

ABSTRACT
Salmonellae are pathogenic bacteria that cause significant morbidity and mortality in humans worldwide. Salmonellae establish infection and avoid clearance by the immune system by mechanisms that are not well understood. We previously showed that l-asparaginase II produced by Salmonella enterica serovar Typhimurium (S. Typhimurium) inhibits T cell responses and mediates virulence. In addition, we previously showed that asparagine deprivation such as that mediated by l-asparaginase II of S. Typhimurium causes suppression of activation-induced T cell metabolic reprogramming. Here, we report that STM3997, which encodes a homolog of disulfide bond protein A (dsbA) of Escherichia coli, is required for l-asparaginase II stability and function. Furthermore, we report that l-asparaginase II localizes primarily to the periplasm and acts together with l-asparaginase I to provide S. Typhimurium the ability to catabolize asparagine and assimilate nitrogen. Importantly, we determined that, in a murine model of infection, S. Typhimurium lacking both l-asparaginase I and II genes competes poorly with wild-type S. Typhimurium for colonization of target tissues. Collectively, these results indicate that asparagine catabolism contributes to S. Typhimurium virulence, providing new insights into the competition for nutrients at the host-pathogen interface.
KEYWORDS: asparaginase, asparagine, catabolism, host response, metabolism, nitrogen metabolism, pathogenesis, Salmonella, T cells, virulence
INTRODUCTION
Salmonella enterica (S. enterica) serovars cause significant morbidity and mortality in humans worldwide (1–5). Clinical syndromes associated with S. enterica serovars include typhoid and paratyphoid fever caused by S. enterica serovar Typhi (S. Typhi) and S. enterica serovar Paratyphi, respectively, gastroenteritis caused by nontyphoidal Salmonella (NTS) serovars such as S. enterica serovar Typhimurium (S. Typhimurium) in immunocompetent individuals, and bacteremia caused by NTS serovars in immunocompromised individuals (6). The different clinical syndromes associated with S. enterica serovars are responsible for a combined global toll of approximately 1 million deaths annually (7), which makes these pathogenic bacteria one of the leading causes of mortality in humans worldwide.
Much of what is known about the pathogenesis of and host response to infections with Salmonella comes from experimental infection of mice with S. Typhimurium, which has served as a useful model for the human disease caused by S. Typhi (8–10). In susceptible strains of mice, S. Typhimurium infection results in acute immunosuppression and delayed onset of protective immune responses (8, 9, 11–13). Immunity to S. Typhimurium that eventually develops requires both humoral and cell-mediated immune responses. T cells, particularly gamma interferon (IFN-γ)-producing CD4+ T cells, play a critical role in the clearance of S. Typhimurium. However, T cell responses to S. Typhimurium are dampened during infection by mechanisms that are not well understood (8, 9, 11).
We previously showed that l-asparaginase II produced by S. Typhimurium inhibits T cell responses and mediates virulence (14). In addition, we previously showed that asparagine deprivation such as that mediated by l-asparaginase II of S. Typhimurium causes inhibition of activation-induced T cell metabolic reprogramming (15).
Here, we show that STM3997, which encodes a putative periplasmic thiol-disulfide oxidoreductase, is required for S. Typhimurium to cause inhibition of T cells and, furthermore, is required for l-asparaginase II stability and function. We also show that l-asparaginase II localizes primarily to the periplasm and acts together with the cytoplasmic asparagine hydrolase l-asparaginase I to provide S. Typhimurium the ability to catabolize asparagine in vitro and during infection. These findings provide new insights into the l-asparaginase-based mechanism used by S. Typhimurium to cause inhibition of T cells and advance our knowledge of the competition for nutrients at the host-pathogen interface.
RESULTS
STM3997 is required for S. Typhimurium to cause inhibition of T cells.
The l-asparaginase II gene STM3106 is required for S. Typhimurium to inhibit T cells and mediate virulence (14). The STM3106 gene was identified in a partial screen of an S. Typhimurium mutant library used to isolate mutants unable to cause down-modulation of T cell receptor beta (TCR-β) surface expression (14). Each S. Typhimurium mutant in the partially screened library carries a targeted deletion of multiple linked, nonessential genes (a total of 3,476 genes are deleted in the entire library [16]). To identify additional genes required for S. Typhimurium to cause inhibition of T cells, we screened the remainder of the targeted multigene deletion library. One mutant, ΔSTM3993–ΔSTM3998 (ΔSTM3993–8) S. Typhimurium, was unable to cause down-modulation of TCR-β surface expression (Fig. 1A and C).
FIG 1.

STM3997 is required for S. Typhimurium to cause inhibition of T cells. (A) Expression of surface TCR-β by anti-CD3ε/CD28-stimulated T cells left uninfected (UI) or cultured with wild-type S. Typhimurium (WT) or ΔSTM3993–8 S. Typhimurium (ΔSTM3993–8). (B) Expression of surface TCR-β by anti-CD3ε/CD28-stimulated T cells left uninfected (UI) or cultured with wild-type S. Typhimurium (WT), ΔSTM3993–8 S. Typhimurium (ΔSTM3993–8), ΔSTM3993–8 S. Typhimurium carrying plasmid pBAD18-Cm (ΔSTM3993–8/Empty Vector) or ΔSTM3993–8 S. Typhimurium carrying derivatives of pBAD18-Cm expressing STM3993 (ΔSTM3993–8/pSTM3993), STM3994 (ΔSTM3993–8/pSTM3994), STM3995 (ΔSTM3993–8/pSTM3995), STM3996 (ΔSTM3993–8/pSTM3996), STM3997 (ΔSTM3993–8/pSTM3997), or STM3998 (ΔSTM3993–8/pSTM3998). (C) Genetic organization of the chromosomal region deleted in ΔSTM3993–8 S. Typhimurium. (D) Expression of surface TCR-β by anti-CD3ε/CD28-stimulated T cells left uninfected (UI) or cultured with wild-type S. Typhimurium (WT), ΔSTM3997 S. Typhimurium (ΔSTM3997), ΔSTM3997 S. Typhimurium carrying pBAD18-Cm (ΔSTM3997/Empty Vector) or ΔSTM3997 S. Typhimurium carrying a derivative of pBAD18-Cm bearing STM3997 (ΔSTM3997/pSTM3997). (E) Blastogenesis of anti-CD3ε/CD28-stimulated T cells treated as described for panel D. (F) IL-2 secretion by anti-CD3ε/CD28-stimulated T cells treated as described for panel D. Data show means with standard errors of the means obtained from at least five independent experiments. Data were analyzed by repeated measures one-way ANOVA with Tukey's multiple comparisons posttest; P values of <0.05 were considered to be statistically significant. Asterisks indicate statistically significant differences for designated posttest comparisons (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; n.s. not significant). GMFI, geometric mean fluorescent intensity.
