

Abstract
On July 10, 2016, Alfred G. Knudson, Jr., MD, PhD, a leader in cancer research, died at the age of 93 years. We deeply mourn his loss. Knudson's two‐hit hypothesis, published in 1971, has been fundamental for understanding tumor suppressor genes and familial tumor‐predisposing syndromes. To understand the molecular mechanism of two‐hit‐initiated tumorigenesis, Knudson used an animal model of a dominantly inherited tumor, the Eker rat. From the molecular identification of Tsc2 germline mutations, the Eker rat became a model for tuberous sclerosis complex (TSC), a familial tumor‐predisposing syndrome. Animal models, including the fly, have greatly contributed to TSC research. Because the product of the TSC2/Tsc2 gene (tuberin) together with hamartin, the product of another TSC gene (TSC1/Tsc1), suppresses mammalian/mechanistic target of rapamycin complex 1 (mTORC1), rapalogs have been used as therapeutic drugs for TSC. Although significant activity of these drugs has been reported, there are still problems such as recurrence of residual tumors and adverse effects. Recent studies indicate that there are mTORC1‐independent signaling pathways downstream of hamartin/tuberin, which may represent new therapeutic targets. The establishment of cellular models, such as pluripotent stem cells with TSC2/Tsc2 gene mutations, will facilitate the understanding of new aspects of TSC pathogenesis and the development of novel treatment options. In this review, we look back at the history of Knudson and animal models of TSC and introduce recent progress in TSC research.
Keywords: Eker rat, retinoblastoma, tuberous sclerosis complex, tumor suppressor gene, two‐hit hypothesis
On July 10, 2016, Alfred G. Knudson, Jr, died at the age of 93 years. He was a man with great insight into cancer genetics and a leader in the cancer research field. He inspired many younger scientists to seek understanding of the mechanisms of tumorigeneis, especially the cell‐of‐origin of cancer, and to strive to prevent this disease. He was a personal mentor of this review's author (O.H.) as well as a valued adviser to cancer geneticists in Japan. We deeply mourn his loss and dedicate this review to him, starting with the story of his research history on cancer genetics and animal models (Fig. 1).1
Figure 1.
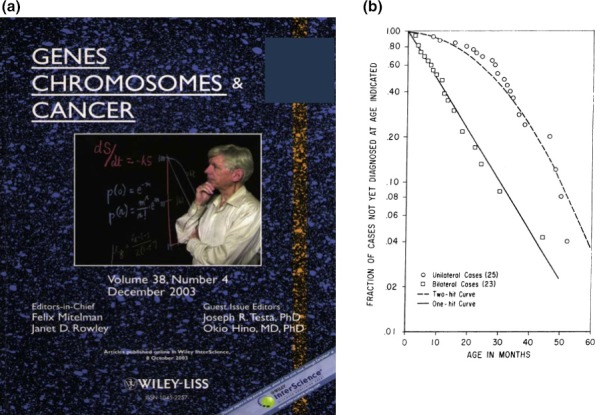
Memorial figures for Knudson. (a) The cover of the special issue of Genes Chromosomes & Cancer on Knudson's 80th birthday (December 2003; vol. 38, issue 4, with the permission of John Wiley & Sons, Inc.). (b) The plot from which Knudson proposed the two‐hit hypothesis (Ref. 2, with the permission of the National Academy of Sciences, USA).
The Two‐hit Hypothesis, a “Driver” in the Development of the Field of CANCER GENETICS
The dominant nature of hereditary cancer had been recognized as a trait long before the basic techniques of molecular biology were established. Although there were reports describing the loss of specific regions of certain chromosomes in particular types of cancer, no information on the driver mutations or specific genes related to carcinogenesis was available in the early 1970s. At that time, Knudson developed the two‐hit hypothesis by statistical methods, without any experimental approaches.2 He simply compared the time at which both eyes would be affected by retinoblastoma if one or two hits were required and predicted the relative chance of this occurring (Fig. 1b).2 Knudson's hypothesis clearly postulated the recessive nature of tumor‐initiating gene mutations and the mode of inheritance in familial cancer. This led to the concept of the existence of tumor suppressor genes and loss‐of‐heterozygosity (LOH) as relevant to carcinogenesis. Notably, Knudson already mentioned the possibility of “delayed mutation” that may correspond to germline‐mosaic mutations.2, 3, 4 Including this possibility, his insights facilitated the development of the field of cancer genetics. The first molecular cloning of the tumor suppressor gene RB1, the predisposing gene for retinoblastoma, was achieved in 1986.5
Preserving an Animal Model of Inherited Tumor: A “Tale” of the Eker Rat
In 1954, a Norwegian pathologist, R. Eker found a rat strain that developed bilateral, multiple and dominantly inheritable renal tumors.6, 7 Later, this new rat strain was named the Eker rat.8 From those days up until the late 1980s, to the best of our knowledge, no other laboratory animal model of inherited tumor with a high penetration was reported. Knudson saw that experimental animal models of hereditary tumors would be useful tools for cancer science. Thus, he sought reports of animal models of hereditary cancers and found information about the Eker rat. He brought these animals to the USA and maintained the mutation on a Long Evans background. By the mid‐1980s to early 1990s, significant progress in research on polymorphic DNA markers had been achieved not only for human but also for rodent genomes. Using syntenic homologies between humans and rats, a positional cloning approach was taken and, finally, a germline insertion was identified in the homolog of the tuberous sclerosis complex (TSC) 2 gene (Tsc2). This was the tumor predisposing mutation of the Eker rat.9, 10, 11, 12
