

ABSTRACT
Staphylococcus aureus is the leading cause of skin and skin structure infections (SSSI). The high frequency of recurring SSSI due to S. aureus, including methicillin-resistant S. aureus (MRSA) strains, despite high titers of specific antibodies and circulating T cells, implies that traditional adaptive immunity imparts incomplete protection. We hypothesized that innate immune memory contributes to the protective host defense against recurring MRSA infection. To test this hypothesis, SSSI was induced in wild-type and rag1−/− mice in the BALB/c and C57BL/6 backgrounds. Prior infection (priming) of wild-type and rag1−/− mice of either background afforded protection against repeat infection, as evidenced by reduced abscess severities and decreased CFU densities compared to those in naive controls. Interestingly, protection was greater on the previously infected flank than on the naive flank for wild-type and rag1−/− mice. For wild-type mice, protective efficacy corresponded to increased infiltration of neutrophils (polymorphonuclear leukocytes [PMN]), macrophages (MΦ), Langerin+ dendritic cells (LDC), and natural killer (NK) cells. Protection was associated with the induction of interleukin-17A (IL-17A), IL-22, and gamma interferon (IFN-γ) as well as the antimicrobial peptides CRAMP and mβD-3. Priming also protected rag1−/− mice against recurring SSSI, with increased MΦ and LDC infiltration and induction of IL-22, CRAMP, and mβD-3. These findings suggest that innate immune memory, mediated by specific cellular and molecular programs, likely contributes to the localized host defense in recurrent MRSA SSSI. These insights support the development of targeted immunotherapeutic strategies to address the challenge of MRSA infection.
KEYWORDS: Staphylococcus aureus, immunity, recurrent infection, skin infection
INTRODUCTION
Staphylococcus aureus is the most common cause of skin and skin structure infections (SSSI), including cellulitis, folliculitis, and furunculosis (1–4). Regardless of prior exposure or antibody status, patients with SSSI due to methicillin-resistant S. aureus (MRSA) exhibit 1-year recurrence rates as high as 27 to 45% (5–7), and these infections often require surgical debridement (8). Skin infection serves as a primary portal of entry for invasion and subsequent hematogenous dissemination. For example, SSSI is a frequent prelude to bacteremia, endocarditis, and osteomyelitis (9, 10). At present, S. aureus is the second most common bloodstream isolate in health care settings, and it is the leading cause of infective endocarditis in developed countries (11–14). MRSA strains now predominate in health care- and community-acquired staphylococcal infections (15). Despite reports of modest declines in rates of MRSA infection in some populations (10), as many as one-third of S. aureus bacteremia patients succumb to this infection, even with gold-standard therapy. As a result, infections due to MRSA represent a leading cause of infection-induced mortality in the United States, resulting in 11,000 to 18,650 deaths per year (16–18). Compounding the above concerns, the increasing use of antibiotics has accelerated the emergence of multidrug-resistant S. aureus strains (19–22). Consequently, resistance to even the most modern antistaphylococcal agents is rising at an alarming rate (23, 24). The public health impact of this trend is of urgent concern, especially given the 15 to 40% mortality rate associated with invasive MRSA infection (25, 26).
A key concern is the observed high rate of recurring S. aureus infection among otherwise healthy individuals who have no known immune deficiencies or risk factors (27–31). In addition, S. aureus is a principal cause of recurring infection in patients with specific immune dysfunctions in the innate and/or adaptive immune response (32). For example, patients with chronic granulomatous disease (CGD) are at increased risk of disseminated staphylococcal infection because their phagocytes have deficient trafficking and oxidative burst responses (33). In comparison, because they have a defective Th17 response, patients with Job's syndrome (hyper-IgE syndrome) and atopic dermatitis are at increased risk of SSSI, but typically less so for systemic S. aureus infection (34–36). Further, patients with acne inversa exhibit staphylococcal persistence in skin abscesses due to deficiencies in the cutaneous interleukin-22 (IL-22) response (37, 38). Thus, different aspects of the host immune system are required to defend cutaneous versus hematogenous compartments against S. aureus infection.
The relative contributions of innate and adaptive immune responses to the defense against recurrent SSSI and ensuing invasive S. aureus infections remain poorly understood. In humans, antibodies generated against many S. aureus antigens are prevalent and long-lasting (39). However, considerable discordance exists between the humoral response and protective immunity to S. aureus. For example, persistent carriers have significantly higher IgG levels for staphylococcal exotoxins than those of noncarriers (40, 41). However, epidemiological studies have demonstrated that persistent carriers are at increased risk for recurrent MRSA infections (42, 43), suggesting that antibodies directed against S. aureus virulence factors are not sufficient for protection (44). Consistent with this view, individuals with defects in humoral immunity are not necessarily at increased risk for S. aureus infections (45). Furthermore, the lack of efficacy of vaccines targeting the humoral response suggests that antibody may not be sufficient for protection against recurrent MRSA SSSI (reviewed in reference 46). Moreover, approximately 5.7% of circulating memory T cells in both carriers and noncarriers are reactive to S. aureus, suggesting that these cells are not sufficient to mediate immune protection against MRSA infection by themselves (47). Taken together, these data suggest that classical adaptive immunity alone is insufficient for optimal host defense against recurrent MRSA infections.
Based on these insights, it is apparent that innate immunity plays a role in protection against recurring S. aureus SSSI. The findings of Montgomery et al. (48) suggested that immune mechanisms relying on antibody and IL-17A are important for protection against recurring skin infection in a mouse model. We previously described the importance of adaptive immune responses, including IL-17A, IL-22, and gamma interferon (IFN-γ), in promoting innate immune effectors of the host defense against SSSI due to MRSA (49, 50). In this respect, we posited in the current studies that innate immune mechanisms are enhanced by prior exposure to S. aureus and contribute to protective efficacy in recurring infection. To test this hypothesis, we used our established mouse model of SSSI to compare immune responses during primary and recurrent S. aureus infections. The extent of infection and the associated immune responses were studied at the site of infection as well as at organs that are targets of hematogenous seeding. The impacts of innate versus adaptive immune mechanisms on protective efficacy were compared in distinct mouse backgrounds with neutral (BALB/c) and proinflammatory (C57BL/6) biases by using wild-type versus rag1−/− mice, the latter of which lack both T cell and B cell functions.
RESULTS
Infection model and natural history.
With our established mouse model of SSSI (49, 50), recurring infection caused by MRSA was studied in wild-type and rag1−/− mice in the BALB/c and C57BL/6 backgrounds. Following initial priming infection in the right flank with MRSA strain USA300, lesions were allowed to resolve fully over a 6-week period until the overlying skin appeared normal. Next, the same organism was inoculated subcutaneously into both the left and right flanks (at 6 weeks post-priming infection), and lesion development was monitored over 7 days. At the study endpoint, the CFU burdens in comparative tissues were quantified, immune cell profiles in secondary lymphoid organs were characterized, and molecular and cellular immune mediators at sites of infection were analyzed by immunohistochemistry. This strategy enabled quantitative and qualitative comparisons of immune responses relative to protective efficacy in primary versus recurring SSSI and in the presence versus absence of adaptive immunity.
Localized infection and immune response. (i) Innate immune memory contributes to host protection against cutaneous abscesses during recurrent MRSA SSSI.