To identify the gene(s) deleted in the mutant required for this phenotype, we generated ΔSTM3993–8 S. Typhimurium strains with a plasmid carrying STM3993, STM3994, STM3995, STM3996, STM3997, or STM3998. Only the plasmid carrying STM3997 (pSTM3997) complemented the phenotype of ΔSTM3993–8 S. Typhimurium (Fig. 1B). Consistent with these results, S. Typhimurium carrying a targeted deletion of STM3997 (ΔSTM3997) was unable to cause down-modulation of TCR-β surface expression (Fig. 1D). Similar results were obtained when we measured the ability of ΔSTM3997 S. Typhimurium to cause inhibition of T cell blastogenesis (Fig. 1E) and interleukin-2 (IL-2) secretion (Fig. 1F). Furthermore, the presence of STM3997 in trans reversed the phenotype of ΔSTM3997 S. Typhimurium (Fig. 1D to F). Collectively, these results indicate that STM3997, like STM3106 (14), is required for S. Typhimurium to cause inhibition of T cells.
STM3997 encodes a periplasmic thiol-disulfide oxidoreductase required to maintain the steady-state level of l-asparaginase II.
The STM3997 gene of S. Typhimurium encodes a homolog of disulfide bond protein A (DsbA) of Escherichia coli. DsbA is a periplasmic thiol-disulfide oxidoreductase involved in the formation of structural disulfide bonds that are essential for the stability and function of many proteins, particularly those that are secreted (17). Given the function of DsbA and our published results indicating that l-asparaginase II produced by S. Typhimurium is necessary and sufficient to cause inhibition of T cells (14), we investigated whether STM3997 is required to maintain the steady-state level of l-asparaginase II. Consistent with the hypothesis that DsbA is involved in the production of functional l-asparaginase II, the steady-state level of l-asparaginase II was reduced significantly in ΔSTM3997 S. Typhimurium (Fig. 2A and B). Furthermore, the presence of STM3997 in trans restored the steady-state level of l-asparaginase II in ΔSTM3997 S. Typhimurium (Fig. 2A and B). As expected, l-asparaginase II gene expression was not affected in ΔSTM3997 S. Typhimurium (see Fig. S1 in the supplemental material). These results indicate that STM3997 is required to maintain the steady-state level of l-asparaginase II.
FIG 2.
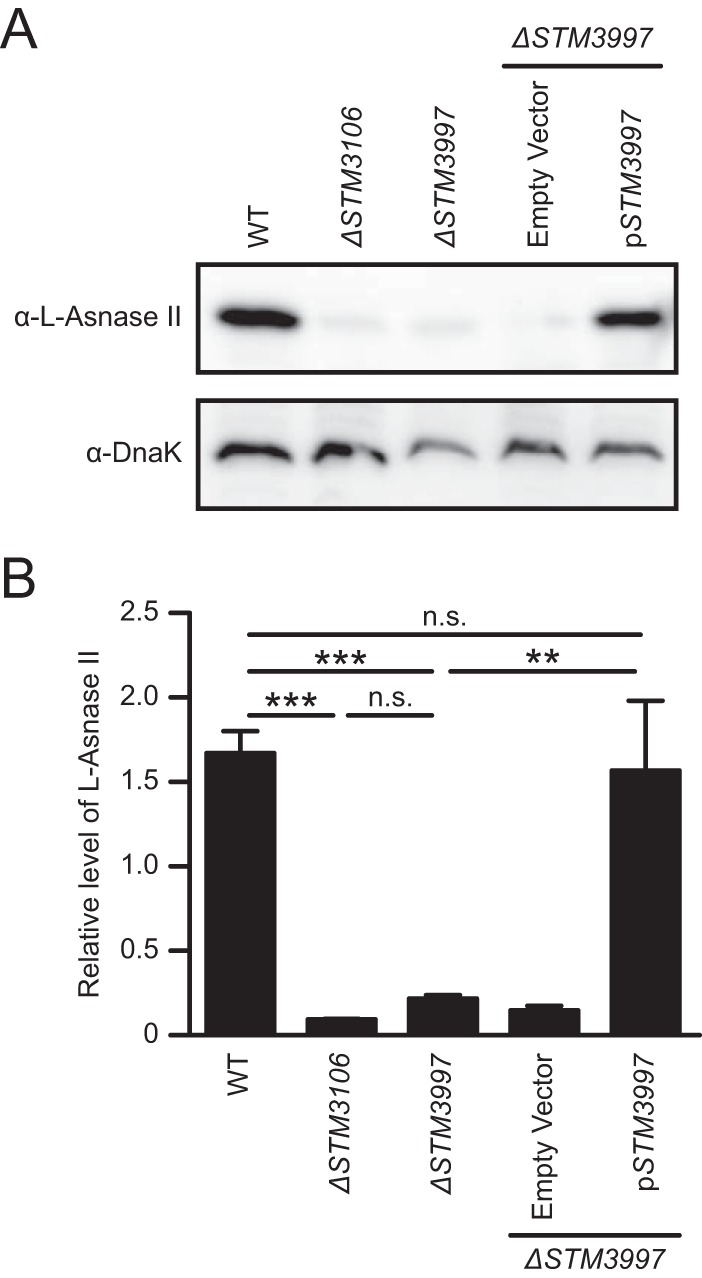
STM3997 encodes a periplasmic thiol-disulfide oxidoreductase required to maintain the steady-state level of l-asparaginase II. (A) Western blot analysis of whole-cell lysates of wild-type S. Typhimurium (WT), S. Typhimurium lacking the l-asparaginase II gene STM3106 (ΔSTM3106), S. Typhimurium lacking the DsbA gene STM3997 (ΔSTM3997), ΔSTM3997 S. Typhimurium carrying plasmid pBAD18-Cm (ΔSTM3997/Empty Vector), or ΔSTM3997 S. Typhimurium carrying a derivative of pBAD18-Cm expressing STM3997 (ΔSTM3997/pSTM3997) using anti-l-asparaginase II antibody (α-l-Asnase-II) or anti-DnaK antibody (α-DnaK). (B) Quantification of Western blot data shown in panel A. DnaK was used as a loading control for Western blotting. Electrophoretic densitometry was used to quantify the relative levels of l-asparaginase II normalized to DnaK. Data are representative of (A) or show means with standard errors of the means obtained from (B) four independent experiments. Data in panel B were analyzed by repeated measures one-way ANOVA with Tukey's multiple comparisons posttest; P values of <0.05 were considered to be statistically significant. Asterisks indicate statistically significant differences for designated posttest comparisons (***, P < 0.001; **, P < 0.01; n.s. not significant).
Cysteine residues at positions 99 and 127 are essential for l-asparaginase II stability and function.
