

ABSTRACT
Emerging pandemic infectious threats, inappropriate antibacterial use contributing to multidrug resistance, and increased morbidity and mortality from diagnostic delays all contribute to a need for improved diagnostics in the field of infectious diseases. Historically, diagnosis of infectious diseases has relied on pathogen detection; however, a novel concept to improve diagnostics in infectious diseases relies instead on the detection of changes in patterns of gene expression in circulating white blood cells in response to infection. Alterations in peripheral blood gene expression in the infected state are robust and reproducible, yielding diagnostic and prognostic information to help facilitate patient treatment decisions.
KEYWORDS: diagnostics, gene expression
BACKGROUND
In infectious diseases, rapid and accurate identification of the etiologic agent driving a given clinical syndrome is crucial for guiding triage decisions, the application of appropriate infection control protocols, and the direction of proper antimicrobial treatment. The wide array of potential pathogens causing a given clinical presentation (e.g., fever and cough) makes direct pathogen identification challenging, and existing diagnostic approaches are often limited by sensitivity, specificity, and/or a prolonged time to available results. However, most infectious disease diagnostics today focus on pathogen detection as the “gold standard,” but this approach exhibits certain pitfalls. The detection of pathogens alone does not necessarily imply the presence of disease but can indicate asymptomatic carriage in colonized individuals. Furthermore, some individuals suffer from disease due to multiple pathogens simultaneously (such as concomitant influenza and bacterial pneumonia), where the detection of a single pathogen still omits critical information related to patient care. Diagnostic tests for some pathogens simply do not yet exist or may not detect all clinically relevant strains, especially with emerging infections, such as in 2009, when existing molecular assays failed to effectively detect a novel pandemic influenza virus strain (1). Fortunately, a burgeoning field of new data suggests that many of these limitations can potentially be addressed by harnessing data surrounding the host response to infectious states.
Analysis of the host immune response triggered in an infectious process as a means to aid diagnosis is not a novel concept. There are many host biomarkers, quantifiable indicators of a biological state, with clinical utility today. Single-analyte biomarkers, such as the erythrocyte sedimentation rate and C-reactive protein, have been utilized for decades as general markers of inflammation (2, 3). However, while each biomarker has its diagnostic niche, most single-analyte biomarkers are associated with limited sensitivity and specificity and demonstrate efficacy only in highly focused clinical syndromes. Perhaps the most useful single-analyte biomarker is procalcitonin (PCT), which demonstrates strong clinical utility for indicating the presence or absence of bacterial infection in both sepsis and lower respiratory tract infections (4, 5). However, in these syndromes, PCT does not offer identification of additional pathogen classes that may be present (including fungi and viruses) and is not broadly generalizable, as PCT has performed less well at identifying bacterial infections in other settings, such as prosthetic joint infection (6). Larger, more complex multianalyte markers of the host response to infectious perturbations may offer the potential for greater specificity and broader applicability to a wide array of clinical settings. The additional complexity of multianalyte biomarkers, however, brings its own challenges, including a need for more diverse computational approaches both for the derivation of biomarker panels as well as in the interpretation of the test output.
There are several examples of multianalyte biomarkers that have already demonstrated diagnostic utility, including tests that measure gene expression, protein panels, metabolite panels, cytokine panels, and others. This review focuses on one such modality, “transcriptomics,” defined as the analysis of host gene expression through RNA transcripts. The sequencing of the human genome, new techniques for RNA stabilization and amplification, multiplexing of PCR and hybridization-based technologies, and breakthroughs in the computation and statistical analysis of massive data sets have brought these methodologies from basic research to being accessible to the clinical realm. Since gene expression is rapidly altered in many cell types in response to a variety of exposures (including infection), utilizing this information has several advantages. For example, host-based gene expression has been proven to be able to distinguish active infection from colonization (7), to distinguish among broad pathogen classes (such as distinguishing viral from bacterial pathogens), and to provide prognostic information and disease severity prediction (8, 9). Furthermore, the widespread availability of quantitative reverse transcriptase PCR (qRT-PCR) platforms in clinical laboratories allows gene expression-based diagnostics to be more easily and directly translatable to patient care. In this brief review, we outline the current process for the design and development of gene expression-based biomarker classifiers, describe examples of the current state of the art in this field, and highlight relevant challenges to the field as it moves forward.
