

ABSTRACT
Escherichia coli and Shigella species are closely related and genetically constitute the same species. Differentiating between these two pathogens and accurately identifying the four species of Shigella are therefore challenging. The organism-specific bioinformatics whole-genome sequencing (WGS) typing pipelines at Public Health England are dependent on the initial identification of the bacterial species by use of a kmer-based approach. Of the 1,982 Escherichia coli and Shigella sp. isolates analyzed in this study, 1,957 (98.4%) had concordant results by both traditional biochemistry and serology (TB&S) and the kmer identification (ID) derived from the WGS data. Of the 25 mismatches identified, 10 were enteroinvasive E. coli isolates that were misidentified as Shigella flexneri or S. boydii by the kmer ID, and 8 were S. flexneri isolates misidentified by TB&S as S. boydii due to nonfunctional S. flexneri O antigen biosynthesis genes. Analysis of the population structure based on multilocus sequence typing (MLST) data derived from the WGS data showed that the remaining discrepant results belonged to clonal complex 288 (CC288), comprising both S. boydii and S. dysenteriae strains. Mismatches between the TB&S and kmer ID results were explained by the close phylogenetic relationship between the two species and were resolved with reference to the MLST data. Shigella can be differentiated from E. coli and accurately identified to the species level by use of kmer comparisons and MLST. Analysis of the WGS data provided explanations for the discordant results between TB&S and WGS data, revealed the true phylogenetic relationships between different species of Shigella, and identified emerging pathoadapted lineages.
KEYWORDS: DNA sequencing, Escherichia coli, MLST, Shigella, identification, kmer
INTRODUCTION
Escherichia coli and Shigella species are closely related; the aggregate biochemical reactions of members of these two genera are similar, and the lipopolysaccharide (LPS) O antigens of known serotypes of Shigella (except Shigella sonnei) are shared with one or more of the many O antigen groups of E. coli (1, 2). Although the relatedness of DNAs from E. coli and Shigella indicate that they constitute a single species (3), they are maintained as separate entities in the interests of epidemiology and clinical medicine (4). There is often a clinical or public health requirement to differentiate between E. coli and Shigella and/or to specify which species of Shigella has been isolated (5, 6).
Traditionally, at the Gastrointestinal Bacterial Reference Unit (GBRU) of Public Health England (PHE), a combination of biochemistry and serotyping was used to differentiate E. coli from Shigella and to identify the four species of Shigella (1). Generally, shigellae are less active biochemically than E. coli, react with a limited set of antisera, and harbor a combination of pathogenicity genes shared only with the enteroinvasive E. coli (EIEC) group (7). EIEC strains are notoriously difficult to identify, and confirmation of the identification requires serological evidence. Within the genus Shigella, S. sonnei is the most reactive species biochemically but has only one LPS O antigen, so it cannot be serotyped. S. flexneri and S. boydii have similar biochemical profiles and are differentiated serologically (1). With the exception of serotype 6 strains, all S. flexneri strains share a polysaccharide backbone composed of a linear tetrasaccharide, and the different serotypes arise as a result of glucosylation and/or O-acetylation of this polysaccharide backbone at various positions (8). S. boydii and S. dysenteriae share the same O antigen structure as that of E. coli, and the majority of isolates have a well-defined and limited set of O antigens (2). Using the traditional set of sugars, S. dysenteriae is generally the least reactive species of Shigella and is characterized by the inability to ferment mannitol (1).
There are exceptions to the traditional biochemistry and serotyping schema rules, as well as examples of serotypes of Shigella being misidentified historically. For example, it is well known that S. flexneri serotype 6 is not genetically related to the other S. flexneri serotypes and that it clusters phylogenetically with S. boydii (9–11). On occasion, the decision of whether to designate an isolate as belonging to a specific Shigella species is based on one or two biochemical reactions and may appear arbitrary. The complexity of the Shigella species schema was exemplified by Ewing in 1986 (1), when he explained that it is based “partly on biochemistry, partly on serology and partly on tradition.”