The structure of l-asparaginase II of E. coli has been solved, revealing the existence of an intramolecular disulfide bond between the thiol groups of cysteine residues at positions 99 and 127 (18). These cysteine residues are conserved in l-asparaginase II of S. Typhimurium (Fig. S2). To determine the importance of these cysteine residues with respect to l-asparaginase II stability and function, we introduced site-specific mutations into a plasmid carrying STM3106 that resulted in alanine substitutions for cysteine residues at positions 99 and 127. The resulting constructs expressing mutant l-asparaginase II proteins with alanine substitutions for cysteine residues at positions 99 (pSTM3106_C99A), 127 (pSTM3106_C127A), or both 99 and 127 (pSTM3106_C99A/C127A) were transformed into ΔSTM3106 S. Typhimurium.
Consistent with the hypothesis that the cysteine residues at positions 99 and 127 are required for the formation of a stabilizing structural disulfide bond, steady-state levels of l-asparaginase II were reduced significantly in ΔSTM3106 S. Typhimurium carrying pSTM3106_C99A, pSTM3106_C127A, or pSTM3106_C99A/C127A compared to the level in ΔSTM3106 S. Typhimurium carrying pSTM3106 (Fig. 3A and B). Furthermore, ΔSTM3106 S. Typhimurium carrying pSTM3106_C99A, pSTM3106_C127A, or pSTM3106_C99A/C127A was unable to cause down-modulation of TCR-β surface expression relative to the level of ΔSTM3106 S. Typhimurium carrying pSTM3106 (Fig. 3C and data not shown). Collectively, these results indicate that the cysteine residues at positions 99 and 127 are essential for the stability and function of l-asparaginase II of S. Typhimurium.
FIG 3.

Cysteine residues at positions 99 and 127 are essential for l-asparaginase II stability and function. (A) Western blot analysis of whole-cell lysates of ΔSTM3106 S. Typhimurium carrying plasmid pSTM3106 (ΔSTM3106/pSTM3106 strain) or derivatives of pSTM3106 containing site-specific mutations in STM3106 that result in alanine substitutions for cysteine residues at positions 99 (ΔSTM3106/pSTM3106_C99A strain), 127 (ΔSTM3106/pSTM3106_C127A strain), or both 99 and 127 (ΔSTM3106/pSTM3106_C99A/C127A strain). Expression of l-asparaginase II and DnaK was detected with anti-l-asparaginase II antibody (α-l-Asnase-II) and anti-DnaK antibody (α-DnaK), respectively. DnaK was used as a loading control for Western blotting. (B) Quantification of Western blot data shown in panel A. Electrophoretic densitometry was used to quantify the relative levels of l-asparaginase II normalized to DnaK. (C) Expression of surface TCR-β by anti-CD3ε/CD28-stimulated T cells left uninfected (UI) or cultured with the ΔSTM3106/pSTM3106, ΔSTM3106/pSTM3106_C99A, or ΔSTM3106/pSTM3106_C127A strain. Data are representative of (A) or show means with standard errors of the means obtained from (B and C) at least four independent experiments. Data were analyzed by repeated measures one-way ANOVA with Tukey's multiple comparisons posttest (B and C); P values of <0.05 were considered to be statistically significant. Asterisks indicate statistically significant differences for designated posttest comparisons (****, P < 0.0001; **, P < 0.01; *, P < 0.05; n.s. not significant). See also Fig. S1 in the supplemental material.
l-Asparaginase II localizes primarily to the periplasm and is used by S. Typhimurium to catabolize asparagine and assimilate nitrogen.
The primary structure of l-asparaginase II of S. Typhimurium contains a predicted signal peptide and signal peptide cleavage site (Fig. S2), suggesting that l-asparaginase II is exported across the inner membrane by use of the general secretory pathway. To determine the subcellular localization of l-asparaginase II of S. Typhimurium, we obtained whole-cell lysates, cytoplasmic and periplasmic fractions, and precipitated proteins from cell-free culture supernatants. We subjected aliquots of these samples to Western blot analysis. Consistent with published evidence indicating that l-asparaginase II of E. coli localizes to the periplasm (19), we found l-asparaginase II of S. Typhimurium to be present primarily in the periplasmic fraction (Fig. 4A). These results indicate that l-asparaginase II of S. Typhimurium is a secreted protein that localizes primarily to the periplasm, where it may function to hydrolyze nutritional asparagine.
FIG 4.
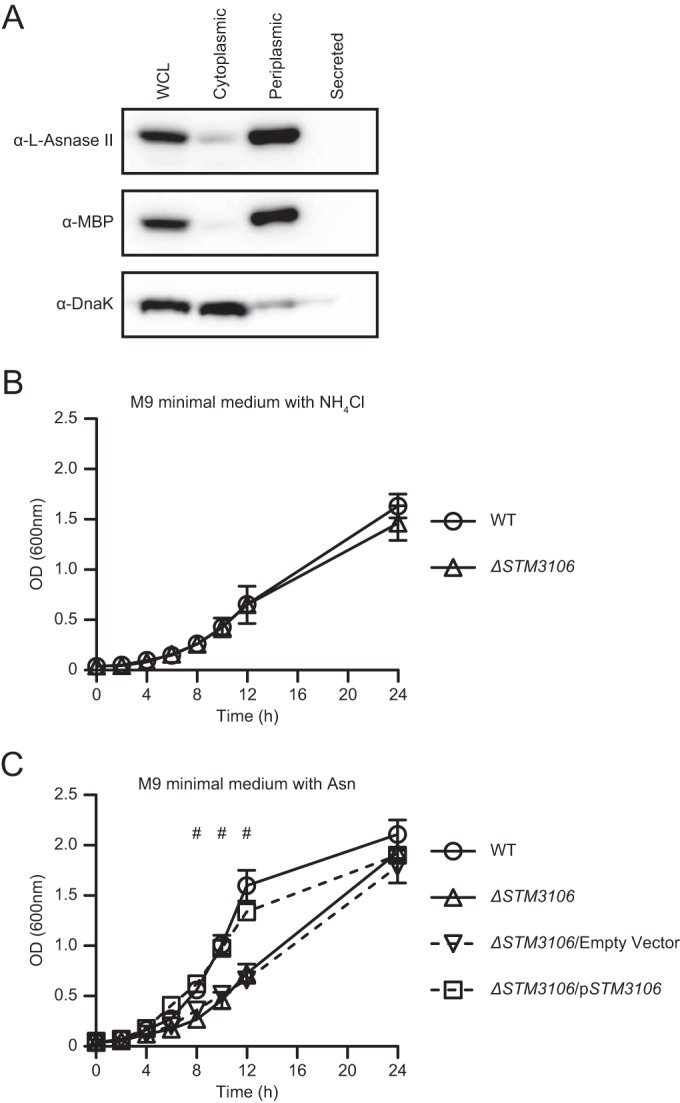
l-Asparaginase II localizes primarily to the periplasm and is used by S. Typhimurium to catabolize asparagine and assimilate nitrogen. (A) Western blot analysis of whole-cell lysates (WCL) of S. Typhimurium, cytoplasmic or periplasmic fractions of S. Typhimurium, or TCA-precipitated proteins from cell-free culture supernatants of S. Typhimurium (Secreted) grown in lysogeny broth. Expression of l-asparaginase II, MBP, and DnaK was detected with anti-l-asparaginase II antibody (α-l-Asnase-II), anti-MBP antibody (α-MBP), and anti-DnaK antibody (α-DnaK), respectively. MBP and DnaK were used as fractionation controls. (B) Growth of wild-type S. Typhimurium (WT) and ΔSTM3106 S. Typhimurium (ΔSTM3106) in M9 minimal medium with ammonium chloride (NH4Cl) as the sole nitrogen source. (C) Growth of wild-type S. Typhimurium (WT), ΔSTM3106 S. Typhimurium (ΔSTM3106), ΔSTM3106 S. Typhimurium carrying plasmid pBAD18-Cm (ΔSTM3106/Empty Vector), and ΔSTM3106 S. Typhimurium carrying a derivative of pBAD18-Cm bearing STM3106 (ΔSTM3106/pSTM3106) in M9 minimal medium with asparagine (Asn) as the sole nitrogen source. Data are representative of (A) or show means with standard errors of the means obtained from (B and C) at least three independent experiments. Data were analyzed by repeated measures two-way ANOVA with Sidak (B) or Tukey's (C) multiple comparisons posttest; P values of <0.05 were considered to be statistically significant. Number (#) symbols indicate statistically significant differences for designated posttest comparisons between WT and ΔSTM3106 S. Typhimurium at indicated time points. See also Fig. S1 in the supplemental material.