DEVELOPING GENE EXPRESSION-BASED CLASSIFIERS OF DISEASE
Disease classifiers can be thought of as patterns of biomarker perturbations capable of categorizing individuals into defined clinical groups (10). The process for the development of a gene expression-based disease classifier can be visualized through several broad steps, including a discovery phase to evaluate gene expression changes in the disease state of interest, a classifier generation step, and a platform development step that incorporates the classifier into a clinically usable diagnostic test.
Study Design.
Consideration of a targeted clinical scenario during classifier development is crucial. While comparison of infected hosts to healthy controls provides meaningful information on disease pathogenesis, this comparison (sick versus completely healthy) is uncommon in clinical practice and thus less useful for classifier development. The ultimate goal of a diagnostic test is to distinguish disease states in individuals with similar clinical presentations. Therefore, experimental derivation of a classifier is ideally performed by using an infected cohort and controls with similar phenotypic features to truly be useful as a diagnostic test in a clinical setting (such as viral versus bacterial respiratory infection and systemic inflammatory response syndrome [SIRS] versus sepsis, etc.).
Classifier Generation.
For most studies, peripheral blood is an ideal source (Fig. 1), as it is easily accessible, is commonly acquired in most clinical settings, and contains abundant cellular RNA, and circulating white blood cells are often directly responding to the myriad immune signals cascading from remote primary sites of infection. Historically, microarray technology has been utilized for gene expression quantification due to its relatively low cost, ease of data generation, established standardized methods for analysis, and good quantitative accuracy, and many (if not all) of the currently approved gene expression-based tests were initially developed on this platform. However, a notable limitation of microarrays is that they are restricted to the detection of sequences that are complementary to the probes included on the array, thus precluding the detection of many splice and sequence variants. Newer technology using next-generation RNA sequencing (RNA-Seq), initially impeded by cost and analytic complexity, has been rapidly replacing microarray technology due to decreasing prices and improved data management and analysis capabilities. RNA-Seq provides a snapshot view of the entire transcriptome at the time of sample acquisition and is not limited by the specific probes present on a microarray (11). RNA-Seq offers a number of additional advantages, including greater sensitivity and a less biased view of the transcriptome while simultaneously having the capability of detecting expressed sequence variants and splice variants.
FIG 1.
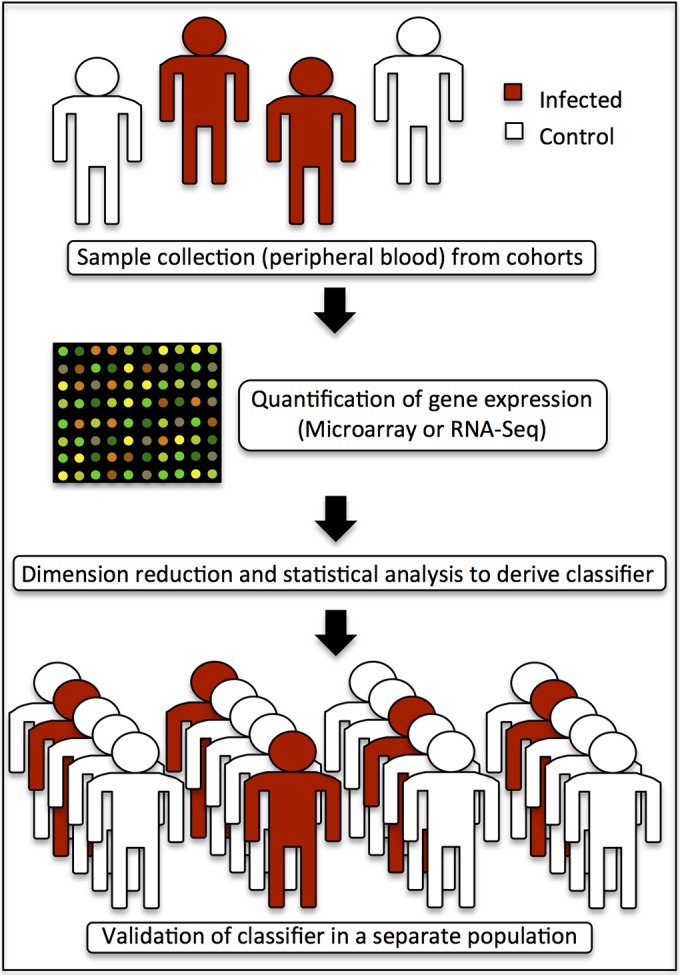
Development of a biomarker signature.