There is a number of similarities between human TSC and the Eker rat, such as the dominant inheritance and LOH of TSC2/Tsc2 region in tumorous lesions. However, unlike the epithelial renal tumors of the Eker rat, tumorous lesions in TSC patients are mainly hamartomas, such as renal angiomyolipoma (AML), lung lymphangioleiomyomatosis (LAM), and subependymal giant cell astrocytoma (SEGA), etc.13 Our studies on the somatic, intragenic Tsc2 mutations (second hits) in tumors and suppression of phenotypes by transgenic expression of wild‐type Tsc2 confirmed that the Tsc2 is a bona fide predisposing gene in the Eker rat.14, 15
TSC: A Multi‐Organ Disorder with a Defect in the Tumor Suppressor Complex
Tuberous sclerosis complex is caused by a mutation either in TSC1 or TSC2 and is characterized by tumorous lesions and neuropsychiatric disorders. Symptoms in patients with either TSC gene mutation are similar, although those with the TSC2 mutation may have more severe neurological phenotypes.13 Both genes are tumor suppressor genes and their products, hamartin (TSC1) and tuberin (TSC2), form a functional complex. In addition to the Eker rat, mice with either TSC gene homolog knocked out have now been established and the development of renal and other tumors was reported.16, 17, 18, 19 Using those animal models, embryonic lethality of homozygous mutants of TSC genes was shown, and various cell lines deficient for TSC genes were established. In addition, several tissue‐specific conditional gene‐targeting systems have been established.20 Although the detailed explanation of each system is beyond the scope of this review, suffice it to say that the functions of TSC genes in various tissues and recapitulation of TSC‐related pathology, especially for brain lesions, have been documented using such conditional knockout systems.20
Good news from the fly and bad news after treatment
The importance of the GAP (GTPase activating protein)‐related domain of tuberin in tumor suppression in vivo was identified using a transgenic Eker rat system.21 A functional link between the hamartin/tuberin complex and the mammalian/mechanistic target of rapamycin (mTOR) kinase and the target of GAP‐related domain, Rheb, was revealed through studies of Drosophila.22 The Rheb‐mTORC1 pathway activates mRNA translation by stimulating p70S6K‐rpS6‐ and eIF4E‐mediated signals. Hamartin/tuberin negatively regulates mTORC1 as does GAP for Rheb (Fig. 2). Tumors in animal models and TSC patients exhibit mTORC1‐related pathway activation.23 Indeed, the growth of TSC gene‐deficient tumors can be suppressed in vivo by rapamycin, concomitant with the downregulation of mTORC1.24, 25 Rapamycin and its homologs (rapalogs) have been clinically used for the treatment of SEGA, AML and LAM, and substantial efficacy has been reported in many cases.26 However, the effects of rapalogs are not cytotoxic but rather cytostatic.25, 26 The complete elimination of tumor cells is impossible and the volume of tumor increases after therapy is stopped.26 Also, there are considerable adverse effects with rapalogs. Thus, other treatment options with different drug targets are needed now.
Figure 2.
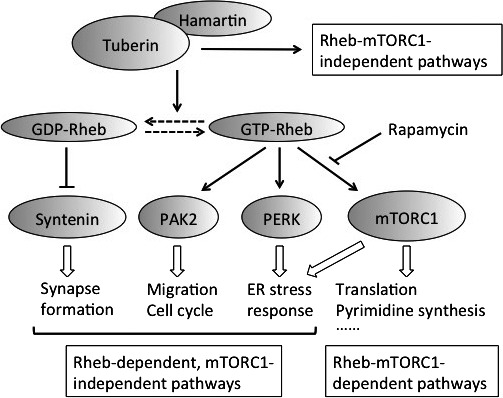
mTORC1‐dependent‐ and ‐independent pathways downstream of hamartin/tuberin. There are two categories of mTORC1‐independent pathways according to their dependence on Rheb. Only representative pathways are shown.
New downstream pathways of mTORC1
Many research groups have tried to identify novel pathways regulated by mTORC1 by means of comprehensive gene expression analysis and proteomics. Over several years recently, the de novo pyrimidine synthesis pathway has been characterized as such a candidate.27, 28 The key enzyme of this pathway, CAD (carbamoyl‐phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase), is phosphorylated and activated by S6K in an mTORC1‐dependent manner. Thus, hyper‐activation of mTORC1 induces pyrimidine synthesis to promote cell growth and cell cycle progression. The mTORC1‐independent binding of CAD to GTP‐bound Rheb was also reported, suggesting that CAD is regulated at multiple levels in TSC gene‐deficient tumors.29
Many other important phenomena downstream of mTORC1, such as autophagy and ER stress, have now been characterized.30, 31, 32, 33, 34, 35, 36 Pathways involved in those phenomena are already being exploited to develop new therapeutic targets and combination treatments with inhibitors of such pathways and rapalogs have been tested at the basic research level. A detailed understanding of the different pathways downstream of mTORC1 is important for developing therapeutic options encompassing independent modes of inhibition of each pathway. This will facilitate the establishment of personalized medication avoiding the adverse effects of rapalogs.