We have shown previously that ΔSTM3106 S. Typhimurium grows as well as wild-type S. Typhimurium (14). Consistent with this notion, ΔSTM3106 S. Typhimurium grew normally in M9 minimal medium with ammonium chloride as the sole nitrogen source (Fig. 4B). However, ΔSTM3106 S. Typhimurium grew significantly less well than wild-type S. Typhimurium in M9 minimal medium with asparagine as the sole nitrogen source (Fig. 4C). The presence of STM3106 in trans reversed the phenotype of ΔSTM3106 S. Typhimurium (Fig. 4C). Collectively, these results indicate that l-asparaginase II is a predominantly periplasmic protein used by S. Typhimurium to catabolize asparagine and assimilate nitrogen.
l-Asparaginase II and l-asparaginase I act together to provide S. Typhimurium the ability to assimilate nitrogen.
The partial growth defect of ΔSTM3106 S. Typhimurium in M9 minimal medium when asparagine is the sole nitrogen source (Fig. 4C) suggests that S. Typhimurium encodes at least one other asparagine hydrolase capable of functionally complementing l-asparaginase II. STM1294 encodes a putative cytoplasmic asparagine hydrolase that is 94.67% identical to l-asparaginase I of E. coli at the amino acid level (Fig. S3A) and is syntenic. To establish the role of this putative asparagine hydrolase in nitrogen assimilation, we generated and tested an S. Typhimurium strain lacking STM1294 (ΔSTM1294 strain). This strain grew normally in M9 minimal medium with ammonium chloride or asparagine as the sole nitrogen source (Fig. S3B and C), suggesting that, under these conditions, STM1294 is not required for S. Typhimurium to assimilate nitrogen.
To examine the possibility that STM1294 and STM3106 encode functionally overlapping asparagine hydrolases, we generated and tested an S. Typhimurium strain lacking both STM1294 and STM3106 (ΔSTM1294 ΔSTM3106 strain). This strain grew normally in M9 minimal medium with ammonium chloride as the sole nitrogen source (Fig. 5A) but grew poorly relative to wild-type S. Typhimurium when asparagine was available as the sole nitrogen source (Fig. 5B). In fact, the ΔSTM1294 ΔSTM3106 double mutant exhibited a much stronger growth phenotype than either the ΔSTM1294 single mutant or the ΔSTM3106 single mutant (Fig. 4C and S3B and C). Finally, the presence of either STM1294 or STM3106 in trans reversed the phenotype of the ΔSTM1294 ΔSTM3106 double mutant (Fig. 5B). These results indicate that STM1294 and STM3106 encode functionally overlapping asparagine hydrolases that can act together to provide S. Typhimurium the ability to catabolize asparagine and assimilate nitrogen.
FIG 5.
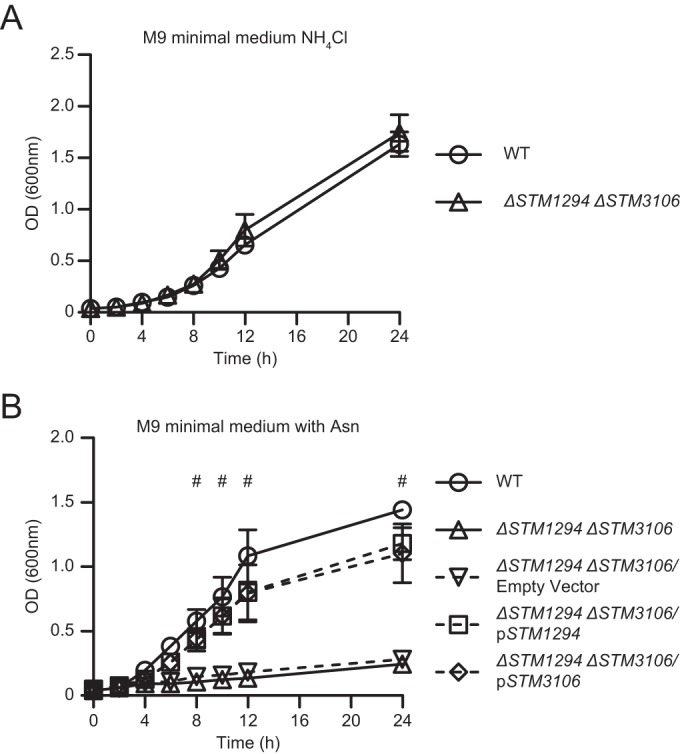
l-Asparaginase II and l-asparaginase I act together to provide S. Typhimurium the ability to assimilate nitrogen. (A) Growth of wild-type S. Typhimurium (WT) and ΔSTM1294 ΔSTM3106 S. Typhimurium (ΔSTM1294 ΔSTM3106) in M9 minimal medium with ammonium chloride (NH4Cl) as the sole nitrogen source. (B) Growth of wild-type S. Typhimurium (WT), ΔSTM1294 ΔSTM3106 S. Typhimurium (ΔSTM1294 ΔSTM3106), ΔSTM1294 ΔSTM3106 S. Typhimurium carrying plasmid pBAD18-Cm (ΔSTM1294 ΔSTM3106/Empty Vector), ΔSTM1294 ΔSTM3106 S. Typhimurium carrying a derivative of pBAD18-Cm expressing STM1294 (ΔSTM1294 ΔSTM3106/pSTM1294), and ΔSTM1294 ΔSTM3106 S. Typhimurium carrying a derivative of pBAD18-Cm expressing STM3106 (ΔSTM1294 ΔSTM3106/pSTM3106) in M9 minimal medium with asparagine (Asn) as the sole nitrogen source. Data show means with standard errors of the means obtained from at least three independent experiments. Data were analyzed by repeated measures two-way ANOVA with Sidak (A) or Tukey's (B) multiple comparisons posttest; P values of <0.05 were considered to be statistically significant. Number (#) symbols indicate statistically significant differences for designated posttest comparisons between WT and ΔSTM1294 ΔSTM3106 S. Typhimurium at indicated time points. See also Fig. S2 in the supplemental material.