Skin abscesses in naive and infection-primed mice were monitored for lesion severity at days 1, 3, 5, and 7 postinfection. Consistent with our previous reports (49, 50), skin lesions developed rapidly and reached a maximal size at ∼4 days in naive wild-type and rag1−/− mice in the BALB/c and C57BL/6 backgrounds (Fig. 1A to D). Interestingly, lesions were not significantly more severe in rag1−/− mice than in wild-type mice, indicating that classical adaptive responses were not required for clearance of the primary infection. Importantly, priming of the immune system by primary infection resulted in smaller abscesses at all time points and significantly reduced the MRSA CFU density at day 7 postinfection for both wild-type and rag1−/− mice compared to that for the respective naive mice (Fig. 1E to L). However, priming of wild-type mice resulted in a greater decrease in MRSA CFU in skin abscesses than that seen in primed rag1−/− mice (see Fig. S1 in the supplemental material). These findings support the concept that innate and adaptive immune memory responses synergize in defense against recurring SSSI due to MRSA. Interestingly, on comparing the naive and reinfected flanks of individual mice, a highly localized protective response was observed for wild-type C57BL/6 mice and BALB/c and C57BL/6 rag1−/− mice. These mice had smaller lesions on the previously infected (right) flank than on the naive (left) flank (Fig. 1M and N). For example, for wild-type C57BL/6 mice, but not BALB/c mice, smaller lesions were observed on the primed flank than on the naive flank (P = 0.049) (Fig. 1M). For the rag1−/− mice, the same finding was observed for BALB/c mice (P = 0.049), and there was a trend toward smaller lesions on the primed flank for C57BL/6 mice (P = 0.07) (Fig. 1N). Histochemical analysis of the infected tissues demonstrated that S. aureus macroabscesses formed in the subcutaneous tissues of naive mice and that these abscesses were smaller in the primed BALB/c (Fig. 2A, panels i to x) and C57BL/6 (Fig. 2B, panels i to x) mice.
FIG 1.

Priming provides protection from dermonecrosis severity and MRSA burden in abscesses of BALB/c and C57BL/6 mice. Mice were preinfected (primed) with USA300 in the right flank, followed by resolution of infection over 6 weeks. Both naive and primed groups then received USA300 in both flanks, and the abscesses were monitored for 7 days. The mean dermonecrosis area (square millimeters) was measured at selected time points over 7 days postinfection. Visualization of skin abscesses on days 3, 5, and 7 in wild-type (WT) (A and C) and rag1−/− (B and D) mice showed smaller lesions in primed mice than in naive mice for both the BALB/c (A and B) and C57BL/6 (C and D) backgrounds. The dermonecrosis area was significantly reduced in primed mice compared to naive infected mice for both wild-type (E and I) and rag1−/− (G and K) mice. The number of MRSA CFU was significantly reduced in both wild-type (F and J) and rag1−/− (H and L) primed mice compared to naive mice. Left and right abscess areas were compared at day 5 postinfection (day of greatest abscess severity) for both wild-type (M) and rag1−/− (N) mice of both backgrounds. The lower limit of detection for bacterial densities was approximately 20 CFU/abscess. The statistical significance of differences was determined by comparison to naive infected mice (for wild-type mice, n ≥ 16; for rag1−/− mice, n ≥ 8), using Student's t test and one-way ANOVA (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Data presented are means ± SD.
FIG 2.
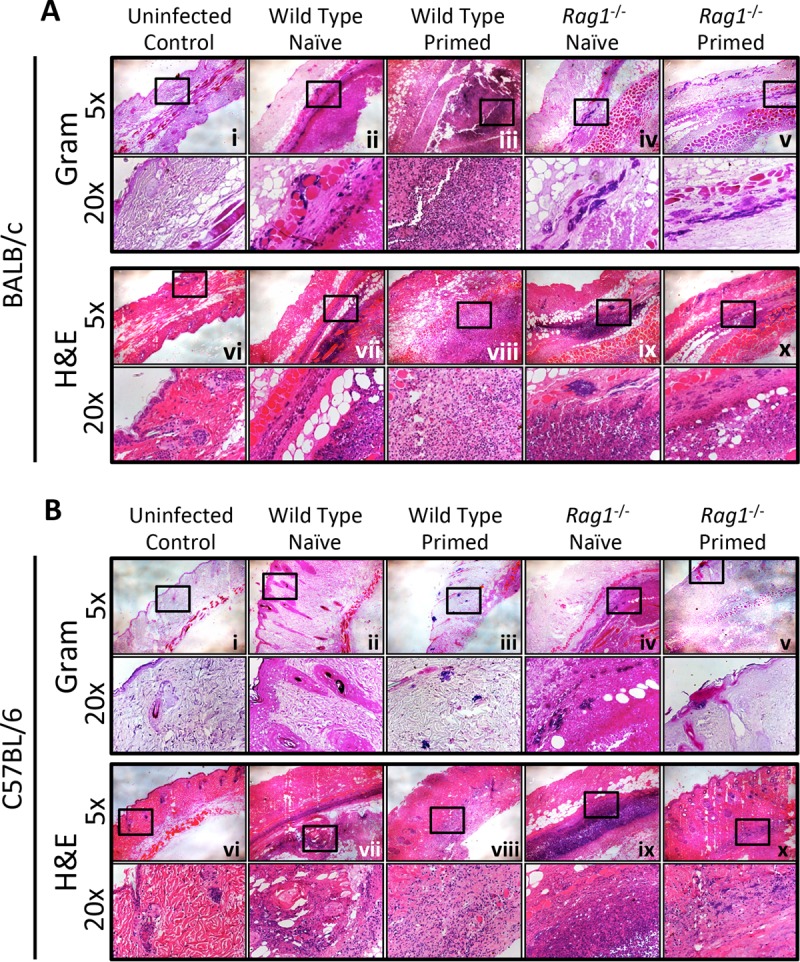
Histopathology profiles of skin lesions were used to show macroabscesses and infiltrating immune cells. Tissue staining of the epidermal surface (top) and the deeper tissue parenchyma is shown for uninfected control and infected mice for both the BALB/c (A) and C57BL/6 (B) backgrounds. Gram staining (i to v) and hematoxylin and eosin (H&E) staining (vi to x) show the locations of macroabscesses and bacteria (S. aureus cells are seen as cocci stained violet in Gram-stained sections).
(ii) Immune cell subsets in draining inguinal lymph nodes are influenced by MRSA infection.
At day 7 postinfection, the lymphocyte populations from draining inguinal lymph nodes of wild-type and rag1−/− mice were characterized by flow cytometry. In wild-type naive and primed BALB/c mice, MRSA skin infection resulted in equivalent increases in T and B lymphocyte populations (Fig. 3A). In these mice, there was a similar increase in CD4+ T lymphocyte populations, but no increase in CD8+ T lymphocytes, compared to those in the uninfected control mice (Fig. 3B). Infection did not result in a significant increase in dendritic cells, macrophages (MΦ), or NK cells (Fig. 3C). As expected, for rag1−/− mice, the populations of detectable T and B cells in lymph nodes were negligible, and no increase was found in either naive or primed groups compared to uninfected controls (Fig. 3D). Due to the low frequency of T lymphocytes detected, CD4+ and CD8+ subset populations could not be analyzed (data not shown). Interestingly, for naive and primed rag1−/− mice, significantly fewer MΦ and NK cells were detected in the lymph nodes than those for uninfected controls in the BALB/c background (Fig. 3E). These results indicate that the protection afforded through priming of BALB/c mice was not due to changes in lymphoid cell populations.
FIG 3.

Priming does not influence cell population changes in the draining inguinal lymph nodes. On day 7 postinfection, the draining inguinal lymph nodes were analyzed for cell population changes. Draining inguinal lymph nodes of BALB/c wild-type (A to C) and rag1−/− (D and E) mice as well as C57BL/6 wild-type (F to H) and rag1−/− (I and J) mice were analyzed for adaptive (A, D, F, and I) and innate (C, E, H, and J) immune cell profiles. Specific T cell subset profiles for wild-type mice were also assessed (B and G). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (versus uninfected mice); +, P < 0.05; ++, P < 0.01 (versus naive mice) (Student's t test; for wild-type mice, n ≥ 16; for rag1−/− mice, n ≥ 8). Data presented are means and SD. ns, not significant.