mTORC1‐independent downstream pathways of the hamartin/tuberin complex
If rapamycin‐insensitive, i.e. mTORC1‐independent, signaling pathways are regulated by hamartin/tuberin complexes, they could be good candidates for new molecular targeting therapeutic drugs. Recently, an increasing number of hamartin/tuberin‐regulated but rapamycin‐insensitive pathways have been identified. These can be classified into two categories according to their dependence on Rheb (Fig. 2).
By kinome analysis, Alves et al. found that PAK2 activity is increased in Tsc2‐deficient MEFs in a Rheb‐dependent, but mTORC1‐independent manner.37 They also found increases in PAK2 phosphorylation in brain lesions from TSC patients. Interestingly, Yamagata et al. reported that GDP‐bound Rheb is not merely an inactive form but selectively binds to and degrades syntenin in a proteasome‐dependent manner.38, 39 In neuronal dendrites of Tsc2 heterozygous mice, the reduction of tuberin renders Rheb into a more GTP‐bound state and causes accumulation of syntenin, which, in turn, disrupts spine structures.38, 39 Although the functional relationship between hamartin/tuberin and syntenin has not been elucidated in other tissues and tumorous lesions, syntenin‐mediated cytoskeletal regulation might be a candidate drug target for TSC. Paradoxically, considering the stimulatory function of mTORC1 on translation, Tyagi et al. reported inhibition of global translation in Rheb‐overexpressing HEK293 cells as well as in Tsc2‐deficient MEFs.40 Rheb enhances phosphorylation of eIFα, thereby inhibiting translation, and directly binds to and stimulates the kinase activity of PERK in vitro.40 Thus, Rheb may directly regulate the balance between two major translation‐controlling pathways dissociated from mTORC1.
There are many reports describing the mTORC1‐independent dysregulation of gene expression.41, 42, 43, 44 However, only some of these indicated that the dysregulation was Rheb‐independent. Thus, a more detailed understanding of Rheb‐independent pathways downstream of hamartin/tuberin would be expected to increase options for developing novel therapeutic approaches to TSC.
Towards next generation treatment for TSC
Because mTORC1 plays a pivotal role in the integration of multiple cellular reactions, its chronic activation seems to influence many downstream pathways, facilitating vulnerabilities and adaptive reprogramming of metabolism. During such reprogramming, cell type‐specific addiction to particular metabolites or reactions frequently occurs.34, 45 Using high‐throughput drug screening and/or recently‐developed genome editing techniques, researchers have explored pathways related to such addictive factors in TSC gene‐deficient cells. To identify growth‐suppressive gene mutations (downregulations) synergistic with TSC gene‐deficiency, Housden et al.46 established a system consisting of CRISPR‐Cas9‐mediated targeting and RNAi screens in Drosophila cells. Confirming their data using Tsc2‐deficient MEFs and TSC AML cells, they identified three candidates, RNGTT, CDK11 and CCNT1, as synthetic interacting genes.46 Notably, RNGTT and CDK11 are implicated in mRNA synthesis and the formation of cap structures, suggesting that increased cap‐dependent translation caused by mTORC1 activation additionally requires the higher rate of mRNA synthesis in TSC gene‐deficient cells. As Houdson et al. focused on knocking out kinases and phosphatases in their study, it should be possible to identify other synthetic lethal pathways with targets other than global knockouts.
Employing a screening approach using many inhibitors of metabolism and signaling pathways, Blenis and colleagues found that the simultaneous inhibition of HSP90 and glutaminase, i.e. induction of ER stress and reduction of glutathione level, induces apoptosis in TSC gene‐deficient cells.47 The effect of this combined inhibition is minimal on TSC gene‐proficient cells, suggesting its usefulness for the treatment of TSC.