Asparagine catabolism contributes to S. Typhimurium virulence.
S. Typhimurium is a facultative intracellular pathogen that thrives inside phagocytes, where access to nutrients is limited. To determine if S. Typhimurium, like Mycobacterium tuberculosis (20) and Francisella tularensis (21), catabolizes asparagine to survive and replicate inside phagocytes, we infected bone marrow-derived macrophages cultured from 129X1/SvJ mice with wild-type S. Typhimurium, the ΔSTM1294 single mutant, the ΔSTM3106 single mutant, or the ΔSTM1294 ΔSTM3106 double mutant. After 18 h of infection, we recovered intracellular bacteria and found reduced numbers of the ΔSTM1294 mutant and the ΔSTM1294 ΔSTM3106 mutant relative to the level of the wild-type S. Typhimurium, while the ΔSTM3106 mutant followed the same trend (Fig. 6A and S4A). These results suggest that S. Typhimurium uses asparagine catabolism while surviving and replicating inside macrophages.
FIG 6.

Asparagine catabolism contributes to S. Typhimurium virulence. (A) Survival and growth of wild-type S. Typhimurium (WT), ΔSTM1294 S. Typhimurium (ΔSTM1294), ΔSTM3106 S. Typhimurium (ΔSTM3106) or ΔSTM1294 ΔSTM3106 S. Typhimurium (ΔSTM1294 ΔSTM3106) inside bone marrow-derived macrophages cultured from 129X1/SvJ mice. (B to D) Competitive indices of the ΔSTM1294 ΔSTM3106 strain versus wild-type S. Typhimurium recovered from mesenteric lymph nodes (MLN; B), livers (C) and spleens (D) of 129X1/SvJ mice (n = 6 to 8 per group per time point) at various times after intragastric inoculation with 1 × 108 CFU of a 1:1 mixture of differentially marked bacterial strains. Data were analyzed by repeated measures two-way ANOVA with Tukey's multiple comparisons posttest (A), repeated-measures one-way ANOVA with Dunnett's multiple comparisons posttest (B to D; data were compared to the expected 1:1 output ratio of the ΔSTM1294 ΔSTM3106 strain versus WT S. Typhimurium). P values of <0.05 were considered to be statistically significant. Asterisks indicate statistically significant differences for designated posttest comparisons (***, P < 0.001; **, P < 0.01; *, P < 0.01; n.s. not significant). See also Fig. S3 in the supplemental material.
While S. Typhimurium must access nutrients to survive and replicate inside mammalian hosts, the exact nutrients exploited by S. Typhimurium during infection remain largely unknown. To establish the role of asparagine catabolism in S. Typhimurium virulence, we inoculated 129X1/SvJ mice intragastrically with 1 × 108 CFU of a 1:1 mixture of the wild-type strain and the ΔSTM1294 ΔSTM3106 double mutant. At various times after inoculation, we performed organ burden assays (Fig. S4B to D) and found that the wild-type strain had outcompeted the mutant for colonization of mesenteric lymph nodes (Fig. 6B), liver (Fig. 6C), and spleen (Fig. 6D). Collectively, these results establish that asparagine catabolism promotes survival and replication of S. Typhimurium inside macrophages and contributes to S. Typhimurium virulence.
DISCUSSION
We report that asparagine catabolism contributes to S. Typhimurium virulence. Specifically, we determined that STM3997, which encodes a homolog of DsbA of E. coli, is required to maintain the steady-state level of l-asparaginase II and that cysteine residues at positions 99 and 127 are essential for l-asparaginase II stability and function (Fig. 1 to 3). In addition, we determined that l-asparaginase II localizes primarily to the periplasm and, together with l-asparaginase I, enables S. Typhimurium to catabolize asparagine (Fig. 4 and 5). Importantly, we determined that, in a murine model of infection, S. Typhimurium lacking both l-asparaginase I and II genes competes poorly with the wild-type strain for colonization of target tissues (Fig. 6). Collectively, these results provide new insights into the competition for nutrients at the host-pathogen interface.
We previously showed that l-asparaginase II produced by S. Typhimurium inhibits T cell responses and mediates virulence (14) and that asparagine deprivation such as that mediated by l-asparaginase II of S. Typhimurium causes inhibition of activation-induced T cell metabolic reprogramming (15). These previous findings and the findings described in this study are consistent with the notion that nutrient availability, including amino acid availability, is a key determinant that affects the outcome of infection. We propose that S. Typhimurium competes with the host for nutritional asparagine, causing the local depletion of a key resource required for the induction of a protective T cell response (Fig. 7).
FIG 7.

Model showing the competition for nutritional asparagine between S. Typhimurium and T cells. Our published findings (14, 15) and results described in the present study support a model in which S. Typhimurium competes with the host for nutritional asparagine, causing the local depletion of a key resource required for T cell activation. See the text for details.
Pathogens require biosynthetic precursors to survive and replicate inside the mammalian host. To access these precursors, pathogens must express appropriate transporters and enzymes in key metabolic pathways that permit precursor use or biosynthesis.
Asparagine catabolism appears to be a key metabolic pathway that promotes S. Typhimurium virulence. Carbon and nitrogen are essential components of many biomolecules, and we suggest that during infection S. Typhimurium accesses both by catabolizing asparagine (Fig. 7). The mechanism(s) S. Typhimurium uses to import nutritional asparagine during infection is not well characterized. Recent studies have shown that asparagine biosynthesis is also important for S. Typhimurium virulence. S. Typhimurium bacteria lacking both asparagine biosynthesis genes asnA (STM3877) and asnB (STM0680) are attenuated for survival and replication inside host cells (22), and are unable to compete with wild-type S. Typhimurium for colonization of murine spleen (23). Furthermore, it has been reported that S. Typhimurium exploits fructose-asparagine for growth in the inflamed intestine (24). Understanding the different mechanisms used by S. Typhimurium to metabolize (i.e., catabolize and synthesize) asparagine during infection will require further study.