Recently, PHE implemented whole-genome sequencing (WGS) for the routine typing of gastrointestinal pathogens for public health surveillance (12–14). The organism-specific bioinformatics WGS typing pipelines at PHE are dependent on the initial identification of the bacterial species by use of a kmer-based approach. Subsequently, for E. coli, S. sonnei, S. flexneri, S. boydii, and S. dysenteriae, the multilocus sequence type (MLST) and, where appropriate, the serotype are derived from the genome sequence (13, 15). The aim of this study was to compare the results of traditional biochemical and serological methods with the WGS kmer-based comparisons and WGS-derived MLST results for the identification of E. coli and Shigella species.
RESULTS AND DISCUSSION
Comparison of identifications of Shigella species by use of traditional methods and a kmer-based method.
Of the 1,982 isolates in this study, 1,957 (98.4%) were designated the same species by both traditional biochemistry and serology (TB&S) and the kmer identification (kmer ID) derived from the WGS data. All isolates identified as S. sonnei (n = 335) and S. flexneri (n = 350) by TB&S were concordant by use of the kmer ID. One hundred sixty of the 161 isolates identified as S. dysenteriae by TB&S were also identified as S. dysenteriae by kmer ID. The mismatched isolate was identified as S. flexneri by the kmer-based method. For the 159 isolates identified as S. boydii by TB&S, 145 results were concordant with the identifications obtained using the kmer-based approach. Of the 14 mismatched isolates, eight were identified as S. flexneri and six were identified as S. dysenteriae by use of the WGS data (Table 1). Finally, 967 of 977 isolates of E. coli identified by TB&S were concordant by use of the kmer ID. The 10 mismatched isolates were identified as EIEC by TB&S and as S. flexneri (n = 3) or S. boydii (n = 7) by the kmer-based approach (Table 1).
TABLE 1.
Comparison of identifications of E. coli and Shigella species by use of traditional methods and kmer ID
| TB&S ID | No. of isolates with kmer ID |
|||||
|---|---|---|---|---|---|---|
| E. coli | EIEC | S. sonnei | S. flexneri | S. boydii | S. dysenteriae | |
| E. coli | 923 | 0 | 0 | 0 | 0 | 0 |
| EIEC | 0 | 44 | 0 | 3 | 7 | 0 |
| S. sonnei | 0 | 0 | 335 | 0 | 0 | 0 |
| S. flexneri | 0 | 0 | 0 | 350 | 0 | 0 |
| S. boydii | 0 | 0 | 0 | 8 | 145 | 6 |
| S. dysenteriae | 0 | 0 | 0 | 1 | 0 | 160 |
Population structure of Shigella species by sequence type.
In order to determine the reasons for mismatches between the two methods, a minimum spanning tree was constructed to analyze the population structure of the data set by MLST (Fig. 1). The isolates belonging to the four Shigella species in this data set belonged to four major clonal complexes (CCs). All isolates of S. sonnei belonged to CC152 (highlighted in yellow in Fig. 1), comprising sequence type 152 (ST152) (n = 331) and a single-locus variant (SLV), ST1503 (n = 4). The majority of the isolates identified as S. flexneri by TB&S (highlighted in blue in Fig. 1) (including those of serotypes 1a, 1b, 2a, 2b, 3a, 3b, 4c, X, and Y) belonged to CC245 (n = 322). The STs associated with CC245 are listed in Table 2. All isolates of S. flexneri serotype 6 (n = 28) belonged to ST145 within CC145 (Fig. 1 and Table 2). S. flexneri serotype 6 was misidentified historically and is more closely related to S. boydii (9–11).
FIG 1.