The use of either microarrays or RNA-Seq to measure quantitative levels of gene expression yields massive amounts of data detailing the up- or downregulation of genes across the entire genome. One of the necessary steps for classifier development is the ability to accurately and reproducibly analyze these vast amounts of complex data. While gene expression analysis is typically carried out on a small sample size of subjects, there are often tens of thousands (microarray) or millions (RNA-Seq) of data points for comparison between individuals. Dimensionality reduction is often used to understand and work with the data generated from each sample. Complex mathematical models, including techniques such as sparse factor modeling (12), Bayesian constructions of the elastic net (13), and others, are frequently utilized to construct classifiers, but the novelty of these high-dimensional data sets lends itself to the de novo creation of entirely new mathematical methodologies to deal with these unique statistical problems. Details of these models are beyond the scope of this review, but these dimensionality reduction methods and complex statistical modeling assist in matching phenotypes to transcriptomic profiles and allow statistical predictions and classifier development and optimization.
Diagnostic Platform.
In order to implement host gene expression-based classifiers as a diagnostic tool, there must be an available technological platform that can measure and interpret relevant subsets of genes that drive the host response. At present, platforms that measure very large numbers of genes (microarrays, RNA-Seq, and others) are excellent for discovery science but are too costly and too time-consuming, and interpretation of their results is too analytically demanding to be useful in most clinical scenarios. Therefore, genomic classifiers must focus on a limited number of important genes, preferably the smallest number required to maintain discriminative ability. A more clinically tractable platform (such as RT-PCR) can then be used to evaluate the expression of these critical genes in order to yield valuable diagnostic results. The current limitations of technological platforms capable of utilizing gene expression-based classifiers are discussed in greater detail below.
CURRENT GENE EXPRESSION-BASED DISEASE CLASSIFIERS
With current technology, individual pathogen-specific biomarkers within the context of a specific clinical syndrome have been difficult to derive. Transcriptomic classifiers that focus on defining the clinical syndrome as being driven by a given pathogen class (i.e., bacterial versus viral), rather than trying to identify the precise organism, have tended to be more robust and offer broader clinical utility. This type of classification scheme can facilitate treatment decisions such as directing appropriate antibiotic usage in respiratory infections or guiding the institution of proper empirical antimicrobial therapy in a febrile hospitalized patient. Here we briefly discuss reported gene expression and host-based diagnostics research as it has been applied to bloodstream and respiratory infections (Table 1).
TABLE 1.
Performance characteristics of selected host gene expression signatures for bloodstream and respiratory infections
| Reference | Disease state | Disease state(s) of comparative control group | No. of genes in signature | No. of subjects in validation group | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|
| 14 | Sepsis | Noninfectious SIRS | 4 | 59 | 0.86 | 0.79 | 0.91 |
| 17 | Viral infection (influenza) | Bacterial infection (S. pneumoniae) | 30 | 33 | 0.93 | —a | — |
| 8 | Viral infection (influenza) | Bacterial infection (E. coli, S. pneumoniae) | 35 | 75 | 0.91 | — | — |
| 8 | Gram-positive bacterial infection (S. pneumoniae) | Gram-negative bacterial infection (E. coli) | 30 | 40 | 0.85 | — | — |
| 18 | Viral infection (RSV) | Viral infection (HRV, influenza virus) | 70 | 137 | 0.95 | 0.94 | 0.98 |
| 26 | Fungal infection (Candida albicans) | Bacterial infection (S. aureus) and uninfected | 67 | 72 | 0.97 | 0.98 | 0.96 |
Sensitivity and specificity not provided in reference.
Sepsis.
Distinguishing whether SIRS is due to an infectious (i.e., sepsis) or a noninfectious etiology is of immediate and critical importance. To date, peripheral blood-derived gene expression-based classifiers have proven capable of distinguishing sepsis from SIRS with excellent diagnostic accuracy, including a 4-gene signature that demonstrated a sensitivity of 79% and a specificity of 91% (14). After distinguishing sepsis from SIRS, the next important task for clinicians is to subclassify septic patients across the spectrum of pathogen classes and degrees of clinical severity in order to personalize treatment decisions. Transcriptomic analyses of critically ill patients with sepsis have revealed that gene expression classifiers are capable of sorting patients into groups with respect to the degree of organ failure, length of intensive care unit (ICU) stay, and overall mortality (15, 16). These studies, along with others in the literature, indicate that biomarker-based clinical tools predicated on peripheral blood gene expression offer promise in helping clinicians understand the underlying pathophysiological differences in the presentation of sepsis and facilitate earlier, more tailored treatment decisions for this challenging patient population.