Beyond the Two‐Hit Hypothesis: Pathogenesis Initiated by One‐Hit On a Tumor Suppressor Gene Revisited
There have been many reports concerning haploinsufficiencies of tumor suppressor genes.48, 49 There may be cases where cells with a heterozygous mutation of a tumor suppressor gene do initiate tumorigenesis, without a second hit, in a cell‐autonomous manner.50 Moreover, it is likely that the heterozygosity of tumor suppressor genes in the cells of the tumor microenvironment promotes tumor development through interactions with tumor cells in a non‐cell‐autonomous manner, as reported using the mouse homolog of the neurofibromatosis type 1 (NF1) gene (Nf1).51, 52 The consequences of haploinsufficiency may be tissue‐specific. Apart from tumorigenesis, various abnormal conditions, such as via effects on cell differentiation and in systemic metabolism, have been documented as consequences of the haploinsufficiency of tumor suppressor genes.53, 54, 55, 56
tuberous sclerosis complex patients often suffer from neuropsychiatric symptoms, such as autism and epilepsy, in addition to their tumorous lesions.13 Animal models of TSC also exhibit defects in cognitive ability and/or social interaction, without any macroscopic pathology in the brain.57, 58, 59, 60, 61 These phenotypes seem to be caused, at least in part, by haploinsufficiency of TSC genes. The administration of rapamycin partly corrects such neuropsychiatric symptoms, suggesting that activated mTORC1 is involved in such haploinsufficiency‐induced pathogenesis (Fig. 3). Whether there is a second hit in TSC‐associated small lesions in the brain is controversial.62, 63, 64 In the animal models of TSC, tumor‐related pathology without a second hit to any predisposing gene has been reported.65, 66 Indeed, no aberrant activation of mTORC1 has been detected in those second hit‐negative lesions.65, 66 Thus, some mTORC1‐independet downstream pathways of hamartin/tuberin may contribute to the effects of haploinsufficiency of TSC genes. Sporadic‐ or TSC‐associated LAM lesions and TSC‐associated angiofibromas are mixed populations of differentiated cells.67, 68 Some of these cells harbor two‐hit mutations and interact with heterozygous mutant‐ or wild‐type cells (in the sporadic cases).67, 68 As is true for Nf1, heterozygous mutations in cells of the microenvironment may promote pathogenesis initiated by a second hit. In addition, like PTEN, TSC genes may show tissue‐specific haploinsufficiency affecting metabolism, as revealed by the Eker rat.69 To determine the molecular basis of haploinsufficiency, Knudson and colleagues compared the gene expression profile of non‐tumorous tissue between TSC patients and those with other inherited syndromes, and control subjects.70, 71 Through the understanding of detailed molecular mechanism of haploinsufficiency, new targets for therapeutic intervention in inherited tumor syndromes including TSC may be found. In terms of the endpoint of treatment, it is ideal that haploinsufficiency‐induced‐, as well as two hit‐initiated‐tumors can be completely eliminated by drug administration for a particular term. In contrast, to control haploinsufficiency‐induced non‐tumorous symptoms, such as neuropsychiatric and metabolic ones, continuous treatment of patients during all of their life might be necessary. Thus, the strategies and endpoint to develop treatment might be different between ones for tumorous and non‐tumorous symptoms.
Figure 3.
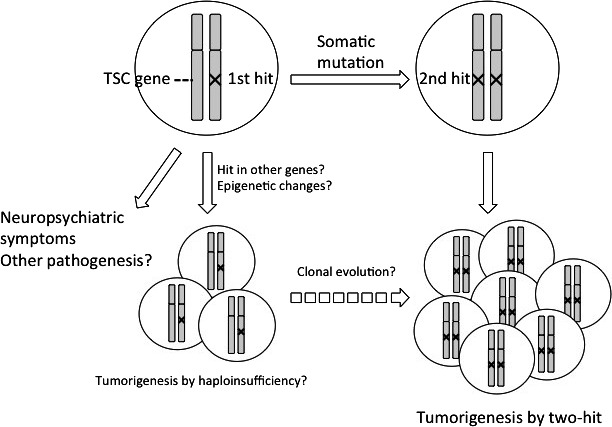
Haploinsufficiency‐ and two‐hit‐initiated pathologies in tuberous sclerosis complex (TSC). Germline mutations (1st hit) cause neuropsychiatric symptoms during infancy without a 2nd hit. Other lesions, such as cortical tubers in brain may be caused by haploinsufficiceny of TSC genes. The majority of tumorous lesions develop after two hits of TSC genes. In animal models, tumorigenesis without a 2nd hit has been reported. Although not depicted in this figure, there are interactions between two‐hit‐initiated tumorous cells and surrounding non‐tumorous cells to form lesions.
Exploring Cell Type‐Specific Tumorigenesis Using Pluripotent Stem Cells
The tissue‐specific nature of different inherited tumors reflects the cell type‐specific effects of mutations in oncogenes and tumor suppressor genes. Tumorigenesis is tightly linked to abnormalities in the differentiation process of stem or precursor cells. To explore the cell type‐specific and cell fate‐related mechanism of tumorigenesis, in vitro differentiation models using pluripotent stem cells (PSCs) are useful. Once such a model has been established, it can be used for high‐throughput screening of drug candidates. Recently, an in vitro model of osteoblast differentiation using induced PSCs (iPSCs) from a Li‐Fraumeni syndrome family member was reported.72 Unexpectedly, p53‐mutant osteoblasts induced from patient‐derived iPSCs exhibited defective differentiation and increased tumorigenicity, associated with aberrant regulation of H19. These results suggest that p53 deficiency induces osteosarcomas by affecting the program of osteogenic differentiation. In another report, Yamada and colleagues established iPSCs from EWS‐FLI1‐driven mouse osteosarcomas and demonstrated their impaired differentiation to osteogenic cells irrespective of EWS‐FLI1 expression.73 These results suggest that the genetic and/or epigenetic alterations generated in osteosarcomas affect osteogenic differentiation. Moreover, osteosarcomas were induced from such osteosarcoma‐derived iPSCs when EWS‐FLI1 is expressed, suggesting cooperation between driver gene alterations.73, 74 There have also been attempts to establish embryonic stem cells (ESCs) from families with inherited tumor‐predisposing syndromes. NF1‐heterozygous ESCs derived from an affected family member show hyperpigmentation when differentiated into the melanocyte lineage.75 This is a typical model of haplosufficiency‐induced pathology.