Population structure of E. coli, highlighting CCs and STs associated with all four Shigella species and with EIEC. Isolates are colored with respect to their identification by traditional biochemistry and serology (yellow, S. sonnei; blue, S. flexneri; green, S. boydii; red, S. dysenteriae; pale blue, EIEC; and white, E. coli), and anomalies are labeled. (A) Misidentified as S. boydii by traditional tests because of a nonfunctional wzx1–5 gene. (B) Part of CC288 (a CC comprising mostly S. dysenteriae isolates) but biochemically and serologically identified as S. boydii. (C) Biochemically and serologically identified as S. boydii. (D) Belongs to ST243 (an ST comprising S. boydii and S. flexneri serotype 6) but biochemically and serologically identified as S. dysenteriae. (E) Identified as EIEC by biochemistry, serology, and PCR.
TABLE 2.
Clonal complexes of S. sonnei, S. flexneri, S. boydii, and S. dysenteriae, associated STs, and relationships between STs and serotypes for STs within CC145 and CC288a
| Species | CC152 (n = 335) |
CC245 (n = 330) |
CC145 (n = 262) |
CC288 (n = 76) |
Minor CCs (n = 1) |
||||
|---|---|---|---|---|---|---|---|---|---|
| ST | ST | Serotype(s) | ST | Serotype(s) | ST | Serotype(s) | ST | Serotype(s) | |
| S. sonnei | ST152 (331) | ||||||||
| ST1503 (4) | |||||||||
| S. flexneri | ST245 (301) | F1 to F5, FX, FY | ST145 (28) | F6 | |||||
| ST628 (2) | |||||||||
| ST630 (4) | |||||||||
| ST631 (3) | |||||||||
| ST651 (5) | |||||||||
| ST1025 (7) | |||||||||
| ST1753 (8) | |||||||||
| S. boydii | ST145 (50) | B2, B4 | ST149 (1) | B5 | ST1767 (1) | B12 | |||
| ST243 (74) | B1, B8, B10, B18, B19 | ST250 (2) | B15 | ||||||
| ST1749 (1) | B7 | ||||||||
| ST1273 (11) | B14 | ST1751 (5) | B9 | ||||||
| ST1743 (2) | B3 | ST1765 (4) | B11 | ||||||
| S. dysenteriae | ST148 (68) | D3, D12, D14, E112707 | ST147 (14) | D2 | |||||
| ST273 (6) | D2 | ||||||||
| ST243 | D7b | ST288 (32) | D2 | ||||||
| ST252 (25) | D4, D9, E670 | ST1759 (11) | D2 | ||||||
| ST1739 (4) | D13 | ||||||||
Numbers in parentheses show the numbers of isolates of the indicated STs. Data in bold were associated with mismatched results.
The isolate was associated with S. boydii ST243 but had acquired the D7 O antigen (labeled “D” in Fig. 1).
CC145 comprised S. flexneri serotype 6 (ST145) and S. boydii (highlighted in green in Fig. 1) and S. dysenteriae (highlighted in red in Fig. 1) serotypes. S. dysenteriae STs within CC145 were ST148 (n = 68), ST252 (n = 25), and ST1739 (n = 4), and the S. boydii STs included ST145 (n = 50), ST243 (n = 74), ST1273 (n = 11), and ST1743 (n = 2) (Fig. 1 and Table 2). The majority of isolates associated with CC288 were S. dysenteriae isolates of ST147 (n = 14), ST273 (n = 6), ST288 (n = 32), and ST1759 (n = 11). S. boydii isolates within CC288 were isolates of ST149 (n = 1), ST250 (n = 2), ST1749 (n = 1), ST1751 (n = 5), and ST1765 (n = 4). One isolate (S. boydii serotype 12) was typed as ST1767, and this ST did not belong to either CC145 or CC288. The relationships between S. flexneri, S. boydii, and S. dysenteriae serotypes and STs are shown in Table 2.
Resolution of mismatches.