Results for lymph node populations in the C57BL/6 wild-type background were similar to those for the BALB/c mice. Priming did not result in increased T cell populations in the draining inguinal lymph nodes of wild-type C57BL/6 mice. Interestingly, priming actually led to a slight decrease in B cells compared to the levels in naive mice (Fig. 3F and G). No differences in dendritic cell, MΦ, or NK cell populations were found in naive versus primed infections (Fig. 3H). For C57BL/6 rag1−/− mice, T and B cells were barely detectable, similar to those of BALB/c rag1−/− mice (Fig. 3I). Interestingly, priming resulted in increased NK cell percentages in the lymph nodes compared to those for naive mice, but there was no change in dendritic cells and MΦ (Fig. 3J). Taken together, these results suggest that NK cells in the lymph nodes may play a role in protection during recurrent MRSA SSSI in the C57BL/6 background.
(iii) Priming increases infiltration of innate effector cells into skin abscesses in recurrent MRSA SSSI.
To determine changes in infiltrating cell populations at the site of infection, immunohistochemical analysis and quantification were performed on skin sections as detailed in Materials and Methods. Skin infection of primed wild-type BALB/c mice induced significantly increased polymorphonuclear leukocyte (PMN), MΦ, Langerin+ dendritic cell (LDC), and NK cell infiltration at the abscess site compared to that in naive mice (Fig. 4A and B). However, in BALB/c rag1−/− mice, priming resulted in increased infiltration of MΦ and LDC, but not PMN or NK cells, compared to that in naive mice (Fig. 4C). Importantly, however, priming resulted in a substantially greater intensification of PMN targeting to the immediate proximity of S. aureus abscesses in rag1−/− mice than in naive controls (Fig. 4A, panels iv and v).
FIG 4.

Increased immune cells infiltrate into skin abscesses of primed and naive wild-type and rag1−/− mice. At day 7 postinfection, abscesses were dissected, fixed in zinc formalin, and embedded in paraffin. Skin sections from BALB/c (A) and C57BL/6 (D) mice were stained with antibodies against Ly6G for neutrophils (PMN) (i to v), F4/80 for macrophages (MΦ) (vi to x), CD207 for Langerin+ dendritic cells (LDC) (xi to xv), and CD49b for NK cells (NK) (xvi to xx). ImageJ software was used to quantify immune cell infiltration at abscess sites of wild-type (B and E) and rag1−/− (C and F) mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (versus naive mice via Student's t test; n ≥ 10 images per sample). The infiltration index was calculated by determining the expression levels of samples (% of uninfected control levels), normalized to 106 CFU MRSA in abscesses of corresponding samples as previously described (53). Data presented are mean infiltration indices and SD.
For C57BL/6 mice, priming resulted in outcomes similar to those for BALB/c mice. MRSA skin infection in primed wild-type C57BL/6 mice led to significantly increased PMN, MΦ, LDC, and NK cell infiltration proximate to macroabscesses compared to that in naive mice (Fig. 4D and E). In the rag1−/− C57BL/6 background, priming resulted in significantly increased PMN, MΦ, and LDC infiltration versus that in naive mice (Fig. 4D and F). These results suggest that MΦ and LDC may play a key role in protection against reinfection independently of adaptive immunity. Also, in wild-type mice, NK cells may coordinate with T and/or B cells to provide protection from recurrent MRSA SSSI. Further, PMN also likely play a role in the enhanced host defense of primed mice.
(iv) Priming influences IL-17A, IL-22, and IFN-γ expression in MRSA skin abscesses.
At the experimental endpoint, cytokines involved in protection against MRSA SSSI were measured in proximity to the abscesses (49, 50). In the BALB/c background, priming of wild-type mice resulted in significantly increased IL-17A, IL-22, and IFN-γ levels in skin abscesses compared to those of naive mice (Fig. 5A and B). It is of great interest that priming in rag1−/− mice resulted in increased IL-22 levels in skin abscesses compared to those of naive mice during MRSA skin infection (Fig. 5A and C). No significant differences in IL-17A or IFN-γ levels were observed in primed versus naive mice in the rag1−/− background.
FIG 5.

Priming induces greater cytokine expression in skin proximal to macroabscesses in primed wild-type and rag1−/− mice. Paraffin-embedded skin sections from BALB/c (A) and C57BL/6 (D) mice were immunohistochemically stained with antibodies targeting IL-17A (i to v), IL-22 (vi to x), and IFN-γ (xi to xv). Quantification for immunohistochemically stained skin sections compared the expression of cytokines in the wild-type (B and E) and rag1−/− (C and F) backgrounds. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (versus naive infected mice via Student's t test; n ≥ 10 images per sample). The cytokine index was calculated by determining the expression levels of samples (% of uninfected control levels), normalized to 106 CFU MRSA in abscesses of corresponding samples as previously described (53). Data presented are mean cytokine indices and SD.
Cytokine responses of C57BL/6 mice were similar to those of BALB/c mice. Priming in wild-type mice resulted in significantly increased IL-17A, IL-22, and IFN-γ levels compared to those of naive mice (Fig. 5D and E). In rag1−/− mice, skin abscesses had significantly increased IL-22 or IFN-γ levels compared to those in naive mice (Fig. 5D and F). Taken together, these data suggest that IL-17A, IL-22, and IFN-γ play significant roles in the enhanced protection against SSSI in wild-type mice. In contrast, in rag1−/− mice lacking adaptive immunity, IL-22 and IFN-γ appear to mediate protective efficacy during reinfection.
(v) Priming induces local HDP elaboration.
On day 7 postinfection, host defense peptides (HDPs) with efficacy against MRSA were measured in skin abscesses (51). In both the BALB/c and C57BL/6 backgrounds, priming of wild-type mice resulted in significantly increased mβD-3 and CRAMP expression compared to that in naive mice during MRSA SSSI (Fig. 6). Similarly, in the rag1−/− background, priming also increased mβD-3 and CRAMP levels compared to those of naive mice, but to a lesser extent than that for wild-type mice. These results suggest that cutaneous HDPs contribute to protection during recurrent MRSA SSSI. Importantly, this response can occur in the absence of adaptive immunity, although it is enhanced in the context of functional T/B lymphocytes.
FIG 6.

Priming induces greater antimicrobial peptide expression in skin abscesses of primed wild-type and rag1−/− mice. Skin sections from BALB/c (A) and C57BL/6 (D) mice were stained with antibodies against CRAMP (i to v) and mβD-3 (vi to x). ImageJ quantification showed antimicrobial peptide expression at abscess sites of wild-type (B and E) and rag1−/− (C and F) mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (versus naive infected mice via Student's t test; n ≥ 10 images per sample). The host defense peptide (HDP) index was calculated by determining the expression levels of samples (% of uninfected control levels), normalized to 106 CFU MRSA in abscesses of corresponding samples as previously described (53). Data presented are mean antimicrobial peptide indices and SD.
Disseminated infection and splenic response. (i) Priming is minimally protective against hematogenously disseminated MRSA infection.
After 7 days of SSSI due to MRSA, quantitative culture of kidney, liver, and spleen homogenates indicated that hematogenous dissemination had occurred, albeit at a low level (Fig. 7). In wild-type BALB/c mice, the bacterial burdens of the kidneys, liver, and spleen in primed and naive mice were similar, indicating that priming did not have an impact on disseminated infection (Fig. 7A to C). Likewise, priming did not protect rag1−/− mice from disseminated infection. Surprisingly, priming appeared to increase the S. aureus burden in the kidneys, but not the liver and spleen, compared to that for naive infection in this background (Fig. 7F).