Other Acute Bacterial and Viral Infections.
In addition to distinguishing sepsis from noninfectious SIRS, gene expression-based classifiers have also shown utility in distinguishing Gram-positive from Gram-negative bacterial infections. Pediatric patients infected with two bacterial pathogens, Escherichia coli and Staphylococcus aureus, develop markedly different gene expression profiles in peripheral blood (8). A 30-gene classifier based on these gene expression changes was developed and successfully distinguished between these two bacteria with 85% accuracy in a pediatric cohort, thus highlighting the specificity of the host immune response to the invading pathogen and suggesting the possibility that host-based gene expression diagnostics could be used to facilitate more targeted antibiotic selection in appropriate patient populations. Furthermore, the genes upregulated in S. aureus infection compared to E. coli infection made sense from a pathophysiological perspective, as they were related mostly to neutrophil activity, including neutrophil chemoattractants such as CXCL1 and PPIB (8).
Distinguishing between viral and bacterial respiratory infections is a common clinical dilemma. One model developed to detect biomarkers of viral infection involves experimental infection of healthy human volunteers with live human rhinovirus (HRV), respiratory syncytial virus (RSV), or influenza A virus (17). A 30-gene classifier developed in this setting separated symptomatically virally infected and uninfected volunteers with 93% accuracy, and it was subsequently validated in a separate cohort of patients, where it classified naturally acquired infections as being either viral or bacterial infections with 80% accuracy (17). Gene expression-based classifiers have also been shown to differentiate between specific viral etiologies, as demonstrated by a 70-gene RSV transcriptional profile that successfully separated infants with RSV lower respiratory tract infection from those with HRV or influenza virus infection with 94% sensitivity and 98% specificity (18). In addition to successfully distinguishing viral and bacterial infections, a gene expression-based classifier of viral infection has also demonstrated the ability to diagnose subjects very early, during the presymptomatic phase, and to allow eventual severity prediction (9).
Other classifiers capable of specifically distinguishing viral and bacterial infections have also been developed. Gene expression profiles from pediatric patients infected with influenza A virus were compared to those from patients with bacterial infections due to Staphylococcus aureus, Streptococcus pneumoniae, and Escherichia coli, and a 35-gene classifier was developed and successfully discriminated between patients with influenza A virus and those with bacterial infections with 91% accuracy (8). These findings were further confirmed in an adult cohort of hospitalized patients, where gene expression-based classifiers were shown to be superior to a single-analyte biomarker, procalcitonin (sensitivity of 95% versus 38% and specificity of 92% versus 91%, respectively) (19). More recent work led to the development of an RT-PCR-based classifier that accurately separates acute upper respiratory illness into bacterial infection, viral infection, bacterium-virus coinfection, or noninfectious causes (20).
As interest in host transcriptomic technologies increases, the capabilities of this type of analysis are being explored for a number of additional important bacterial diseases, including the development of classifiers that can identify tuberculosis (TB) based on the extent of disease, predict the response to treatment (21, 22), and distinguish TB from other similar pulmonary diseases such as lung cancer, sarcoidosis, and community-acquired pneumonia with 88% sensitivity and 94% specificity (23). There are also preliminary data suggesting a role for gene expression-based classifiers in identifying cases of infection by less common bacterial pathogens such as Bacillus anthracis, Yersinia pestis, Francisella tularensis, Brucella melitensis, and many others (24, 25), and we anticipate that the number of potential clinical uses will continue to grow.
Fungal Infections.