In the case of TSC, studies with PSCs are ongoing. Ito et al. established Tsc2 −/− ESCs from Eker rat embryos and found that they have the potential to differentiate into three germ layers, despite mTORC1 being hyper‐activated.76 This result is in contrast to the case of the adenomatous polyposis coli gene (Apc) in which mouse Apc −/− ESCs fail to differentiate effectively in the teratoma formation assay.77 Thus, each tumor suppressor gene mutation might give rise to different effects at different levels of pluripotent and cellular differentiation states. Interestingly, among Tsc2 −/− teratoma tissues, tumor‐like aberrant structures are seen, which are suppressed by administration of rapamycin.78 These results may be relevant to renal tumorigenesis in the Eker rat. In another paper, the importance of Tsc2 in the regulation of pluripotency in mouse ESC was also reported.79 Very recently, a human ESC model of TSC was established by targeting TSC2 gene with a zinc‐finger nuclease technique.80 As in the case of rat ESCs, mTORC1 activity is increased in human TSC2 −/− ESCs relative to wild‐type and TSC2 +/− ESCs. Upon neuronal differentiation experiments, TSC2 −/− ESCs exhibit abnormalities in proliferation and morphology from the early stage as well as in synaptic function at the later stage. TSC2 +/− ESCs also exhibit abnormalities in synaptic function, but not in proliferation and morphology. These phenotypes are reversed by rapamycin, clearly indicating the dosage‐dependent, differentiation stage‐specific effects of mTORC1 (Fig. 4).
Figure 4.
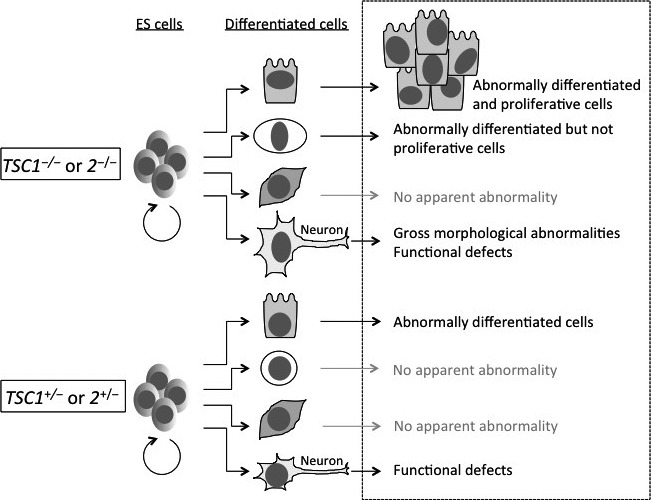
A model of differentiation abnormalities in tuberous sclerosis complex (TSC) using pluripotent stem cells (PSCs). There are tissue‐specific mechanisms in the pathogenesis of TSC. Modeling of differentiation abnormalities related to TSC (in the box delineated by dotted lines) will be useful to explore the mechanism of pathogenesis and to establish high‐throughput screening systems. Upon differentiation, compared with wild‐type cells (not depicted in this figure) homozygous PSCs mutant for TSC genes (TSC1 −/− or 2 −/−) may show differentiation abnormalities with or without aberrant proliferation in some specific lineages. In the case of neurons, heterozygous cells (TSC1 +/− or 2 +/−) may cause functional defects related to neuropsychiatric symptoms of TSC. For neuronal differentiation of human TSC2‐mutant ES cells, refer to Costa, V, et al.80
The Ultimate Goal: Prevention of Pathogenesis in Familial Tumor‐Predisposing Syndromes
In this review, we introduced and discussed familial tumor predisposing syndromes, taking TSC as the main example. As mentioned earlier, the tumor phenotype of Eker rat is different from that of TSC patients. To advance research, not only for TSC, but also for other diseases, we must consider the phenotypic difference between human patients and animal models. Although the basic mechanism, such related to the function of tumor suppressor gene product, might be conserved, the “final output” of phenotype is sometimes different. Needless to say, animal models are important to understand the conserved mechanisms in which molecular targets of drugs are involved. However, unraveling the mechanism causing difference, i.e. human‐specific tissue specificity, might be the key to finding highly selective molecular targets for therapy.
The goal of cancer research is to prevent deaths from cancer. The suppression of recurrence and metastasis is the most important objective. For many hereditary tumor‐predisposing syndromes, however, it is also feasible to consider preventing the development of pathology in the first place. For the purpose of chemoprevention and treatment, the primary research target is the target pathway of the drug in question. To minimize adverse effects, it is optimal to find a target pathway that is specific to the cell type from which the tumors initiate. Such pathways need to be elucidated through the understanding of cell type‐specific or differentiation‐related mechanisms of tumorigenesis. In addition, the differences in drug efficacy and adverse effects among patients must be considered. These may be determined by genetic background as already revealed for adverse effects on treatment with irinotecan.81 Large‐scale pharmacogenomic studies and the development of patient‐specific cell or tissue models may facilitate the establishment of methods for personalized prevention.