FIG 7.

Comparative impacts of priming on hematogenous dissemination in wild-type or rag1−/− mice during MRSA SSSI. On day 7 postinfection, CFU densities in kidneys, livers, and spleens from BALB/c wild-type (A to C) and rag1−/− (D to F) mice as well as C57BL/6 wild-type (G to I) and rag1−/− (J to L) mice were assessed for disseminated infection. *, P < 0.05 versus naive mice (for wild-type kidneys, n ≥ 16; for rag1−/− kidneys, n ≥ 8; for wild-type spleens and livers, n ≥ 8; for rag1−/− spleens and livers, n ≥ 4). Data presented are means and SD.
Different results were observed for C57BL/6 mice. In wild-type mice, priming provided protection from disseminated infection of the spleen (P = 0.029) but not the kidney or liver (Fig. 7G to I). In contrast, priming of rag1−/− mice did not protect from disseminated infection in any organ (Fig. 7J to L). Therefore, priming afforded protection against hematogenous infection in only the spleens of C57BL/6 mice.
(ii) Splenic immune cell profiles do not differ in primary versus recurrent MRSA SSSI.
To further characterize the immune response to hematogenous infection, immune cell populations from the spleen were analyzed by flow cytometry. In wild-type BALB/c mice, SSSI resulted in splenic T cell expansion, especially that of CD4+ T cells, in both naive and primed mice versus uninfected controls (Fig. 8A and B). No significant differences in dendritic cell, MΦ, or NK cell populations were observed (Fig. 8C). As expected, no expansion of T or B lymphocytes was observed in spleens of naive or primed rag1−/− mice (Fig. 8D). Similar to the results for the lymph nodes, MΦ abundance in the spleen was decreased to equivalent degrees in naive and primed rag1−/− mice compared to that in uninfected controls. Also similar to the findings observed for the lymph nodes, no differences in cell populations were observed in primed versus naive rag1−/− BALB/c mice (Fig. 8D and E).
FIG 8.

Priming does not influence cell population changes in the spleen. On day 7 postinfection, mouse spleens were analyzed for cell population changes. Spleens of BALB/c wild-type (A to C) and rag1−/− (D and E) mice as well as C57BL/6 wild-type (F to H) and rag1−/− (I and J) mice were assessed for adaptive (A, D, F, and I) and innate (C, E, H, and J) immune cell profiles. (B and G) Specific T cell subset profiles for wild-type mice were also assessed. **, P < 0.01; ***, P < 0.001 (versus uninfected mice); +++, P < 0.001 (versus naive mice) (Student's t test; for wild-type mice, n ≥ 8; for rag1−/− mice, n ≥ 4). Data presented are means and SD.
In C57BL/6 mice, changes in splenic cell populations were similar to those observed in the draining lymph nodes. For wild-type mice, equivalent expansions of CD4+ T cell populations were observed in the spleens of both naive and primed mice compared to those of uninfected controls. However, no changes in cell populations were observed in primed versus naive C57BL/6 wild-type mice (Fig. 8F to H). As expected, for rag1−/− mice, no expansion of T and B lymphocyte populations was observed during MRSA infection (Fig. 8I). Also, priming of rag1−/− mice resulted in increased NK cell populations in the spleen compared to those of naive mice. Although MΦ infiltration was decreased in the spleens of both naive and primed mice compared to those of uninfected controls, no differences in dendritic cell or MΦ populations were detected in primed versus naive rag1−/− mice (Fig. 8J). Collectively, these data suggest that although SSSI induces a systemic T and B cell response, this response does not protect mice against hematogenous seeding of the target organs.
Impact of mouse strain background on immune memory.
To compare the specific contributions of priming to immune memory in distinct mouse strains, differences in immune responses relative to bacterial burdens were analyzed in primed and naive BALB/c versus C57BL/6 mice. Two interesting outcomes were observed in these studies. First, priming of BALB/c mice showed a trend toward greater reductions in MRSA CFU upon recurrent infection than those with priming of C57BL/6 mice for both the wild-type and rag1−/− genotypes (Fig. 9A and B). This reduction was associated with significantly greater increases in immune cell infiltration, cytokine responses, and HDP induction (Fig. 9C, E, and G) during primed infection of wild-type BALB/c mice than those for C57BL/6 mice. With the rag1−/− genotype, primed BALB/c mice showed a trend toward greater increases in infiltrating MΦ, LDC, and NK cells, as well as the induction of IL-17A, IL-22, and CRAMP, than those for primed C57BL/6 mice (Fig. 9D, F, and H).
FIG 9.

Priming in different mouse genetic backgrounds yields differential proinflammatory responses. Differences in MRSA burdens and proinflammatory responses in the skin are shown for naive versus primed mice. Differences in mean log10 CFU between primed and naive mice were calculated for the wild-type (A) and rag1−/− (B) genotypes for both the BALB/c and C57BL/6 mouse backgrounds. The changes in indices of immune cell infiltration (C and D), cytokine expression (E and F), and host defense peptide (HDP) induction (G and H) between naive and primed mice were calculated for the wild-type (C, E, and G) and rag1−/− (D, F, and H) genotypes for both the BALB/c and C57BL/6 backgrounds. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (versus C57BL/6 mice via Student's t test). Data presented are means and SD for index differences between the naive and primed groups.
Second, different molecular programs appear to predominate in innate memory against recurring SSSI in distinct mouse strains. For example, priming of C57BL/6 rag1−/− mice resulted in a greater induction of IFN-γ than that seen with priming of BALB/c mice (Fig. 9F). While they had different magnitudes and effector profiles, the observed immune responses in the rag1−/− background for either mouse strain indicate that the outcomes were not entirely dependent on adaptive memory.
DISCUSSION
The role of immune memory in protection against primary or recurrent S. aureus infection has not been fully understood. The present study assessed four key aspects of protective immunity to recurrent MRSA infection in an SSSI mouse model (49, 50): (i) the impact of immune memory, (ii) the contributions of innate versus adaptive mechanisms to immune memory, (iii) cutaneous versus hematogenous targeting of protective immune memory in relation to adaptive versus innate mechanisms, and (iv) innate memory responses relative to immune-skewing paradigms in distinct mouse strains. Unique to this study was a comparison of immune mechanisms during recurrent infection in wild-type versus rag1−/− mice. The current data indicate that primary SSSI due to MRSA primes the immune system and significantly attenuates the severity of recurrent infection. This efficacy is manifested by a reduced abscess MRSA burden and dermonecrosis area. Importantly, protective immune memory was observed in rag1−/− mice. Because the rag1−/− background is null for recombinase activating gene 1, required for the generation of mature T/B lymphocytes, our findings indicate that innate immune memory is integral to the host defense against recurrent SSSI due to MRSA.
In wild-type mice, innate immune effector cells were intensified at the site of infection in the skin and are likely contributors of protective host memory. For example, priming caused greater PMN, MΦ, LDC, and NK cell infiltration in the skin of wild-type BALB/c and C57BL/6 mice. Importantly, priming of rag1−/− BALB/c and C57BL/6 mice also led to increased MΦ and LDC, but not NK cell, accumulation in skin abscesses. Moreover, cell populations in the draining inguinal lymph nodes were not different between naive and primed mice, suggesting that innate memory targets sites of infection. This pattern of results suggests that MΦ and LDC play important roles in protective innate memory at sites of SSSI due to MRSA.