Fungal organisms trigger a wide array of clinical syndromes and often present marked diagnostic challenges, including differentiating active fungal infection from colonization. Although the bulk of reported research has dealt with gene classifiers for bacterial and viral pathogens, the response to fungal pathogens has been studied on a smaller scale. One study of nonneutropenic medical and surgical ICU patients found a fungal colonization rate of 64% for these patients but noted that the actual rate of invasive mycosis was only 2.0% in the same patients (7). This study highlights that pathogen detection within a host does not always imply the presence of disease and further demonstrates the need for host-based diagnostics to provide answers in these difficult scenarios. The majority of host-based diagnostic research in the fungal arena focuses on Candida albicans, where a 67-gene classifier developed with a murine model of candidemia was found to be 98% sensitive and 96% specific for distinguishing Candida infection from bacterial infection or uninfected controls (26). Furthermore, the Candida classifier was also capable of distinguishing disseminated Candida infection from Staphylococcus aureus sepsis in a separate cohort of mice. However, this gene expression classifier has yet to be validated in human patients with candidemia. Additional promising preliminary work with other pathogenic fungi, such as Aspergillus fumigatus, has been performed (27), but no diagnostic classifiers of acute human disease have yet been developed. As the field of host-based diagnostics continues to grow, pathogenic fungi such as Aspergillus, Cryptococcus, and endemic fungi, which all drive diagnostically challenging clinical syndromes, are prime targets for the development of gene expression-based classifiers of disease.
TECHNOLOGY: MOVING FROM THE LAB TOWARD PATIENT CARE
Following classifier development, the next step in transitioning to the clinical arena is the migration of the classifier onto a testing platform. This step has historically represented a major barrier, as technologies capable of rapidly and quantitatively measuring large, complex, gene-based biomarker classifiers have not been available. PCR-based platforms are the current gold standard for mRNA analysis of targeted gene expression (10) (Fig. 2). However, PCR-based platforms for multiplex mRNA (or gene) amplification from peripheral blood cells historically offer extended turnaround times. With multiple necessary steps, including blood collection, RNA stabilization, RNA purification, qRT-PCR, target detection/quantification, and interpretation of results, this process can take many hours from sample collection to a diagnostic answer, making many platforms impractical for use in a typical clinical setting. The complex steps involved from sample acquisition to simultaneous quantification of multiple mRNAs from large gene-based classifiers have also hindered the widespread applicability of these technologies. Although PCR-based platforms are present in the vast majority of clinical microbiology laboratories, the turnaround time, complexity, and limits on the number of testable gene targets currently limit the practical use of multigene classifiers to research and less-time-critical clinical scenarios.
FIG 2.

Characteristics of a diagnostic test. TLDA, TaqMan low-density array.
Platforms for point-of-care diagnostic applications need improvement beyond currently available technologies in several key areas before widespread implementation in clinical settings is possible. The best reported classifiers based on host gene expression involve the calculation of relative weighted expression levels of subsets of critical genes and thus require some level of quantification of target mRNAs present in a sample, as has been demonstrated by the AlloMap gene expression test for rejection in heart transplant recipients (28). Another desirable component of a clinically relevant platform is an increase in the available multiplex capacity. Small classifiers consisting of only a few genes (i.e., 1 to 10 genes) can be adapted to many platforms, while larger classifiers (with over 100 genes) offer more robust discriminative ability but are less adaptable, since most clinically available platforms are not capable of simultaneously amplifying such a large number of targets. Therefore, the ideal technology would involve a sufficiently large multiplexing capacity (while an ideal gene classifier would simultaneously minimize the number of genes included). Although there are commercial platforms currently available that are capable of performing RT-PCR and detecting up to 100 gene targets, many of these existing assays to date have provided only qualitative answers, stating that a target is either present or absent. In order to be useful as a host-based transcriptomic diagnostic assay, the platform must provide quantitative answers so that the relative abundance of each mRNA can be calculated. However, as interest in this field increases, there is movement by the industry to modify existing platforms to overcome these limitations. Finally, the turnaround time is a critical component for many clinical applications. Availability of results in 24 h might be suitable for a hospitalized patient, but the emergency department and urgent care settings, with high turnover rates, require results in much shorter time frames (on the order of 2 h or less) to be practical for facilitating treatment decisions.
There are also regulatory challenges in translating a gene expression diagnostic test to clinical use. Demonstration of noninferiority or superiority compared to gold standard diagnostics is inherently challenging. Close contact with relevant FDA officials is critical when developing and designing novel disease classifiers for diagnostic use, as their input into acceptable processes for pursuing comparison to existing standards is invaluable. In addition to diagnostic metrics alone, it is often desirable to demonstrate that clinical information generated from a biomarker-based assay actually changes clinician practice and drives improved clinical outcomes, and in actual practice, this is not always the case. Consideration of additional regulatory hurdles such as those described above add complexity and additional layers of nuance and effort to bringing such tests to widespread clinical use but are unavoidable as part of the current regulatory landscape.
