128/JVI.02573-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitamura M. Control of NF-kappaB and inflammation by the unfolded protein response. Int Rev Immunol. 2011;30:4–15. doi: 10.3109/08830185.2010.522281. [DOI] [PubMed] [Google Scholar]
- 38.Hung JH, Su IJ, Lei HY, Wang HC, Lin WC, Chang WT, Huang W, Chang WC, Chang YS, Chen CC, Lai MD. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem. 2004;279:46384–46392. doi: 10.1074/jbc.M403568200. [DOI] [PubMed] [Google Scholar]
- 39.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 40.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kuhn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh S, Ponten A, Fleischmann BK, Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365–373. doi: 10.1093/cvr/cvq005. [DOI] [PubMed] [Google Scholar]
- 43.Norman DA, Yacoub MH, Barton PJ. Nuclear factor NF-kappa B in myocardium: developmental expression of subunits and activation by interleukin-1 beta in cardiac myocytes in vitro. Cardiovasc Res. 1998;39:434–441. doi: 10.1016/s0008-6363(98)00118-7. [DOI] [PubMed] [Google Scholar]
- 44.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mustapha S, Kirshner A, De Moissac D, Kirshenbaum LA. A direct requirement of nuclear factor-κB for suppression of apoptosis in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H939–945. doi: 10.1152/ajpheart.2000.279.3.H939. [DOI] [PubMed] [Google Scholar]
- 47.Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009;82:59–66. doi: 10.1093/cvr/cvp040. [DOI] [PubMed] [Google Scholar]
- 48.Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-κB is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98:6668–6673. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Zuylen WJ, Doyon P, Clement JF, Khan KA, D'Ambrosio LM, Do F, St-Amant-Verret M, Wissanji T, Emery G, Gingras AC, Meloche S, Servant MJ. Proteomic profiling of the TRAF3 interactome network reveals a new role for the ER-to-Golgi transport compartments in innate immunity. PLoS Pathog. 2012;8:e1002747. doi: 10.1371/journal.ppat.1002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 51.Holm GH, Pruijssers AJ, Li L, Danthi P, Sherry B, Dermody TS. Interferon regulatory factor 3 attenuates reovirus myocarditis and contributes to viral clearance. J Virol. 2010;84:6900–6908. doi: 10.1128/JVI.01742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aoyagi T, Matsui T. The Cardiomyocyte as a Source of Cytokines in Cardiac Injury. Journal of cell science & therapy. 2011;2012:003. doi: 10.4172/2157-7013.s5-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LaFramboise WA, Scalise D, Stood ley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–1808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 54.Lindner D, Zietsch C, Tank J, Sossalla S, Fluschnik N, Hinrichs S, Maier L, Poller W, Blankenberg S, Schultheiss HP, Tschope C, Westermann D. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res Cardiol. 2014;109:428. doi: 10.1007/s00395-014-0428-7. [DOI] [PubMed] [Google Scholar]
- 55.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 56.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyamoto SD, Brown RD, Robinson BA, Tyler KL, Long CS, Debiasi RL. Cardiac cell-specific apoptotic and cytokine responses to reovirus infection: determinants of myocarditic phenotype. J Card Fail. 2009;15:529–539. doi: 10.1016/j.cardfail.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu T, Li YJ, Bian AH, Zuo HB, Zhu TW, Ji SX, Kong F, Yin DQ, Wang CB, Wang ZF, Wang HQ, Yang Y, Yoo BC, Cho JY. The regulatory role of activating transcription factor 2 in inflammation. Mediators Inflamm. 2014;2014:950472. doi: 10.1155/2014/950472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baty CJ, Sherry B. Cytopathogenic effect in cardiac myocytes but not in cardiac fibroblasts is correlated with reovirus-induced acute myocarditis. J Virol. 1993;67:6295–6298. doi: 10.1128/jvi.67.10.6295-6298.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarke P, Debiasi RL, Meintzer SM, Robinson BA, Tyler KL. Inhibition of NF-kappa B activity and cFLIP expression contribute to viral-induced apoptosis. Apoptosis. 2005;10:513–524. doi: 10.1007/s10495-005-1881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clarke P, Meintzer SM, Moffitt LA, Tyler KL. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J Biol Chem. 2003;278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- 62.Clarke P, Richardson-Burns SM, DeBiasi RL, Tyler KL. Mechanisms of apoptosis during reovirus infection. Curr Top Microbiol Immunol. 2005;289:1–24. doi: 10.1007/3-540-27320-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M., Jr The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 65.Sherry B. Generating primary cultures of murine cardiac myocytes and cardiac fibroblasts to study viral myocarditis. Methods Mol Biol. 2015;1299:1–16. doi: 10.1007/978-1-4939-2572-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]