These findings support our hypothesis that protective memory in skin involves specific innate effector cells and requires durable changes in innate immune signaling. Such concepts are consistent with recent studies of innate memory (51–56). For example, dermal MΦ are known to play key roles in innate memory (54). These cells enhance recruitment of PMN in response to staphylococci, and abscess resolution relies on CCR2-mediated monocyte/MΦ infiltration (55, 56). In our studies, PMN migrated to MRSA abscesses in primed rag1−/− mice (Fig. 3A, panels iv and v). Thus, MΦ can respond to MRSA infection in the absence of adaptive immunity and likely potentiate the recruitment and/or functions of PMN, thereby enhancing antistaphylococcal efficacy. Together, these data suggest that MΦ and PMN cooperate in innate memory targeting MRSA SSSI. The present results also indicate that LDC are integral to innate memory in the defense against MRSA. These cells accumulated in skin abscesses of primed rag1−/− mice, demonstrating that their recruitment is independent of adaptive immunity. Although dendritic cells typically have poor direct microbicidal activity (57), the current data suggest that LDC contribute to protective immunity via innate mechanisms. For example, LDC activated by bacterial antigens can produce proinflammatory cytokines that enhance effector functions of phagocytes (e.g., MΦ and PMN) to potentiate clearance of S. aureus (58–60).
Our studies also point to a role for molecular effectors in innate memory against SSSI due to MRSA. We analyzed the responses of cytokines and HDPs known to be protective against S. aureus skin infection (49–51). As anticipated, priming of wild-type BALB/c and C57BL/6 mice induced greater IL-17A, IL-22, and IFN-γ expression as well as CRAMP and mβD-3 elaboration. Priming of rag1−/− mice of both the BALB/c and C57/BL6 backgrounds led to increased expression of IL-22 but not IL-17A. Furthermore, unique to the C57BL/6 background, priming of rag1−/− mice also induced greater IFN-γ expression in skin. Thus, our current results imply that IL-22 and IFN-γ are more associated with innate immune memory in this model, whereas IL-17A expression is T cell dependent. These cytokine responses correlated with increased expression of the HDPs CRAMP and mβD-3 in abscesses. However, HDP induction in rag1−/− mice was much lower than that in wild-type backgrounds, suggesting that adaptive immunity also contributes to their expression. Collectively, this pattern of results suggests that T cell-independent pathways of IL-22 and IFN-γ production can induce protective HDPs, consistent with the results of prior studies (51). For example, keratinocytes, epithelial cells, adipocytes, and other cutaneous tissues produce HDPs directly in response to infectious stimuli (51, 61, 62). In support of this concept, our prior studies demonstrated that induction of HDPs in relation to IL-17A and IL-22 in MRSA SSSI contributes to the host defense in primary and vaccine-induced immunity against MRSA SSSI (50). These findings are concordant with known roles for HDPs in protection against cutaneous S. aureus infection (61, 63, 64).
Another important finding was that of differential immune memory responses in mouse strains known to have distinct immune-skewing paradigms. In this respect, the observed differences in efficacy and immune responses due to priming of rag1−/− and wild-type BALB/c versus C57BL/6 mice provided two important new insights. First, innate memory significantly contributes to protective efficacy in both mouse strains. This observation is supported by the fact that certain immune determinants associated with protective efficacy were identified in both BALB/c and C57BL/6 rag1−/− mice. Second, the effectors of such innate memory appear to differ in magnitude and profile in BALB/c versus C57BL/6 mice. For example, while responses were seen in both backgrounds, MΦ and LDC predominated as cellular effectors of innate memory, and IL-17A, IL-22, and CRAMP predominated as molecular effectors, in rag1−/− BALB/c mice. In comparison, IFN-γ responses were of a higher magnitude than those in BALB/c mice during recurrent infection (Fig. 9), and the cellular response was of a lesser magnitude, in C57BL/6 rag1−/− mice. Notably, for primary infection, C57BL/6 mice had significantly lower MRSA burdens in skin than those of BALB/c mice, corresponding to a proinflammatory bias in this strain (see Fig. S2 in the supplemental material). While Th1 versus Th2 immune skewing during primary infection is recognized in these mouse strains (65), the current data provide new insights into programs of innate immunity during recurrent S. aureus infection.
Finally, two outcomes in the current model support the concept of localized targeting in protective immune memory. First, priming of wild-type or rag1−/− mice provided minimal protection from disseminated infection. Second, priming yielded greater protection in the previously infected flank than in the contralateral naive flank of the same mouse in either mouse background. This observation further suggests that immune memory in skin is spatially defined. These results support the concept that mechanisms of innate memory are functional in the cutaneous barrier and preferentially target sites that have previously been infected by the challenge organism.
In summary, the understanding and application of protective immune mechanisms hold great promise for improved prevention and treatment of infections caused by S. aureus and other pathogens. Immune memory has traditionally been attributable exclusively to adaptive immunity as mediated by memory subsets of T/B lymphocytes. Innate memory (“trained immunity”) is a relatively new concept focused on mechanisms of host memory distinct from traditional adaptive immunity. In our current studies, we observed the following characteristics of trained immunity in response to recurrent S. aureus SSSI, as reviewed in reference 66: (i) protective immune memory independent of T/B cell responses, (ii) localized protection, and (iii) molecular and cellular effector contributions to protective efficacy. Although beyond the scope of this paper, ongoing studies in our laboratory aim to specify these determinants of innate immune memory and how they may function in consort with classical adaptive immunity. For example, specific immune effector cells implicated as being integral to innate memory in the current studies might be examined using innovative adoptive transfer studies in genetically defined mouse strains. In addition, ongoing studies are examining the determinants of protective immunity in response to other S. aureus strains and microbial pathogens to assess the degree to which the current findings may be generalizable.
MATERIALS AND METHODS
MRSA strain.
This study utilized MRSA strain LAC, a USA300 strain isolated from an outbreak at the Los Angeles County Jail. USA300 strains represent the most prevalent community-associated MRSA etiologies of SSSI and account for up to 80% of MRSA infections (4, 67, 68). The phenotypic and genotypic profiles of this strain have previously been characterized thoroughly (69). Bacteria were cultured from virulence-validated master cell banks and grown to log phase in brain heart infusion (BHI) medium at 37°C. Resulting cells were harvested, washed, suspended in phosphate-buffered saline (PBS), sonicated, quantified by spectrophotometry, and diluted to the desired CFU inoculum in PBS.
Mouse model of recurrent SSSI.
Animal studies were performed in accordance with approved animal use policies of the Los Angeles Biomedical Research Institute at Harbor-UCLA following NIH guidelines. Male wild-type and rag1−/− [C.129S7(B6)-Rag1tm1Mom/J; B6.129S7-Rag1tm1Mom/J] BALB/c and C57BL/6 mice (20 to 25 g; Jackson Laboratory, Sacramento, CA) were studied using a subcutaneous SSSI model as we previously detailed (49, 50). Despite the presence of CD3+ precursors, rag1−/− mice do not produce functionally mature T or B cells (70, 71). In brief, mice were randomized and anesthetized, and their flanks were shaved and disinfected with 70% ethanol. For primary infection, SSSI was established via inoculation with 1 × 107 CFU MRSA in 100 μl PBS by subcutaneous injection into the right flank. Control animals received an identical volume of PBS alone. In infected animals, abscesses formed along with corresponding dermonecrosis, as anticipated, and they were permitted to resolve completely over 5 weeks. To study immune responses in recurring SSSI, the same previously infected (primed) mice were then reinfected in both the right and left flanks, using an identical regimen. To minimize any chance of residual organisms at previously infected sites, lesions from primary infection were allowed to resolve fully for 6 weeks prior to subsequent reinfection. At that point, lesions had complete skin healing and fur regrowth. This recovery period from priming infection compared favorably to or extended beyond those used in prior studies of recurring S. aureus skin infection (48, 72). Additionally, our prior studies established the loss of detectable MRSA bioluminescence by 14 days postinfection in the same model as that used for the present study (unpublished data). Lesions were then studied over the ensuing 7 days postinfection, at which time the mice were humanely euthanized for tissue analysis.