FUTURE PERSPECTIVES
The interaction between pathogen and host has been shown to produce reliable and reproducible responses in host peripheral blood gene expression across a broad range of infections. The process of utilizing information from the host immune response in the realm of clinical patient care is well under way. The largest barrier to the implementation of this technology involves the development of platforms capable of hosting diagnostic gene expression-based classifiers. Development of a broader armamentarium of classifiers for many more clinical scenarios would help drive the market for platforms capable of measuring these types of analytes in a clinical setting. Furthermore, in order to increase the generalizability and utility of these diagnostic tests, the performance of host-based biomarker classifiers also needs to be defined in immunocompromised hosts, patients with chronic inflammatory illnesses, and patients at extremes of age, since these populations respond differently to similar pathogen challenges. Once gene-based classifier technologies are ready for clinical use, such platforms will require large-scale validation to define the clinical outcomes associated with the implementation of new testing algorithms compared to current standards of care. In conclusion, although a great deal of work remains to be done before host-based peripheral blood-derived gene expression diagnostics become a reality, the available data show the potential of these novel diagnostic modalities to impact the clinical care of acute infectious diseases.
ACKNOWLEDGMENTS
E.L.T., C.W.W., and M.T.M. have pending patents for host-based biomarker signatures involving respiratory viral infections.
Biographies

Zachary E. Holcomb is a medical student at Duke University. He received his B.S. in Chemistry from the University of Virginia College at Wise. Mr. Holcomb has an interest in infectious diseases and has spent the past year exploring host-based gene expression diagnostics with a particular emphasis on fungal infections. He has been recognized as a Stead Research Scholar at Duke University and was selected as a 2016 IDSA Medical Research Scholar.

Ephraim L. Tsalik, M.D., M.H.S., Ph.D., is an Assistant Professor of Medicine in the Center for Applied Genomics and Precision Medicine and the Division of Infectious Diseases at Duke University. He is also a Staff Physician in the Emergency Department at the Durham VA Medical Center. Dr. Tsalik received his M.D. and Ph.D. from Columbia University. He then started a residency in Internal Medicine at Duke University in 2005, followed by a fellowship in Infectious Diseases, where he concurrently earned a master's of Health Sciences. Dr. Tsalik's research has focused on the development, evaluation, and promotion of diagnostics for acute infectious disease. He has made use of systems biology approaches to characterize the host response to infection so as to drive the development of diagnostic and prognostic assays. Dr. Tsalik also serves in the Antibacterial Resistance Leadership Group, where he interfaces with industry and academic partners to advance diagnostics development.

Christopher W. Woods, M.D., M.P.H., F.I.D.S.A., is professor in the Departments of Medicine and Pathology at Duke University, adjunct associate professor in Epidemiology at the UNC—Chapel Hill School of Public Health, and adjunct associate professor in the Emerging Infections Program at the Duke-National University of Singapore Graduate Medical School. He received his M.D. from Duke University and M.P.H. from the University of North Carolina and completed residency in Internal Medicine and fellowships in Infectious Diseases and Medical Microbiology at Duke University. He is Codirector of the Hubert-Yeargan Center for Global Health and Associate Director for Applied Genomics at the Duke Center for Applied Genomics and Precision Medicine (CAGPM). He is Global Health lead for the Duke Tropical Conservation Initiative. Clinically, he is Chief, Infectious Diseases, and hospital epidemiologist for the Durham VA Medical Center. He is board certified in internal medicine, infectious diseases, and medical microbiology and the coauthor of over 130 peer-reviewed articles.

Micah T. McClain, M.D., Ph.D., is an Assistant Professor of Medicine in the Division of Infectious Diseases and the Center for Applied Genomics and Precision Medicine at Duke University and a Staff Physician at the Durham VA Medical Center. Dr. McClain received his M.D. and Ph.D. (Immunology) from the University of Oklahoma Health Science Center before completing Internal Medicine Residency and a Fellowship in Infectious Diseases at Duke University. His current research involves analysis of host genomic and immunologic responses in acute infections, including examining host responses to infectious challenge in vulnerable populations such as elderly and immunosuppressed individuals. Additional ongoing work focuses on defining the host response to fungal infections, tick-borne illnesses, and other emerging infectious diseases as well as the immunologic mechanisms that drive resilience to infection in human hosts.