Asparagine catabolism is important for the virulence of a number of significant human pathogens, including S. Typhimurium (14; also the present study), Mycobacterium tuberculosis (20), Helicobacter pylori (25), Campylobacter jejuni (26), and Francisella tularensis (21). A number of these pathogens can cause chronic or persistent infections, and immunity to these pathogens is dependent, at least in part, on T cells (8, 9, 27–31), which require nutritional asparagine for activation (14, 15). Thus, pathogens competing with the host for nutrients may have evolved so as to exploit asparagine catabolism as a means to limit T cell responses (Fig. 7).
Bacterial l-asparaginases have been a cornerstone of acute lymphoblastic leukemia treatment since the late 1970s. These enzymes are used for remission induction and intensification treatment in all pediatric regimens as well as in the majority of adult treatment protocols and act through a mechanism that likely involves asparagine deprivation (32). Our published findings (14, 15) and the results described in this study indicate that pathogens competing with the host for nutrients may similarly use asparagine deprivation to cause inhibition of protective T cell responses. Therefore, the targeting of asparagine metabolism (e.g., bacterial asparagine catabolism) holds great potential for the development of new strategies to fight infectious diseases.
MATERIALS AND METHODS
Ethics statement.
All procedures that used mice were approved by the Institutional Animal Care and Use Committee at Stony Brook University and were conducted in accordance with the recommendations outlined in the Guide for the Care and Use of Laboratory Animals adopted by the National Institutes of Health (33). Euthanasia of mice was performed by inhalation of carbon dioxide, a method that is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Bacterial strains, strain construction, and culture conditions.
With the use of standard microbiological techniques, bacteria were grown aerobically at 37°C in lysogeny broth or on lysogeny agar except where otherwise noted. Kanamycin (60 μg/ml) was added to the culture medium to select for various mutant strains of S. Typhimurium in the 14028 or IR715 strain background (see below). Chloramphenicol (30 μg/ml) and l-arabinose (0.075%, wt/vol; 5 mM) were added to the culture medium to maintain plasmids in vivo and induce expression of plasmid-encoded l-asparaginase II, respectively. In the context of mouse infections, nalidixic acid (50 μg/ml) or kanamycin was added to the culture medium to select for wild-type or mutant S. Typhimurium in the IR715 strain background (see below).
S. Typhimurium strain 14028 (American Type Culture Collection, Manassas, VA, USA) was used as the wild-type strain. For mouse infections, S. Typhimurium strain IR715 (34), a spontaneous nalidixic acid-resistant derivative of S. Typhimurium strain 14028, was used as the wild-type strain. The library of targeted, multigene deletion mutants of S. Typhimurium strain 14028 has been published previously (16). An isogenic derivative of S. Typhimurium strain 14028 lacking STM3106 (ΔSTM3106 strain), ΔSTM3106 S. Typhimurium carrying the bacterial gene expression vector pBAD18-Cm (American Type Culture Collection) (ΔSTM3106/pBAD strain; here referred to as the ΔSTM3106/empty vector strain), and ΔSTM3106 S. Typhimurium carrying a derivative of pBAD18-Cm bearing STM3106 (ΔSTM3106/pBAD-STM3106 strain; here referred to as the ΔSTM3106/pSTM3106 strain) have also been published previously (14).
Isogenic derivatives of S. Typhimurium strain 14028 lacking STM1294 (ΔSTM1294 strain) or STM3997 (ΔSTM3997 strain) were generated by use of the lambda red recombinase method (35). Briefly, the kanamycin resistance gene carried by plasmid pCLF4 (36) was amplified by use of PCR with primers ΔSTM1294-F and ΔSTM1294-R to delete STM1294 or primers ΔSTM3997-F and ΔSTM3997-R to delete STM3997 (see Table S1 in the supplemental material).
To generate an S. Typhimurium strain lacking both STM1294 and STM3106, plasmid pCP20 (37), which encodes the FLP recombinase, was electroporated into ΔSTM3106 S. Typhimurium. This strain was generated by use of the aforementioned lambda red recombinase method and is resistant to kanamycin. Following FLP recombinase-mediated excision of the kanamycin resistance gene, the resulting strain was cured of plasmid pCP20, yielding a strain with an unmarked deletion of STM3106. Next, the STM1294 deletion mutation was moved from ΔSTM1294 S. Typhimurium into the unmarked ΔSTM3106 S. Typhimurium strain by bacteriophage P22-mediated transduction, yielding an S. Typhimurium strain lacking both STM1294 and STM3106 (ΔSTM1294 ΔSTM3106 strain). A similar approach was used to delete STM1294 in S. Typhimurium strain IR715 lacking STM3106 (14).
The bacterial gene expression vector pBAD18-Cm was used to generate isogenic derivatives of ΔSTM3993–8, ΔSTM1294, and ΔSTM1294 ΔSTM3106 S. Typhimurium strains carrying plasmids bearing the STM1294, STM3993, STM3994, STM3995, STM3996, STM3997, or STM3998 gene. The open reading frames corresponding to these genes were amplified from the chromosome of S. Typhimurium strain 14028 by PCR with gene-specific primer pairs (Table S1). Restriction sites for endonucleases NheI and XbaI were incorporated into the primer sequences to facilitate cloning of the PCR products into pBAD18-Cm immediately downstream of the arabinose-inducible araC promoter. The resulting constructs (or empty vector) were verified by DNA sequencing and introduced into the ΔSTM3993–8, ΔSTM1294, or ΔSTM1294 ΔSTM3106 S. Typhimurium strain by electroporation. Transformants were selected for on lysogeny agar supplemented with chloramphenicol. Isolated single colonies were picked, and the presence of the plasmids was confirmed. The resulting strains grew normally in lysogeny broth (supplemented with chloramphenicol) following induction with l-arabinose (data not shown).
Site-directed mutagenesis was used to introduce site-specific mutations into STM3106, resulting in an alanine substitution for cysteine at position 99 or 127. Site-directed mutagenesis was performed on plasmid pBAD-STM3106 (pSTM3106) by use of Q5 DNA polymerase (New England BioLabs, Ipswich, MA, USA) and primer pair STM3106.C99A-F and STM3106.C99A-R (for position 99) or primer pair STM3106.C127A-F and STM3106.C127A-R (for position 127). After PCR, the parental DNA was digested by use of DpnI endonuclease (Table S1). The resulting constructs, pSTM3106_C99A and pSTM3106_C127A, were verified by DNA sequencing and introduced into ΔSTM3106 S. Typhimurium by electroporation. Transformants were selected for on lysogeny agar supplemented with chloramphenicol. Isolated single colonies were picked, and the presence of the plasmids was confirmed. The resulting strains grew normally in lysogeny broth (supplemented with chloramphenicol) following induction with l-arabinose (data not shown). To introduce two site-specific mutations into STM3106, resulting in alanine substitutions for cysteine residues at positions 99 and 127, site-directed mutagenesis was performed on plasmid pSTM3106_C127A by use of the aforementioned PCR-based approach and primers STM3106.C99A-F and STM3106.C99A-R.