Skin lesion severity.
Dermonecrosis areas of each flank were measured for every mouse over the postchallenge study periods. Mice were anesthetized and the dermonecrosis area (in square millimeters, determined using the lesion length [l] and width [w]) quantified for each lesion as previously reported (49, 50).
MRSA tissue burden.
At the 7-day study endpoint, mice were humanely euthanized and their tissues processed for quantitative culture. Both flanks were aseptically dissected, and abscesses were removed and separated into two equivalent sections. One-half of each section was homogenized in 1 ml sterile PBS and quantitatively cultured (the other half was reserved for immunohistochemistry as detailed below). Resultant colonies were enumerated, and the data were expressed as the number of CFU per abscess. To assess hematogenous dissemination, the kidneys and spleen from each mouse were processed in a manner similar to that for quantitative culture. Likewise, a representative lateral lobe from the liver of each mouse was dissected, weighed, and processed for quantitative culture. All cultures were incubated at 37°C for 24 h on sheep blood agar (Hardy Diagnostics, Santa Maria, CA), and resulting colonies were enumerated as log10 CFU per abscess, kidney, or spleen or log10 CFU per milligram of liver. The lower limit of detection for quantitative cultures was considered to be 20 CFU per sample.
Immune cell subset profiles.
At time of sacrifice, the inguinal lymph nodes and one-half of the spleen of each mouse were aseptically dissected and placed into 5 ml RPMI with 5% fetal bovine serum (FBS). The tissue preparations were mechanically dissociated through a 70-μm cell strainer to isolate cells. The resulting cell suspension was treated with red blood cell lysis buffer (Thermo Fisher, Grand Island, NY), washed, and counted. One hundred thousand (1 × 105) cells per tissue sample were stained with fluorochrome-conjugated antibodies directed against CD3 (pan-T lymphocytes), CD4 (CD4+ T lymphocytes), CD8 (CD8+ T lymphocytes), Nk1.1 (natural killer [NK] cells), CD45 (lymphocyte common antigen), CD11c (dendritic cells), F4/80 (macrophages), major histocompatibility complex class II (MHCII) (antigen-presenting cell activation marker), or CD19 (B lymphocytes). Specific antibodies used in the immunophenotyping experiments are detailed in Table S1 in the supplemental material. Cell subsets were analyzed by multicolor flow cytometric analysis, and results are presented as percentages of positively stained cells relative to the total cell population.
Immunohistochemistry.
Immunological determinants associated with the host defense against MRSA were assessed in skin abscesses at the study endpoint as we previously described (49, 50, 52). In brief, samples were aseptically dissected from animals, fixed in 10% zinc formalin, and embedded in paraffin. Next, 3-μm sections were cut, dewaxed, and rehydrated, followed by heat-induced antigen retrieval (Dako, Carpinteria, CA). Samples were blocked with dual endogenous enzyme block (Dako) and 2 to 10% normal serum corresponding to the secondary antibody. Sections were then incubated with a primary antibody targeting a specific molecule or cell of interest, followed by staining with a secondary antibody conjugated to horseradish peroxidase or biotin (Santa Cruz Biotechnology, Santa Cruz, CA). Colorimetric development was achieved by reaction with streptavidin-horseradish peroxidase (Dako) and 3,3′-diaminobenzidine (DAB; Vector Laboratories, Burlingame, CA), and the samples were counterstained with hematoxylin. Primary antibodies targeting immune cells, cytokines, and antimicrobial peptides used for immunohistochemistry are detailed in Table S1 in the supplemental material. Images were visualized using a Zeiss BX43 microscope with a DP21 digital camera and quantitatively analyzed using ImageJ software (http://imagej.nih.gov/ij/) as previously described (50). Outcomes were enumerated as an integral of staining in a given tissue sample compared to that for its respective comparators (control or primed group). A minimum of 10 (range, 10 to 30) images with identical total areas were quantified for each study group; areas containing no tissue were subtracted as background to afford internal control. Cellular infiltration, cytokine, and host defense peptide expression indices were calculated by comparison to uninfected control values, normalized to 106 CFU MRSA in abscesses of corresponding samples (53). Data are presented as mean infiltration indices ± standard deviations (SD).
Statistical analyses.
Differences in experimental results were compared by Student's t test and analysis of variance (ANOVA), where appropriate. Data are presented as means ± standard deviations. P values of <0.05 were considered statistically significant. Graphs were generated using GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported in part by grants U01 AI-124319-01, R21 AI-111661-02, and R33 AI-111661-01 (to M.R.Y.) from the U.S. National Institutes of Health and by grant 16POST31160035 (to L.C.C.) from the American Heart Association.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00876-16.
REFERENCES
- 1.Dryden MS. 2010. Complicated skin and soft tissue infection. J Antimicrob Chemother 65(Suppl 3):S35–S44. [DOI] [PubMed] [Google Scholar]
- 2.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng AG, DeDent AC, Schneewind O, Missiakas D. 2011. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol 19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shastry L, Rahimian J, Lascher S. 2007. Community-associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections in men who have sex with men in New York City. Arch Intern Med 167:854–857. doi: 10.1001/archinte.167.8.854. [DOI] [PubMed] [Google Scholar]
- 6.Skiest D, Brown K, Hester J, Moore T, Crosby C, Mussa HR, Hoffman-Roberts H, Cooper T. 2006. Community-onset methicillin-resistant Staphylococcus aureus in an urban HIV clinic. HIV Med 7:361–368. doi: 10.1111/j.1468-1293.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- 7.Anderson EJ, Hawkins C, Bolon MK, Palella FJ Jr. 2006. A series of skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus in HIV-infected patients. J Acquir Immune Defic Syndr 41:125–127. doi: 10.1097/01.qai.0000192004.08153.ea. [DOI] [PubMed] [Google Scholar]
- 8.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. 2004. IL-22 increases the innate immunity of tissues. Immunity 21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Tattevin P, Schwartz BS, Graber CJ, Volinski J, Bhukhen A, Bhukhen A, Mai TT, Vo NH, Dang DN, Phan TH, Basuino L, Perdreau-Remington F, Chambers HF, Diep BA. 2012. Concurrent epidemics of skin and soft tissue infection and bloodstream infection due to community-associated methicillin-resistant Staphylococcus aureus. Clin Infect Dis 55:781–788. doi: 10.1093/cid/cis527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landrum ML, Neumann C, Cook C, Chukwuma U, Ellis MW, Hospenthal DR, Murray CK. 2012. Epidemiology of Staphylococcus aureus blood and skin and soft tissue infections in the US military health system, 2005–2010. JAMA 308:50–59. doi: 10.1001/jama.2012.7139. [DOI] [PubMed] [Google Scholar]
- 11.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 12.Fowler VG Jr, Miro JM, Hoen B, Cabell CH, Abrutyn E, Rubinstein E, Corey GR, Spelman D, Bradley SF, Barsic B, Pappas PA, Anstrom KJ, Wray D, Fortes CQ, Anguera I, Athan E, Jones P, van der Meer JT, Elliott TS, Levine DP, Bayer AS. 2005. Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA 293:3012–3021. doi: 10.1001/jama.293.24.3012. [DOI] [PubMed] [Google Scholar]