Eight of the isolates identified as S. boydii by TB&S were identified as S. flexneri by using the kmer data and belonged to ST1753, a double-locus variant of ST245 (labeled “A” in Fig. 1). The kmer ID therefore provided the correct identification. S. flexneri and S. boydii cannot be distinguished biochemically, and their differentiation is achieved by serology (1). The isolates belonging to ST1753 did not agglutinate with any of the S. flexneri antisera and were therefore initially identified as S. boydii by TB&S. Further analysis of the WGS data from these eight isolates showed that the wzx1–5 gene, which is common to all S. flexneri isolates except those of serotype 6, was detected but nonfunctional due to an early stop codon, thus explaining the aberrant serology result and the incorrect TB&S identification.
Six isolates identified as S. boydii by TB&S were identified as S. dysenteriae by using the WGS data and belonged to either ST1751 (n = 5) or ST1767 (n = 1) (labeled “B” and “C,” respectively, in Fig. 1). Biochemically, all these isolates utilized mannitol (which is characteristic of S. boydii but not S. dysenteriae) (1) and agglutinated with antisera raised against S. boydii serotype 9 (ST1751) and S. boydii serotype 12 (ST1767). ST1751 is part of CC288, which is predominantly an S. dysenteriae complex, and analysis of the single nucleotide differences in the core genome illustrated the relationships between ST1751 and the other STs in CC288 (Fig. 2). ST1751 is positioned between two branches, one comprising isolates of S. dysenteriae and the other associated with S. boydii. ST1767 does not belong to either of the two main S. boydii or S. dysenteriae CCs and appears as an outlier in the MLST population structure (labeled “C” in Fig. 1).
FIG 2.
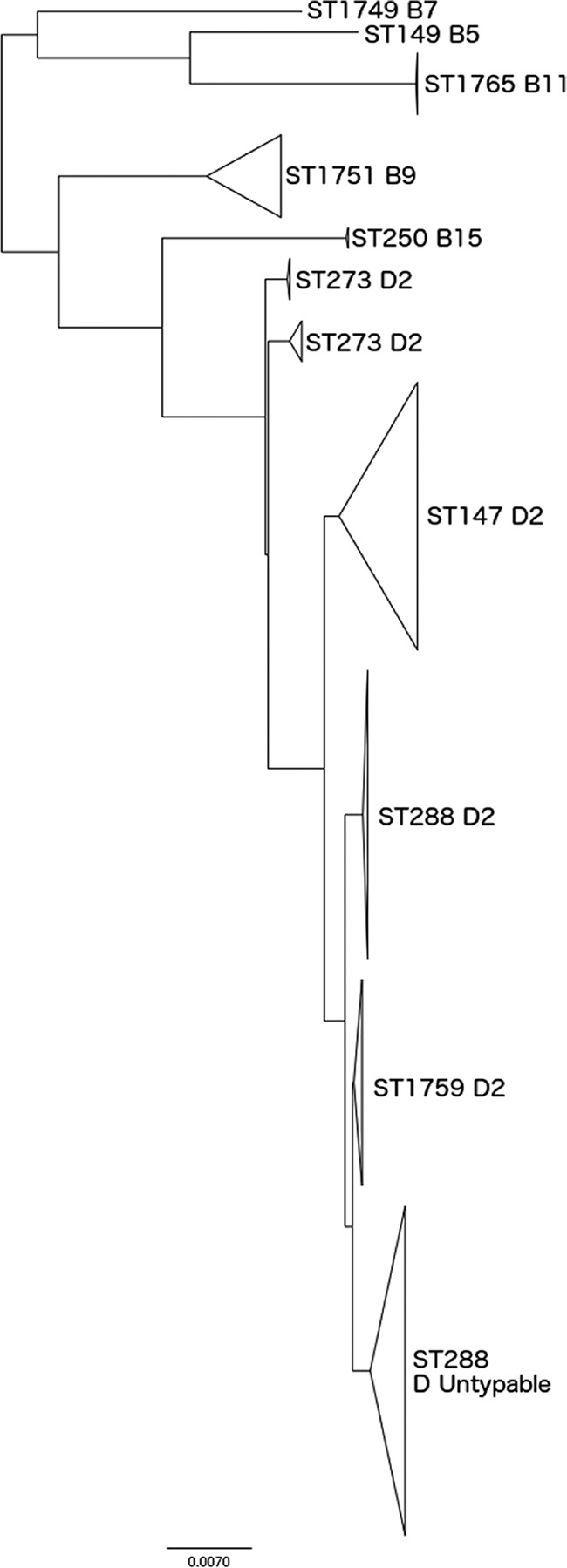
Phylogenetic analysis of CC288 based on core genome SNPs, highlighting the relationship between S. boydii and S. dysenteriae strains within this CC.