Bacterial growth curve assays.
Overnight cultures were washed twice with sterile water and diluted (1:200) into fresh lysogeny broth. Chloramphenicol and l-arabinose were added to the culture medium to maintain plasmids in vivo and to induce expression of plasmid-encoded l-asparaginase II, respectively. Bacteria were grown aerobically at 37°C in a rotary shaker incubator (250 rpm). Bacterial growth was monitored over time by measuring the change in absorbance of the cultures at an optical density of 600 nm (OD600). M9 minimal medium was used where indicated to perform bacterial growth assays under nutrient-limiting conditions. M9 minimal medium was prepared with glycerol (0.2%) as the primary carbon source and ammonium chloride (18.7 mM) or asparagine (12 mM) as the sole nitrogen source.
Bacterial transcript analysis.
Overnight cultures were diluted (1:200) into 3 ml of fresh lysogeny broth. Bacteria were grown aerobically at 37°C until the OD600 reached 0.5, when l-arabinose (5 mM) was added to the cultures to induce expression of plasmid-encoded DsbA. After 1 h of induction, RNA was isolated by use of an RNeasy Protect bacteria minikit (Qiagen, Valencia, CA, USA) and TRI reagent (Molecular Research Center, Cincinnati, OH, USA). A total of 100 ng of RNA was converted into cDNA by use of a Verso cDNA synthesis kit (Thermo Fisher Scientific, Grand Island, NY, USA). Quantitative PCR was performed by use of an ABI 7300 real-time PCR system (Thermo Fisher Scientific), PerfeCTa SYBR green FastMix ROX (QuantaBio, Beverly, MA, USA), and the primer pair qSTM3106-F and qSTM3106-R (for STM3106) or the pair qSTM3835-F and qSTM3835-R (for STM3835). The 2−ΔΔCT (where CT is threshold cycle) method was used to analyze the data and determine relative expression of STM3106 (ansB) normalized to that of STM3835 (gyrB).
T cell enrichment and T cell assay.
Splenocytes harvested from naive C57BL/6J mice (6- to 8-week-old females; The Jackson Laboratory, Bar Harbor, ME, USA) were used as a source of T cells. Following the lysis of red blood cells in ACK lysing buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA, pH 7.2 to 7.4), CD90.2 MicroBeads and magnetic cell separation technology (Miltenyi Biotec, San Diego, CA, USA) were used to enrich for CD90.2+ cells. CD90.2 is a pan-T cell marker expressed by both CD4+ and CD8+ T cells. Enriched populations of T cells were suspended in RP-10 medium (RPMI 1640 medium [Gibco, Grand Island, NY, USA] supplemented with 10% fetal bovine serum, 0.2 M glutamine, 0.1 M HEPES, and 50 μM 2-mercaptoethanol) and used in T cell assays.
To measure the effect of S. Typhimurium on TCR-β surface expression, blastogenesis, and IL-2 secretion, enriched populations of T cells suspended in RP-10 medium supplemented with 1 μg/ml anti-CD28 monoclonal antibody (MAb) (clone E18; BioLegend, San Diego, CA, USA) were seeded at 1 × 105 cells per well into flat-bottom 96-well tissue culture plates coated with 5 μg/ml anti-CD3ε MAb (clone 145-2C11; BioLegend). The T cells were then cultured in the absence or presence of S. Typhimurium at a multiplicity of infection of ∼60. l-Arabinose was added to the cultures for continued induction of plasmid-encoded l-asparaginase II expression by l-arabinose-pretreated S. Typhimurium. After 2 h of incubation at 37°C in 5% CO2, the T cells were pelleted by centrifugation and resuspended in RP-10 medium supplemented with gentamicin (50 μg/ml) and with penicillin and streptomycin (2%) to kill all remaining bacteria. After an additional 20 h of incubation at 37°C in 5% CO2, the T cells were harvested, stained, and analyzed by flow cytometry. Additionally, the culture supernatants were collected, and IL-2 concentrations were determined by IL-2 enzyme-linked immunosorbent assay (ELISA).
Cell staining and analysis by flow cytometry.
All antibodies and reagents described in this section were purchased from BioLegend. Cells were stained in the presence of Fc block (clone 93) with allophycocyanin-conjugated anti-mouse CD90.2 MAb (clone 30-H12), phycoerythrin-conjugated anti-mouse CD25 MAb (clone PC61), and fluorescein isothiocyanate-conjugated anti-mouse TCR-β MAb (clone H57-597). Data were acquired on a FACSCalibur with CellQuest Pro software (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed with FlowJo, version X, software (Tree Star, Ashland, OR, USA).
Live cells expressing CD90.2 and CD25 were identified as anti-CD3ε- and anti-CD28-stimulated T cells. Size and granularity of live cells were analyzed by use of forward and side scatter. Blastogenesis was calculated by determining the percentage of live cells that, in response to treatment with anti-CD3ε and anti-CD28 MAb, had transformed from small lymphocytes into larger, more granular cells resembling blast cells.
IL-2 ELISA.
IL-2 concentrations in collected T cell supernatants were determined by use of a mouse IL-2 ELISA MAX Deluxe kit (BioLegend).
Bacterial fractionation.
Overnight cultures were diluted (1:200) into 55 ml of fresh lysogeny broth and grown aerobically at 37°C until the OD600 reached ∼0.5, after which the cultures were placed on ice to stop bacterial growth. Next, the cultures (50 ml) were subjected to centrifugation (10,000 × g for 10 min at 4°C). To obtain secreted protein fractions, proteins in the resulting supernatants were precipitated with trichloroacetic acid (TCA) and suspended in 1 ml of 1× Laemmli sample buffer. To obtain cytoplasmic and periplasmic fractions, the bacterial pellets were washed in 20 mM Tris (pH 8), suspended in 1 ml of TSE buffer (200 mM Tris, pH 8, 500 mM sucrose, 1 mM EDTA) (38), and incubated for 1 h on ice, after which the samples were subjected to centrifugation (10,000 × g for 20 min at 4°C). The supernatants were collected, yielding periplasmic fractions, and the pellets were resuspended in 1 ml of TSE buffer. The resuspended pellets were sonicated while on ice water by use of a Microson Ultrasonic Cell Disruptor XL (4 cycles of 15 s on and 15 s off; power, 5 [Misonix, Farmingdale, NY, USA]) and subjected to centrifugation (10,000 × g for 10 min at 4°C). The supernatants were collected, yielding cytoplasmic fractions. Laemmli sample buffer (6×) was added to the cytoplasmic and periplasmic fractions at a final concentration of 1×. Aliquots (5 ml) of the bacterial cultures were subjected to centrifugation, and the resulting pellets were suspended in 0.1 ml of 1× Laemmli sample buffer to ultimately yield whole-cell lysates (see below). All samples containing Laemmli sample buffer were then heated to 95°C for 30 min and subjected to PAGE and Western blot analysis.