- 13.Hoen B, Duval X. 2013. Clinical practice. Infective endocarditis. N Engl J Med 368:1425–1433. doi: 10.1056/NEJMcp1206782. [DOI] [PubMed] [Google Scholar]
- 14.Leroy O, Georges H, Devos P, Bitton S, De Sa N, Dedrie C, Beague S, Ducq P, Boulle-Geronimi C, Thellier D, Saulnier F, Preau S. 2015. Infective endocarditis requiring ICU admission: epidemiology and prognosis. Ann Intensive Care 5:45. doi: 10.1186/s13613-015-0091-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 17.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, Fridkin S. 2013. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med 173:1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. 2012. Predictors of mortality in Staphylococcus aureus bacteremia. Clin Microbiol Rev 25:362–386. doi: 10.1128/CMR.05022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amin AN, Cerceo EA, Deitelzweig SB, Pile JC, Rosenberg DJ, Sherman BM. 2014. Hospitalist perspective on the treatment of skin and soft tissue infections. Mayo Clin Proc 89:1436–1451. doi: 10.1016/j.mayocp.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Hanaki H, Cui L, Ikeda-Dantsuji Y, Nakae T, Honda J, Yanagihara K, Takesue Y, Matsumoto T, Sunakawa K, Kaku M, Tomono K, Fukuchi K, Kusachi S, Mikamo H, Takata T, Otsuka Y, Nagura O, Fujitani S, Aoki Y, Yamaguchi Y, Tateda K, Kadota J, Kohno S, Niki Y. 2014. Antibiotic susceptibility survey of blood-borne MRSA isolates in Japan from 2008 through 2011. J Infect Chemother 20:527–534. doi: 10.1016/j.jiac.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez R, Paz LI, Rosato RR, Rosato AE. 2014. Ceftaroline is active against heteroresistant MRSA clinical strains despite associated mutational mechanisms and intermediate levels of resistance. Antimicrob Agents Chemother 58:5736–5746. doi: 10.1128/AAC.03019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saravolatz SN, Martin H, Pawlak J, Johnson LB, Saravolatz LD. 2014. Ceftaroline-heteroresistant Staphylococcus aureus. Antimicrob Agents Chemother 58:3133–3136. doi: 10.1128/AAC.02685-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera AM, Boucher HW. 2011. Current concepts in antimicrobial therapy against select gram-positive organisms: methicillin-resistant Staphylococcus aureus, penicillin-resistant pneumococci, and vancomycin-resistant enterococci. Mayo Clin Proc 86:1230–1243. doi: 10.4065/mcp.2011.0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucher H, Miller LG, Razonable RR. 2010. Serious infections caused by methicillin-resistant Staphylococcus aureus. Clin Infect Dis 51(Suppl 2):S183–S197. doi: 10.1086/653519. [DOI] [PubMed] [Google Scholar]
- 25.Selton-Suty C, Celard M, Le Moing V, Doco-Lecompte T, Chirouze C, Iung B, Strady C, Revest M, Vandenesch F, Bouvet A, Delahaye F, Alla F, Duval X, Hoen B. 2012. Preeminence of Staphylococcus aureus in infective endocarditis: a 1-year population-based survey. Clin Infect Dis 54:1230–1239. doi: 10.1093/cid/cis199. [DOI] [PubMed] [Google Scholar]
- 26.Bae IG, Federspiel JJ, Miro JM, Woods CW, Park L, Rybak MJ, Rude TH, Bradley S, Bukovski S, de la Maria CG, Kanj SS, Korman TM, Marco F, Murdoch DR, Plesiat P, Rodriguez-Creixems M, Reinbott P, Steed L, Tattevin P, Tripodi MF, Newton KL, Corey GR, Fowler VG Jr. 2009. Heterogeneous vancomycin-intermediate susceptibility phenotype in bloodstream methicillin-resistant Staphylococcus aureus isolates from an international cohort of patients with infective endocarditis: prevalence, genotype, and clinical significance. J Infect Dis 200:1355–1366. doi: 10.1086/606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mascitti KB, Gerber JS, Zaoutis TE, Barton TD, Lautenbach E. 2010. Preferred treatment and prevention strategies for recurrent community-associated methicillin-resistant Staphylococcus aureus skin and soft-tissue infections: a survey of adult and pediatric providers. Am J Infect Control 38:324–328. doi: 10.1016/j.ajic.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stevens DL, Bisno AL, Chambers HF, Everett ED, Dellinger P, Goldstein EJ, Gorbach SL, Hirschmann JV, Kaplan EL, Montoya JG, Wade JC. 2005. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 41:1373–1406. doi: 10.1086/497143. [DOI] [PubMed] [Google Scholar]
- 29.Shallcross LJ, Hayward AC, Johnson AM, Petersen I. 2015. Evidence for increasing severity of community-onset boils and abscesses in UK general practice. Epidemiol Infect 143:2426–2429. doi: 10.1017/S0950268814003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shallcross LJ, Hayward AC, Johnson AM, Petersen I. 2015. Incidence and recurrence of boils and abscesses within the first year: a cohort study in UK primary care. Br J Gen Pract 65:e668–e676. doi: 10.3399/bjgp15X686929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Giudice P, Bes M, Hubiche T, Roudiere L, Blanc V, Lina G, Vandenesch F, Etienne J. 2011. Clinical manifestations and outcome of skin infections caused by the community-acquired methicillin-resistant Staphylococcus aureus clone ST80-IV. J Eur Acad Dermatol Venereol 25:164–169. doi: 10.1111/j.1468-3083.2010.03731.x. [DOI] [PubMed] [Google Scholar]
- 32.Song E, Jaishankar GB, Saleh H, Jithpratuck W, Sahni R, Krishnaswamy G. 2011. Chronic granulomatous disease: a review of the infectious and inflammatory complications. Clin Mol Allergy 9:10. doi: 10.1186/1476-7961-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaposzta R, Marodi L. 1995. Chronic neutropenia and defect in superoxide generation of granulocytes in two patients: enhancement of bactericidal capacity and respiratory burst activity by treatment with recombinant human granulocyte colony-stimulating factor. Pediatr Res 37:50–55. doi: 10.1203/00006450-199501000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Freeman AF, Holland SM. 2008. The hyper-IgE syndromes. Immunol Allergy Clin North Am 28:277–291. doi: 10.1016/j.iac.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. 2012. An update on the hyper-IgE syndromes. Arthritis Res Ther 14:228. doi: 10.1186/ar4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heimall J, Freeman A, Holland SM. 2010. Pathogenesis of hyper IgE syndrome. Clin Rev Allergy Immunol 38:32–38. doi: 10.1007/s12016-009-8134-1. [DOI] [PubMed] [Google Scholar]
- 37.Matusiak L, Bieniek A, Szepietowski JC. 2014. Bacteriology of hidradenitis suppurativa—which antibiotics are the treatment of choice? Acta Derm Venereol 94:699–702. doi: 10.2340/00015555-1841. [DOI] [PubMed] [Google Scholar]
- 38.Wolk K, Warszawska K, Hoeflich C, Witte E, Schneider-Burrus S, Witte K, Kunz S, Buss A, Roewert HJ, Krause M, Lukowsky A, Volk HD, Sterry W, Sabat R. 2011. Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol 186:1228–1239. doi: 10.4049/jimmunol.0903907. [DOI] [PubMed] [Google Scholar]
- 39.Stentzel S, Sundaramoorthy N, Michalik S, Nordengrun M, Schulz S, Kolata J, Kloppot P, Engelmann S, Steil L, Hecker M, Schmidt F, Volker U, Roghmann MC, Broker BM. 2015. Specific serum IgG at diagnosis of Staphylococcus aureus bloodstream invasion is correlated with disease progression. J Proteomics 128:1–7. doi: 10.1016/j.jprot.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 40.Verkaik NJ, de Vogel CP, Boelens HA, Grumann D, Hoogenboezem T, Vink C, Hooijkaas H, Foster TJ, Verbrugh HA, van Belkum A, van Wamel WJ. 2009. Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J Infect Dis 199:625–632. doi: 10.1086/596743. [DOI] [PubMed] [Google Scholar]