One isolate was identified as S. dysenteriae by TB&S and as S. flexneri by kmer ID (labeled “D” in Fig. 1). This isolate did not metabolize mannitol and reacted with antisera raised against S. dysenteriae serotype 7, hence the initial identification by TB&S. The kmer ID was S. flexneri, as the closest match to this isolate in the kmer database was S. flexneri serotype 6 (misidentified historically and phylogenetically related to S. boydii). Analysis of the WGS data showed that the O antigen cassette encoded the S. dysenteriae serotype 7 O antigen (2). Therefore, this isolate had acquired an S. dysenteriae O antigen in an S. boydii phylogenetic background with kmers that most closely matched S. flexneri serotype 6. This example demonstrates the complexity associated with identification of Shigella isolates to the species level.
Ten isolates identified as EIEC by TB&S were identified as Shigella species by kmer ID, although the similarities to the nearest reference genome were relatively low (80.1 to 80.4%) (labeled “E” in Fig. 1). All 10 isolates belonged to ST270 and had ipaH. However, they also had biochemical and serological characteristics of E. coli. Biochemically, all isolates utilized lactose and ortho-nitrophenyl-β-galactoside and had ornithine decarboxylase activity. Serologically, all isolates had E. coli O antigen biosynthesis genes (see Table S1 in the supplemental material). EIEC ST270 may represent a novel emerging Shigella pathotype (16).
Summary.
The comparison analysis in this study showed that >98% of the kmer ID results were concordant with the results derived by traditional methods. The isolates of S. flexneri belonging to ST1753, misidentified by TB&S as S. boydii due to a dysfunctional wzx1–5 gene, highlight one of the problems with using serology to identify Shigella to the species level (15). Historically, S. boydii and S. dysenteriae were differentiated biochemically by the mannitol test, and the relationship between these two species within CC145 and CC288 demonstrates that separating species based on one test can be misleading with respect to their true phylogenetic relationship. Both CC145 and CC288 contain S. dysenteriae and S. boydii STs, and it was expected that this close phylogenetic relationship would confound attempts to use a kmer-based approach to differentiate these two species. In fact, with the exception of S. boydii serotype 9 (ST1751) isolates that were in close proximity to STs comprising isolates of S. dysenteriae serotype 2, the kmer ID results correlated well with the TB&S identification results.
In this study, we showed that Shigella can be differentiated from E. coli and accurately identified to the species level from WGS data by use of a kmer-based approach. Analysis of the WGS data provided explanations for the confounding factors associated with identifying the Shigella species by using TB&S. Most importantly, moving forward, WGS data will enable us to determine the true relationships between the different species of Shigella and will facilitate the surveillance of emerging pathoadapted lineages.
MATERIALS AND METHODS
Bacterial isolates.