Disclosure Statement
The authors have no conflict of interest to disclose.
Acknowledgments
This work was supported in part by JSPS KAKENHI (Grant Numbers JP16K12983 and JP26460398) and AMED (16ek0109015s0303). We deeply apologize to researchers whose work is not referred in this review because of limited space.
Cancer Sci 108 (2017) 5–11
Funding Information
Japan Society for the Promotion of Science, (Grant / Award Number: ‘JP16K12983’, ‘JP26460398’) Japan Agency for Medical Research and Development, (Grant / Award Number: ‘16ek0109015s0303’).
References
- 1. Knudson AG. Cancer genetics through a personal retrospectroscope. Genes Chrom Cancer 2003; 38: 288–91. [DOI] [PubMed] [Google Scholar]
- 2. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Auerbach C. A possible case of delayed mutation in man. Ann Hum Genet 1956; 20: 266–9. [DOI] [PubMed] [Google Scholar]
- 4. Knudson AG Jr, Hethcote HW, Brown BW. Mutation and childhood cancer: a probabilistic model for the incidence of retinoblastoma. Proc Natl Acad Sci USA 1975; 72: 5116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friend SH, Bernards R, Rogelj S et al A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986; 323: 643–6. [DOI] [PubMed] [Google Scholar]
- 6. Eker R. Familial renal adenomas in Wistar rats. Acta Path Microbiol Scand 1954; 34: 554–62. [DOI] [PubMed] [Google Scholar]
- 7. Eker R, Mossige J. A dominant gene for renal adenomas in the Eker rat. Nature 1961; 189: 858–9.13780066 [Google Scholar]
- 8. Waynforth HB, Hunter‐Craig CJ. The Eker renal tumor rat: tumor transplantability and a re‐evaluation of tumor histology. Neoplasma 1981; 28: 309–15. [PubMed] [Google Scholar]
- 9. Hino O, Klein‐Szanto AJP, Feed JJ et al Spontaneous and radiation‐induced renal tumrs in the Eker rat model of dominantly inherited cancer. Proc Natl Acad Sci USA 1993; 90: 327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hino O, Kobayashi T, Tsuchiya H et al The predisposing gene of the Eker rat inherited cancer syndrome is tightly linked to the tuberous sclerosis (TSC2) gene. Biochem Biophys Res Commun 1994; 203: 1302–8. [DOI] [PubMed] [Google Scholar]
- 11. Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG. Predisposition to renal carcinoma in the Eker rat is determined by germ‐line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci USA 1994; 91: 11413–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kobayashi T, Hirayama Y, Kobayashi E, Kubo Y, Hino O. A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat Genet 1995; 9: 70–4. [DOI] [PubMed] [Google Scholar]
- 13. Thiele EA, Jóźwiak S. Natural history of tuberous sclerosis complex and overview of manifestations In: Kwiatkowski DJ, Whittemore VH, Thiele EA, eds. Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics, 1st edn Weinheim, Germany: WILEY‐VCH Verlag GmbH & Co. KGaA, 2010; 11–20. [Google Scholar]
- 14. Kobayashi T, Urakami S, Hirayama Y et al Intragenic Tsc2 somatic mutations as Knudson's second hit in spontaneous and chemically induced renal carcinomas in the Eker rat model. Jpn J Cancer Res 1997; 88: 254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobayashi T, Mitani H, Takahashi R et al Transgenic rescue from embryonic lethality and renal carcinogenesis in the Eker rat model by introduction of a wild‐type Tsc2 gene. Proc Natl Acad Sci USA 1997; 94: 3990–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ‐line Tsc2 mutation in mice. Cancer Res 1999; 59: 1206–11. [PubMed] [Google Scholar]
- 17. Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2 +/− mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 1999; 104: 687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi T, Minowa O, Sugitani Y et al A germ‐line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA 2001; 98: 8762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwiatkowski DJ, Zhang HB, Bandura JL et al A mouse model of TSC1 reveals sex‐dependent lethality from liver hemangiomas, and up‐regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet 2002; 11: 525–34. [DOI] [PubMed] [Google Scholar]
- 20. Kwiatkowski DJ. Rat and mouse models of tuberous sclerosis In: Kwiatkowski DJ, Whittemore VH, Thiele EA, eds. Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics, 1st edn Weinheim, Germany: WILEY‐VCH Verlag GmbH & Co. KGaA, 2010; 117–43. [Google Scholar]
- 21. Momose S, Kobayashi T, Mitani H et al Identification of the coding sequences responsible for Tsc2‐mediated tumor suppression using a transgenic rat system. Hum Mol Genet 2002; 11: 2997–3006. [DOI] [PubMed] [Google Scholar]
- 22. Pan D. Animal models of TSC: insights from Drosophila In: Kwiatkowski DJ, Whittemore VH, Thiele EA, eds. Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics, 1st edn Weinheim, Germany: WILEY‐VCH Verlag GmbH & Co. KGaA, 2010; 145–58. [Google Scholar]
- 23. Kenerson HL, Aicher LD, True LD, Yeung RS. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res 2002; 62: 5645–50. [PubMed] [Google Scholar]
- 24. Kobayashi T, Adachi H, Mitani H, Hirayama Y, Hino O. Toward chemotherapy for Tsc2‐mutant renal tumor. Proc Jpn Acad 2003; 79B: 22–5. [Google Scholar]
- 25. Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res 2005; 57: 67–75. [DOI] [PubMed] [Google Scholar]
- 26. Bissler JJ, McCormack FX, Young LR et al Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008; 358: 140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robitaille AM, Christen S, Shimobayashi M et al Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013; 339: 1320–3. [DOI] [PubMed] [Google Scholar]