- 41.Clarke SR, Brummell KJ, Horsburgh MJ, McDowell PW, Mohamad SA, Stapleton MR, Acevedo J, Read RC, Day NP, Peacock SJ, Mond JJ, Kokai-Kun JF, Foster SJ. 2006. Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J Infect Dis 193:1098–1108. doi: 10.1086/501471. [DOI] [PubMed] [Google Scholar]
- 42.Kluytmans J, van Belkum A, Verbrugh H. 1997. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 44.Fowler VG Jr, Proctor RA. 2014. Where does a Staphylococcus aureus vaccine stand? Clin Microbiol Infect 20(Suppl 5):S66–S75. doi: 10.1111/1469-0691.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ballow M. 2002. Primary immunodeficiency disorders: antibody deficiency. J Allergy Clin Immunol 109:581–591. doi: 10.1067/mai.2002.122466. [DOI] [PubMed] [Google Scholar]
- 46.Proctor RA. 2012. Challenges for a universal Staphylococcus aureus vaccine. Clin Infect Dis 54:1179–1186. doi: 10.1093/cid/cis033. [DOI] [PubMed] [Google Scholar]
- 47.Kolata JB, Kuhbandner I, Link C, Normann N, Vu CH, Steil L, Weidenmaier C, Broker BM. 2015. The fall of a dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J Infect Dis 212:830–838. doi: 10.1093/infdis/jiv128. [DOI] [PubMed] [Google Scholar]
- 48.Montgomery CP, Daniels M, Zhao F, Alegre ML, Chong AS, Daum RS. 2014. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and IL-17A. Infect Immun 82:2125–2134. doi: 10.1128/IAI.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeaman MR, Filler SG, Chaili S, Barr K, Wang H, Kupferwasser D, Fu Y, Hennessey JP Jr, Schmidt C, Edwards JE Jr, Xiong YQ, Ibrahim AS. 2014. Mechanisms of NDV-3 vaccine efficacy in MRSA skin versus invasive infection. Proc Natl Acad Sci U S A 111:E5555–E5563. doi: 10.1073/pnas.1415610111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan LC, Chaili S, Filler SG, Barr K, Wang H, Kupferwasser D, Edwards JE Jr, Xiong YQ, Ibrahim AS, Miller LS, Schmidt CS, Hennessey JP Jr, Yeaman MR. 2015. Nonredundant roles of interleukin-17A (IL-17A) and IL-22 in murine host defense against cutaneous and hematogenous infection due to methicillin-resistant Staphylococcus aureus. Infect Immun 83:4427–4437. doi: 10.1128/IAI.01061-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahrens K, Schunck M, Podda GF, Meingassner J, Stuetz A, Schroder JM, Harder J, Proksch E. 2011. Mechanical and metabolic injury to the skin barrier leads to increased expression of murine beta-defensin-1, -3, and -14. J Invest Dermatol 131:443–452. doi: 10.1038/jid.2010.289. [DOI] [PubMed] [Google Scholar]
- 53.Park H, Solis NV, Louie JS, Spellberg B, Rodriguez N, Filler SG. 2014. Different tumor necrosis factor alpha antagonists have different effects on host susceptibility to disseminated and oropharyngeal candidiasis in mice. Virulence 5:625–629. doi: 10.4161/viru.29699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quintin J, Cheng SC, van der Meer JW, Netea MG. 2014. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol 29:1–7. doi: 10.1016/j.coi.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Feuerstein R, Seidl M, Prinz M, Henneke P. 2015. MyD88 in macrophages is critical for abscess resolution in staphylococcal skin infection. J Immunol 194:2735–2745. doi: 10.4049/jimmunol.1402566. [DOI] [PubMed] [Google Scholar]
- 56.Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, Hickey MJ, Firth N, Weninger W. 2014. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol 15:45–53. doi: 10.1038/nri3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagl M, Kacani L, Mullauer B, Lemberger EM, Stoiber H, Sprinzl GM, Schennach H, Dierich MP. 2002. Phagocytosis and killing of bacteria by professional phagocytes and dendritic cells. Clin Diagn Lab Immunol 9:1165–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voorhees T, Chang J, Yao Y, Kaplan MH, Chang CH, Travers JB. 2011. Dendritic cells produce inflammatory cytokines in response to bacterial products from Staphylococcus aureus-infected atopic dermatitis lesions. Cell Immunol 267:17–22. doi: 10.1016/j.cellimm.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaffler H, Demircioglu DD, Kuhner D, Menz S, Bender A, Autenrieth IB, Bodammer P, Lamprecht G, Gotz F, Frick JS. 2014. NOD2 stimulation by Staphylococcus aureus-derived peptidoglycan is boosted by Toll-like receptor 2 costimulation with lipoproteins in dendritic cells. Infect Immun 82:4681–4688. doi: 10.1128/IAI.02043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin JO, Zhang W, Du JY, Yu Q. 2014. BDCA1-positive dendritic cells (DCs) represent a unique human myeloid DC subset that induces innate and adaptive immune responses to Staphylococcus aureus infection. Infect Immun 82:4466–4476. doi: 10.1128/IAI.01851-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. 2001. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 62.Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, Gallo RL. 2015. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347:67–71. doi: 10.1126/science.1260972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noore J, Noore A, Li B. 2013. Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob Agents Chemother 57:1283–1290. doi: 10.1128/AAC.01650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burd RS, Furrer JL, Sullivan J, Smith AL. 2002. Murine beta-defensin-3 is an inducible peptide with limited tissue expression and broad-spectrum antimicrobial activity. Shock 18:461–464. doi: 10.1097/00024382-200211000-00013. [DOI] [PubMed] [Google Scholar]
- 65.Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. 2004. Innate immune response in Th1- and Th2-dominant mouse strains. Shock 22:460–466. doi: 10.1097/01.shk.0000142249.08135.e9. [DOI] [PubMed] [Google Scholar]
- 66.Netea MG, Quintin J, van der Meer JW. 2011. Trained immunity: a memory for innate host defense. Cell Host Microbe 9:355–361. doi: 10.1016/j.chom.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 67.Johnson JK, Khoie T, Shurland S, Kreisel K, Stine OC, Roghmann MC. 2007. Skin and soft tissue infections caused by methicillin-resistant Staphylococcus aureus USA300 clone. Emerg Infect Dis 13:1195–1200. doi: 10.3201/eid1308.061575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.David MZ, Mennella C, Mansour M, Boyle-Vavra S, Daum RS. 2008. Predominance of methicillin-resistant Staphylococcus aureus among pathogens causing skin and soft tissue infections in a large urban jail: risk factors and recurrence rates. J Clin Microbiol 46:3222–3227. doi: 10.1128/JCM.01423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burlak C, Hammer CH, Robinson MA, Whitney AR, McGavin MJ, Kreiswirth BN, Deleo FR. 2007. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol 9:1172–1190. doi: 10.1111/j.1462-5822.2006.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Falk I, Potocnik AJ, Barthlott T, Levelt CN, Eichmann K. 1996. Immature T cells in peripheral lymphoid organs of recombinase-activating gene-1/-2-deficient mice. Thymus dependence and responsiveness to anti-CD3 epsilon antibody. J Immunol 156:1362–1368. [PubMed] [Google Scholar]
- 71.Guidos CJ, Williams CJ, Wu GE, Paige CJ, Danska JS. 1995. Development of CD4+CD8+ thymocytes in RAG-deficient mice through a T cell receptor beta chain-independent pathway. J Exp Med 181:1187–1195. doi: 10.1084/jem.181.3.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, Bubeck Wardenburg J. 2014. Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J Infect Dis 210:1012–1018. doi: 10.1093/infdis/jiu223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.