In this study, 1,982 isolates from the GBRU archive, identified as either E. coli (n = 977; 54 isolates were identified as EIEC), S. sonnei (n = 335), S. flexneri (n = 350), S. boydii (n = 159), or S. dysenteriae (n = 161) by traditional biochemistry and serology (TB&S) and by PCR targeting ipaH, were sequenced (see Table S1 in the supplemental material). All isolates were submitted to GBRU for confirmation and typing from local hospital laboratories between 2004 and January 2016. The isolates were selected based on the following two sets of criteria: (i) isolates submitted to GBRU between 2004 and 2014 were selected to cover a range of different serotypes (n = 641), and (ii) all isolates submitted to GBRU from local hospital laboratories between April 2015 and January 2016 and identified as E. coli or Shigella species by TB&S (n = 1,340) were included.
Serotyping, biochemistry, and PCR.
Serotyping was based on the somatic lipopolysaccharide O and flagellar H antigens as detected in agglutination assays with specific rabbit antibodies (17). For E. coli, more than 200 different O antigens and more than 50 H antigens have been identified. Currently, there are 15 established S. flexneri serotypes (F1a, F1b, F1c, F2a, F2b, F3a, F3b, F4a, F4b, F4c, F5a, F5b, FX, FY, and F6). S. boydii and S. dysenteriae have 20 (B1 to B20) and 15 (D1 to D15) recognized serotypes, respectively. There are two provisional S. dysenteriae serotypes in this study, designated E112707 and E670. Biochemistry tests were set up using a combination of appropriate substrates (1). The ability (or inability) of a bacterium to utilize a substrate was recorded as a positive (or negative) result and compared to published tables categorizing the known reactions of E. coli and Shigella species (1, 7). The key biochemical tests included lysine and ornithine decarboxylase activities, indole production, and the utilization of lactose, mannitol, and ortho-nitrophenyl-β-galactoside. DNAs from cultures and fecal extracts were tested by real-time PCR targeting ipaH on a Rotorgene Q machine (Qiagen, United Kingdom), using the following primers and probe: forward primer, 5′-AGGTCGCTGCATGGCTGGAA; reverse primer, 5′-CACGGTCCTCACAGCTCTCA; and probe, AACTCAGTGCCTCTGCGGAGCTTCGACA-6-carboxyfluorescein (FAM). The amplification parameters were 95°C for 5 min followed by 95°C for 15 s and 60°C for 60 s.
Whole-genome sequencing and kmer identification.
Genomic DNA was extracted, fragmented, and tagged for multiplexing by use of Nextera XT DNA sample preparation kits and then sequenced using an Illumina HiSeq 2500 system to produce 100-bp paired-end sequence fragments (Illumina, Cambridge, United Kingdom). FASTQ paired-end reads were quality trimmed using Trimomatic, with bases with PHRED scores of <30 removed from the trailing end. If the read length posttrimming was less than 50 bp, the read and its pair were discarded.
For the purposes of this study, the bioinformatics analysis was divided into three main components: (i) identification to the species level by kmer identification (kmer ID), (ii) sequence type assignment, and (iii) single nucleotide polymorphism (SNP) analysis. The kmer ID was the first step in the generic bioinformatics analysis pipeline and directed the sequencing down the appropriate pathogen pathway. The MLST analysis orientated the isolate in the phylogeny and facilitated the selection of the most appropriate reference genome prior to the highly discriminatory SNP typing analysis.
A kmer (a short string of DNA of length k; in this method, k = 18)-based approach was used to confirm the identity of the sample before organism-specific algorithms were applied. As the kmer length decreases, the percentage of kmers shared by all genomes increases. kmers with lengths shorter than 18 bp were not descriptive enough to distinguish between genomes at the species level. The kmer length was not extended beyond 18 bp because this required additional computation and there was no additional discriminatory benefit observed. The kmer algorithm determined the set of kmers of the fixed length k that were present in the WGS reads of each sample at least twice, using a sliding window with a step size of 1 on each read. kmers containing ambiguous nucleotide codes (codes other than A, C, G, or T) were discarded. The resulting set of kmers was compared to the set of kmers found for a collection of reference genomes. The similarity of the sets was expressed as a percentage. This value indicates the portion of the kmers in the reference genome that were also found in the reads. The closest percentage match was identified and provided initial confirmation of the species. The kmer ID software is available at https://github.com/phe-bioinformatics/kmerid.