- 28. Ben‐Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013; 339: 1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato T, Akasu H, Shimono W et al Rheb protein binds CAD (carbamoyl‐phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) protein in a GTP‐ and effector domain‐dependent manner and influences its cellular localization and carbamoyl‐phosphate synthetase (CPSase) activity. J Biol Chem 2015; 290: 1096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ozcan U, Ozcan L, Yilmaz E et al Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell 2008; 29: 541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parkhitko A, Myachina F, Morrison TA et al Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)‐dependent. Proc Natl Acad Sci USA 2011; 108: 12455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siroky BJ, Yin H, Babcock JT et al Human TSC‐associated renal angiomyolipoma cells are hypersensitive to ER stress. Am J Physiol Renal Physiol 2012; 303: F831–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang N, Zhang C, Dill P et al Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med 2014; 211: 2249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Medvetz D, Priolo C, Henske EP. Therapeutic targeting of cellular metabolism in cells with hyperactive mTORC1: a paradigm shift. Mol Cancer Res 2014; 13: 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson CE, Hunt DK, Wiltshire M et al Endoplasmic reticulum stress and cell death in mTORC1‐overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol Oncol 2015; 9: 675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alayev A, Salamon RS, Sun Y et al Effects of combining rapamycin and resveratrol on apoptosis and growth of TSC2‐deficient xenograft tumors. Am J Respir Cell Mol Biol 2015; 53: 637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alves MM, Fuhler GM, Queiroz KCS et al PAK2 is an effector of TSC1/2 signaling independent of mTOR and a potential therapeutic target for tuberous sclerosis complex. Sci Rep 2015; 5: 14534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yasuda S, Sugiura H, Katsurabayashi S et al Activation of Rheb, but not of mTORC1, impairs spine synapse morphogenesis in tuberous sclerosis complex. Sci Rep 2014; 4: 5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugiura H, Yasuda S, Katsurabayashi S et al Rheb activation disrupts spine synapse formation through accumulation of syntenin in tuberous sclerosis complex. Nat Commun 2015; 6: 6842. [DOI] [PubMed] [Google Scholar]
- 40. Tyagi R, Shahani N, Gorgen L et al Rheb inhibits protein synthesis by activating the PERK‐eIF2α signaling cascade. Cell Rep 2015; 10: 684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karbowniczek M, Zitserman D, Khabibullin D et al The evolutionarily conserved TSC/Rheb pathway activates Notch in tuberous sclerosis complex and Drosophila external sensory organ development. J Clin Invest 2010; 120: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gan B, Sahin E, Jiang S et al mTORC1‐dependent and ‐independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci USA 2008; 105: 19384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee PS, Tsang SW, Moses MS et al Rapamycin‐insensitive up‐regulation of MMP2 and other genes in tuberous sclerosis complex 2‐deficient lymphangioleiomyomatosis‐like cells. Am J Repir Cell Mol Biol 2010; 42: 227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun Y, Gallacchi D, Zhang EY et al Rapamycin‐resistant poly (ADP‐ribose) polymerase‐1 overexpression is a potential therapeutic target in lymphangioleiomyomatosis. Am J Repir Cell Mol Biol 2014; 51: 738–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Young RM, Ackerman D, Quinn ZL et al Dysregulated mTORC1 renders cells critically depend on desaturated lipids for survival under tumor‐like stress. Genes Dev 2013; 27: 1115–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Housden BE, Valvezan AJ, Kelley C et al Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR‐based knockdowns with RNAi. Sci Signal 2015; 8: rs9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li J, Csibi A, Yang S et al Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1‐driven tumor cells. Proc Natl Acad Sci USA 2015; 112: E21–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Venkatachalam S, Shi YP, Jones SN et al Retention of wild‐type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998; 17: 4657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo‐insuffcient for tumor suppression. Nature 1998; 396: 177–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berger AH, Knudson AG, Pandolfi PP. A continuum model of tumor suppression. Nature 2011; 476: 163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: schwann cell origin and role of tumor environment. Science 2002; 296: 920–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang FC, Ingram DA, Chen S et al Nf1‐dependent tumors require a microenvironment containing Nf1 + /– and c‐kit‐dependent bone marrow. Cell 2008; 135: 437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ingram DA, Yang FC, Travers JB et al Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo . J Exp Med 2000; 191: 181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Konishi H, Mohseni M, Tamaki A et al Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci USA 2011; 108: 17773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pal A, Barber TM, Van de Bunt M et al PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med 2012; 367: 1002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feliotter HE, Michel C, Uy P, Bathurst L, Davey S. BRCA1 haploinsufficiency leads to altered expression of genes involved in cellular proliferation and development. PLoS ONE 2014; 9: e100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1 +/− mice in the absence of cerebral lesions and seizures. Ann Neurol 2007; 62: 648–55. [DOI] [PubMed] [Google Scholar]