REFERENCES
- 1.Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 2.Waugh TR. 1923. The blood sedimentation test; its history, technique, nature and clinical application. Can Med Assoc J 13:604–608. [PMC free article] [PubMed] [Google Scholar]
- 3.Tillett WS, Francis T. 1930. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J Exp Med 52:561–571. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. 1993. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 341:515–518. doi: 10.1016/0140-6736(93)90277-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christ-Crain M, Jaccard-Stolz D, Bingisser R, Gencay MM, Huber PR, Tamm M, Muller B. 2004. Effect of procalcitonin-guided treatment on antibiotic use and outcome in lower respiratory tract infections: cluster-randomised, single-blinded intervention trial. Lancet 363:600–607. doi: 10.1016/S0140-6736(04)15591-8. [DOI] [PubMed] [Google Scholar]
- 6.Ettinger M, Calliess T, Kielstein JT, Sibai J, Bruckner T, Lichtinghagen R, Windhagen H, Lukasz A. 2015. Circulating biomarkers for discrimination between aseptic joint failure, low-grade infection, and high-grade septic failure. Clin Infect Dis 61:332–341. doi: 10.1093/cid/civ286. [DOI] [PubMed] [Google Scholar]
- 7.Petri MG, Konig J, Moecke HP, Gramm HJ, Barkow H, Kujath P, Dennhart R, Schafer H, Meyer N, Kalmar P, Thulig P, Muller J, Lode H. 1997. Epidemiology of invasive mycosis in ICU patients: a prospective multicenter study in 435 non-neutropenic patients. Paul-Ehrlich Society for Chemotherapy, Divisions of Mycology and Pneumonia Research. Intensive Care Med 23:317–325. doi: 10.1007/s001340050334. [DOI] [PubMed] [Google Scholar]
- 8.Ramilo O, Allman W, Chung W, Mejias A, Ardura M, Glaser C, Wittkowski KM, Piqueras B, Banchereau J, Palucka AK, Chaussabel D. 2007. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 109:2066–2077. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, Veldman T, Kingsmore SF, Huang Y, Lambkin-Williams R, Gilbert AG, Hero AO III, Ramsburg E, Glickman S, Lucas JE, Carin L, Ginsburg GS. 2013. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS One 8:e52198. doi: 10.1371/journal.pone.0052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaas AK, Burke T, Chen M, McClain M, Nicholson B, Veldman T, Tsalik EL, Fowler V, Rivers EP, Otero R, Kingsmore SF, Voora D, Lucas J, Hero AO, Carin L, Woods CW, Ginsburg GS. 2013. A host-based RT-PCR gene expression signature to identify acute respiratory viral infection. Sci Transl Med 5:203ra126. doi: 10.1126/scitranslmed.3006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baginsky S, Hennig L, Zimmermann P, Gruissem W. 2010. Gene expression analysis, proteomics, and network discovery. Plant Physiol 152:402–410. doi: 10.1104/pp.109.150433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho CM, Chang J, Lucas JE, Nevins JR, Wang Q, West M. 2008. High-dimensional sparse factor modeling: applications in gene expression genomics. J Am Stat Assoc 103:1438–1456. doi: 10.1198/016214508000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Carlson D, Zaas A, Woods CW, Ginsburg GS, Hero AO III, Lucas J, Carin L. 2011. Detection of viruses via statistical gene expression analysis. IEEE Trans Biomed Eng 58:468–479. doi: 10.1109/TBME.2010.2059702. [DOI] [PubMed] [Google Scholar]
- 14.McHugh L, Seldon TA, Brandon RA, Kirk JT, Rapisarda A, Sutherland AJ, Presneill JJ, Venter DJ, Lipman J, Thomas MR, Klein Klouwenberg PM, van Vught L, Scicluna B, Bonten M, Cremer OL, Schultz MJ, van der Poll T, Yager TD, Brandon RB. 2015. A molecular host response assay to discriminate between sepsis and infection-negative systemic inflammation in critically ill patients: discovery and validation in independent cohorts. PLoS Med 12:e1001916. doi: 10.1371/journal.pmed.1001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maslove DM, Tang BM, McLean AS. 2012. Identification of sepsis subtypes in critically ill adults using gene expression profiling. Crit Care 16:R183. doi: 10.1186/cc11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong HR, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, Checchia PA, Lin R, Shanley TP, Bigham MT, Wheeler DS, Doughty LA, Tegtmeyer K, Poynter SE, Kaplan JM, Chima RS, Stalets E, Basu RK, Varisco BM, Barr FE. 2011. Validation of a gene expression-based subclassification strategy for pediatric septic shock. Crit Care Med 39:2511–2517. doi: 10.1097/CCM.0b013e3182257675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaas AK, Chen M, Varkey J, Veldman T, Hero AO III, Lucas J, Huang Y, Turner R, Gilbert A, Lambkin-Williams R, Oien NC, Nicholson B, Kingsmore S, Carin L, Woods CW, Ginsburg GS. 2009. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 6:207–217. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mejias A, Dimo B, Suarez NM, Garcia C, Suarez-Arrabal MC, Jartti T, Blankenship D, Jordan-Villegas A, Ardura MI, Xu Z, Banchereau J, Chaussabel D, Ramilo O. 2013. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med 10:e1001549. doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suarez NM, Bunsow E, Falsey AR, Walsh EE, Mejias A, Ramilo O. 2015. Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J Infect Dis 212:213–222. doi: 10.1093/infdis/jiv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsalik EL, Henao R, Nichols M, Burke T, Ko ER, McClain MT, Hudson LL, Mazur A, Freeman DH, Veldman T, Langley RJ, Quackenbush EB, Glickman SW, Cairns CB, Jaehne AK, Rivers EP, Otero RM, Zaas AK, Kingsmore SF, Lucas J, Fowler VG Jr, Carin L, Ginsburg GS, Woods CW. 2016. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci Transl Med 8:322ra11. doi: 10.1126/scitranslmed.aad6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O'Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff TH, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SH, Walzl G, Aderem A, Hanekom WA. 24 March 2016. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet doi: 10.1016/s0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloom CI, Graham CM, Berry MP, Rozakeas F, Redford PS, Wang Y, Xu Z, Wilkinson KA, Wilkinson RJ, Kendrick Y, Devouassoux G, Ferry T, Miyara M, Bouvry D, Valeyre D, Gorochov G, Blankenship D, Saadatian M, Vanhems P, Beynon H, Vancheeswaran R, Wickremasinghe M, Chaussabel D, Banchereau J, Pascual V, Ho LP, Lipman M, O'Garra A. 2013. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS One 8:e70630. doi: 10.1371/journal.pone.0070630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das R, Hammamieh R, Neill R, Ludwig GV, Eker S, Lincoln P, Ramamoorthy P, Dhokalia A, Mani S, Mendis C, Cummings C, Kearney B, Royaee A, Huang XZ, Paranavitana C, Smith L, Peel S, Kanesa-Thasan N, Hoover D, Lindler LE, Yang D, Henchal E, Jett M. 2008. Early indicators of exposure to biological threat agents using host gene profiles in peripheral blood mononuclear cells. BMC Infect Dis 8:104. doi: 10.1186/1471-2334-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paranavitana C, Pittman PR, Velauthapillai M, Zelazowska E, Dasilva L. 2008. Transcriptional profiling of Francisella tularensis infected peripheral blood mononuclear cells: a predictive tool for tularemia. FEMS Immunol Med Microbiol 54:92–103. doi: 10.1111/j.1574-695X.2008.00456.x. [DOI] [PubMed] [Google Scholar]
- 26.Zaas AK, Aziz H, Lucas J, Perfect JR, Ginsburg GS. 2010. Blood gene expression signatures predict invasive candidiasis. Sci Transl Med 2:21ra17. doi: 10.1126/scitranslmed.3000715. [DOI] [PubMed] [Google Scholar]
- 27.Cortez KJ, Lyman CA, Kottilil S, Kim HS, Roilides E, Yang J, Fullmer B, Lempicki R, Walsh TJ. 2006. Functional genomics of innate host defense molecules in normal human monocytes in response to Aspergillus fumigatus. Infect Immun 74:2353–2365. doi: 10.1128/IAI.74.4.2353-2365.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamani MH, Taylor DO, Haire C, Smedira N, Starling RC. 2007. Post-transplant ischemic injury is associated with up-regulated AlloMap gene expression. Clin Transplant 21:523–525. doi: 10.1111/j.1399-0012.2007.00681.x. [DOI] [PubMed] [Google Scholar]