Reference genomes (n = 1,781) for 59 bacterial genera comprising the majority of human pathogens, commensal bacteria, and common contaminants were downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria. The kmer algorithm compared each sample to the representative genomes for these 59 bacterial genera and returned the most similar genome together with a similarity estimate. First, the genus of the test sample was determined by comparing the sample to three representative genomes for each genus, and based on the results of this comparison, the most likely candidate genera were examined further. The minimum number of candidate genera selected was two, and the maximum number selected was five. A further 40 representative genomes for each candidate genus were compared to the sample. The sample was subsequently compared to all representatives in the top 2 to 5 candidate genomes, and a similarity value was calculated. The top hit among these comparisons was reported.
MLST and single nucleotide polymorphism typing.
Sequence type (ST) assignment was performed using a modified version of SRST, using the MLST database described by Tewolde et al. (18). MOST software (for MLST) is available at https://github.com/phe-bioinformatics/MOST.
For the isolates belonging to clonal complex 288, high-quality Illumina reads were mapped to a SPADES v3.5.0 de novo assembly of strain 140103 by using BWA-MEM (19, 20). SNPs were identified using GATK2 (21) in the unified genotyper mode. Core genome positions, defined as those present in the reference genome and in at least 80% of the isolates, that had a high-quality SNP (>90% consensus; minimum depth, 10×; mapping quality [MQ] ≥ 30) in at least one isolate were extracted, and RaxML (22) was used to derive the maximum likelihood phylogeny for the isolates.
Accession number(s).
FASTQ reads for all sequences in this study can be found under accession number PRJNA315192 at the PHE Pathogens BioProject at the National Center for Biotechnology Information website. The taxonomic names in the BioProject are based on the results of the traditional biochemistry and serology tests.
Supplementary Material
ACKNOWLEDGMENTS
We thank Vivienne do Nascimento and Dawn Hedges for curation of the serotyping scheme at GBRU.
This work was supported by the National Institute for Health Research Health Protection Research Unit in Gastrointestinal Infections.
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health, or Public Health England.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.01790-16.
REFERENCES
- 1.Ewing WH. 1986. The genus Escherichia and the genus Shigella, p 93–172. In Edwards and Ewing's identification of Enterobacteriaceae, 4th ed Elsevier Science Publishing Co, Inc, New York, NY. [Google Scholar]
- 2.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, Reeves PR, Wang L. 2008. Structure and genetics of Shigella O antigens. FEMS Microbiol Rev 32:627–653. doi: 10.1111/j.1574-6976.2008.00114.x. [DOI] [PubMed] [Google Scholar]
- 3.Pettengill EA, Pettengill JB, Binet R. 2016. Phylogenetic analyses of Shigella and enteroinvasive Escherichia coli for the identification of molecular epidemiological markers: whole-genome comparative analysis does not support distinct genera designation. Front Microbiol 6:1573. doi: 10.3389/fmicb.2015.01573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahl JW, Morris CR, Emberger J, Fraser CM, Ochieng JB, Juma J, Fields B, Breiman RF, Gilmour M, Nataro JP, Rasko DA. 2015. Defining the phylogenomics of Shigella species: a pathway to diagnostics. J Clin Microbiol 53:951–960. doi: 10.1128/JCM.03527-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simms I, Field N, Jenkins C, Childs T, Gilbart VL, Dallman TJ, Mook P, Crook PD, Hughes G. 2015. Intensified shigellosis epidemic associated with sexual transmission in men who have sex with men—Shigella flexneri and S. sonnei in England, to end of February 2015. Euro Surveill 20:21097. doi: 10.2807/1560-7917.ES2015.20.15.21097. [DOI] [PubMed] [Google Scholar]