- 58. Ehninger D, Han S, Shilyansky C et al Reversal of learning deficits in a Tsc2 +/− mouse model of tuberous sclerosis. Nat Med 2008; 14: 843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Young DM, Schenk AK, Yang SB, Jan YN, Jan LY. Altered ultrasonic vocalization in a tuberous sclerosis mouse model of autism. Proc Natl Acad Sci USA 2010; 107: 11074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sato A, Kasai S, Kobayashi T et al Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 2012; 3: 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D. Epilepsy and Tsc2 haploinsufficiency lead to autistic‐like social deficit behaviors in rats. Behav Genet 2011; 41: 364–72. [DOI] [PubMed] [Google Scholar]
- 62. Mizuguchi M, Mori M, Nozaki Y et al Absense of allelic loss in cytomegalic neurons of cortical tuber in the Eker rat model of tuberous sclerosis. Acta Neuropathol 2004; 107: 47–52. [DOI] [PubMed] [Google Scholar]
- 63. Qin W, Chan JA, Vinters HV et al Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second‐hit mutations in these genes are rare events. Brain Pathol 2010; 20: 1096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010; 74: 1716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wilson C, Bonnet C, Guy C et al Tsc1 haploinsufficiency without mammalian target of rapamycin activation is sufficient for renal cyst formation in Tsc1 +/− mice. Cancer Res 2006; 66: 7934–8. [DOI] [PubMed] [Google Scholar]
- 66. D'Armiento J, Shiomi T, Marks S, Geraghty P, Sankarasharma D, Chada K. Mesenchymal tumorigenesis driven by TSC2 haploinsufficiency requires HMGA2 and is independent of mTOR pathway activation. Cancer Res 2016; 76: 844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li S, Takeuchi F, Wang J et al Mesenchymal‐epithelial interactions involving epiregulin in tuberous sclerosis complex hamartomas. Proc Natl Acad Sci USA 2008; 105: 3539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Clements D, Dongre A, Krymskaya VP, Johnson SR. Wild type mesenchymal cells contribute to the lung pathology of lymphangioleiomyomatosis. PLoS ONE 2015; 10: e0126025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aizawa Y, Shirai T, Kobayashi T et al The tuberous sclerosis complex model Eker (TSC2+/−) rat exhibits hyperglycemia and hyperketonemia due to decreased glycolysis in the liver. Arch Biochem Biophys 2016; 590: 48–55. [DOI] [PubMed] [Google Scholar]
- 70. Stoyananova R, Clapper ML, Bellacosa A et al Altered gene expression in phenotypically normal renal cells from carriers of tumor suppressor gene mutations. Cancer Biol Ther 2004; 3: 1313–21. [DOI] [PubMed] [Google Scholar]
- 71. Bellacosa A, Godwin AK, Peri S et al Altered gene expression in morphologically normal epitherial cells from heterozygous carriers of BRCA1 or BRCA2 mutations. Cancer Prev Res 2010; 3: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee DF, Su J, Kim HS et al Modeling familial cancer with induced pluripotent stem cells. Cell 2015; 161: 240–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Komura S, Semi K, Itakura F et al An EWS‐FLI1‐induced osteosarcoma model unveiled a crucial role of impaired osteogenic differentiation on osteosarcoma development. Stem Cell Rep 2016; 6: 592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tanaka M, Aisaki K, Kitajima S, Igarashi K, Kanno J, Nakamura T. Gene expression response to EWS‐FLI1 in mouse embryonic cartilage. Genome Data 2014; 2: 296–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Allouche J, Bellon N, Saidani M et al In vitro modeling of hyperpigmentation associated to neurofibromatosis type 1 using melanocytes derived from human embryonic stem cells. Proc Natl Acad Sci USA 2015; 112: 9034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ito Y, Kawano H, Kanai F et al Establishment of Tsc2‐deficient rat embryonic stem cells. Int J Oncol 2015; 46: 1944–52. [DOI] [PubMed] [Google Scholar]
- 77. Kielman MF, Rindapää M, Gaspar C et al Apc modulates embryonic stem‐cell differentiation by controlling the dosage of ß‐catenin signaling. Nat Genet 2002; 32: 594–605. [DOI] [PubMed] [Google Scholar]
- 78. Kawano H, Ito Y, Kanai F et al Aberrant differentiation of Tsc2‐deficient teratomas associated with activation of the mTORC1‐TFE3 pathway. Oncol Rep 2015; 34: 2251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, Smith A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 2013; 153: 335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Costa V, Aigner S, Vukcevic M et al mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep 2016; 15: 86–95. [DOI] [PubMed] [Google Scholar]
- 81. Innocenti F, Undevia SD, Iyer L et al Genetics variants in the UDP‐glucuronosyltransferase 1A1 gene predict the risk of sever neutropenia of irinotecan. J Clin Oncol 2004; 22: 1382–8. [DOI] [PubMed] [Google Scholar]