- 6.Dallman TJ, Chattaway MA, Mook P, Godbole G, Crook PD, Jenkins C. 2016. Use of whole genome sequencing for the public health surveillance of Shigella sonnei in England & Wales, 2015. J Med Microbiol 65:882–884. doi: 10.1099/jmm.0.000296. [DOI] [PubMed] [Google Scholar]
- 7.van den Beld MJ, Reubsaet FA. 2012. Differentiation between Shigella, enteroinvasive Escherichia coli (EIEC) and noninvasive Escherichia coli. Eur J Clin Microbiol Infect Dis 31:899–904. doi: 10.1007/s10096-011-1395-7. [DOI] [PubMed] [Google Scholar]
- 8.Allison GE, Verma NK. 2000. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol 8:17–23. doi: 10.1016/S0966-842X(99)01646-7. [DOI] [PubMed] [Google Scholar]
- 9.Slopek S, Mulczyk M. 1967. Concerning the classification of Shigella flexneri 6 bacilli. Arch Immunol Ther Exp 15:600–603. [PubMed] [Google Scholar]
- 10.Yang J, Nie H, Chen L, Zhang X, Yang F, Xu X, Zhu Y, Yu J, Jin Q. 2007. Revisiting the molecular evolutionary history of Shigella spp. J Mol Evol 64:71–79. doi: 10.1007/s00239-006-0052-8. [DOI] [PubMed] [Google Scholar]
- 11.Connor TR, Barker CR, Baker KS, Weill FX, Talukder KA, Smith AM, Baker S, Gouali M, Pham Thanh D, Jahan Azmi I, Dias da Silveira W, Semmler T, Wieler LH, Jenkins C, Cravioto A, Faruque SM, Parkhill J, Wook Kim D, Keddy KH, Thomson NR. 2015. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. eLife 4:e07335. doi: 10.7554/eLife.07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dallman TJ, Byrne L, Ashton PM, Cowley LA, Perry NT, Adak G, Petrovska L, Ellis RJ, Elson R, Underwood A, Green J, Hanage WP, Jenkins C, Grant KA, Wain J. 2015. Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin Infect Dis 61:305–312. doi: 10.1093/cid/civ318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattaway MA, Dallman TJ, Gentle A, Wright MJ, Long SE, Ashton PM, Perry NT, Jenkins C. 2016. Whole genome sequencing for public health surveillance of Shiga toxin-producing Escherichia coli other than serogroup O157. Front Microbiol 7:258. doi: 10.3389/fmicb.2016.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashton PM, Nair S, Peters TM, Bale JA, Powell DG, Painset A, Tewolde R, Schaefer U, Jenkins C, Dallman TJ, de Pinna EM, Grant KA, Salmonella Whole Genome Sequencing Implementation Group. 2016. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ 4:e1752. doi: 10.7717/peerj.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gentle A, Ashton PM, Dallman TJ, Jenkins C. 2016. Evaluation of molecular methods for serotyping Shigella flexneri. J Clin Microbiol 54:1456–1461. doi: 10.1128/JCM.03386-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazen TH, Leonard SR, Lampel KA, Lacher DW, Maurelli AT, Rasko DA. 2016. Investigating the relatedness of enteroinvasive Escherichia coli to other E. coli and Shigella isolates by using comparative genomics. Infect Immun 84:2362–2371. doi: 10.1128/IAI.00350-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross RJ, Rowe B. 1985. Serotyping of Escherichia coli, p 345–360. In Sussman M. (ed), The virulence of Escherichia coli: reviews and methods. Special publication of the Society for General Microbiology no. 13. Academic Press, London, United Kingdom. [Google Scholar]
- 18.Tewolde R, Dallman T, Schaefer U, Sheppard CL, Ashton P, Pichon B, Ellington M, Swift C, Green J, Underwood A. 2016. MOST: a modified MLST typing tool based on short read sequencing. PeerJ 4:e2308. doi: 10.7717/peerj.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.