PAGE and Western blot analysis.
Aliquots of whole-cell lysates, cytoplasmic and periplasmic fractions, and samples containing proteins precipitated from cell-free culture supernatants mixed with Laemmli/SDS sample buffer were boiled for 15 min, subjected to centrifugation to pellet debris, and loaded onto 16% Tris-tricine polyacrylamide gels with discontinuous buffering systems (39). Following gel electrophoresis, proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA) by use of a Trans-Blot SD Semi-Dry Transfer Cell system (Bio-Rad).
Expression of l-asparaginase II was detected with polyclonal antibody (1:10,000) from rabbits immunized with recombinant l-asparaginase II of E. coli (catalog number 21013; Abcam, Cambridge, MA, USA). Expression of the chaperone protein DnaK was detected with MAb (1:30,000) from mice immunized with recombinant DnaK of E. coli (clone 8E2/2; Abcam). Expression of maltose-binding protein (MBP) was detected with MAb (1:2,000) from rats immunized with recombinant MBP of E. coli (clone YM-2; BioLegend, San Diego, CA, USA).
For detection, horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibodies (catalog number 7074; Cell Signaling Technology, Danvers, MA, USA), goat anti-mouse IgG secondary antibodies (170-5047; Bio-Rad), or goat anti-rat IgG secondary antibodies (112-035-003; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were used (1:10,000) in combination with ECL Western blotting detection reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA). An ImageQuant LAS 500 imaging system (GE Healthcare Life Sciences) was used for chemiluminescence detection and gel documentation. Electrophoretic densitometry (Image Studio Lite; Li-Cor, Lincoln, NE, USA) was used to quantify the relative levels of l-asparaginase II normalized to DnaK.
Macrophage culture and macrophage infection assay.
Bone marrow-derived macrophages were cultured from 129X1/SvJ mice, as described previously (40). Briefly, bone marrow cells isolated from femurs of 129X1/SvJ mice were suspended in medium (Dulbecco's modified Eagle's medium [DMEM] with 4.5 g/liter glucose [Gibco] supplemented with 20% heat-inactivated fetal bovine serum [FBS; Atlanta Biologicals, Lawrenceville, GA, USA], 30% L-cell conditioned medium, 2 mM glutamine, and 1 mM sodium pyruvate) and seeded into 100-mm-diameter petri dishes at 4 × 106 cells per plate. After 5 days of incubation at 37°C in 5% CO2, macrophages were collected and used for infection assays.
Macrophage infection assays were performed by use of a standard gentamicin protection assay. Briefly, bone marrow-derived macrophages were suspended in medium (same as above, except that concentrations of FBS and L-cell conditioned medium were reduced to 10% and 15%, respectively) and seeded at 1.5 × 106 cells per ml per well into a 24-well tissue culture plate. After overnight incubation at 37°C in 5% CO2, the medium was replaced with 0.5 ml of fresh medium, and the macrophages were infected with bacteria at a multiplicity of infection of 1. Upon addition of bacteria, the plate was centrifuged for 5 min at 1,000 rpm to facilitate bacterial contact with the macrophage monolayer. After 20 min of incubation at 37°C in 5% CO2, the wells were washed three times with 1 ml of phosphate-buffered saline (PBS) to remove non-cell-associated bacteria, after which fresh medium supplemented with gentamicin (25 μg/ml) was added to kill all extracellular bacteria. This was referred to as the 0-h time point. After 1 and 18 h of incubation at 37°C in 5% CO2, the wells were washed three times with 1 ml of PBS to remove the gentamicin-containing medium, and the macrophages were lysed by use of 0.5 ml of Triton X-100 (0.1%) to release intracellular bacteria. Viable bacteria were enumerated by plating for CFU onto lysogeny agar. To determine the number of cell-associated bacteria at the 0-h time point, macrophages were lysed immediately following the initial washes with PBS, after which viable bacteria were enumerated by plating for CFU onto lysogeny agar.
Murine infections.
Naive, 8- to 10-week-old female 129X1/SvJ mice (The Jackson Laboratory) were inoculated intragastrically with 1 × 108 CFU of a 1:1 mixture of differentially marked wild-type and ΔSTM1294 ΔSTM3106 S. Typhimurium (in the IR715 strain background) suspended in 0.1 ml of phosphate-buffered saline. To improve the consistency of intragastric infections, food (but not water) was removed 6 to 8 h prior to inoculation. Immediately following inoculation, food was provided ad libitum. Tenfold serial dilutions of the inoculum were plated to confirm the inoculum titer and input ratio. At various times after inoculation, mice were euthanized, and target tissues (i.e., mesenteric lymph nodes, liver, and spleen) were harvested and processed to determine bacterial loads. S. Typhimurium bacteria were enumerated by serial dilution and plating. Competitive indices were calculated by dividing the number of ΔSTM1294 ΔSTM3106 S. Typhimurium (nalidixic acid and kanamycin resistant) bacteria by the number of wild-type S. Typhimurium (nalidixic acid resistant) bacteria normalized to the input ratio.
Statistical analysis.
Statistical analysis was performed with Prism, version 6 (GraphPad Software, La Jolla, CA, USA). Data were analyzed by one-way analysis of variance (ANOVA) with Dunnett's multiple comparisons posttest, repeated-measures one-way ANOVA with Dunnett's or Tukey's multiple comparisons posttest, or repeated measures two-way ANOVA with Sidak or Tukey's multiple comparisons posttest. A P value of <0.05 was considered to be statistically significant (see figure legends for detailed significance levels).
Supplementary Material
ACKNOWLEDGMENTS
We thank James Bliska, Nicholas Carpino, Wali Karzai, Hao Shen, and members of the van der Velden laboratory for helpful discussions. We also thank Jorge Benach and David Thanassi for constructive feedback on the manuscript.
This research was supported by NIH grant R01AI101221. In addition, NIH provided funding to A.W.M.V.D.V. under grants R21AI092165 and P01AI055621, to H.A.-P. under grants R01AI083646 and R21AI083964, and to H.A.-P. and M.M. under grant R01AI075093. USDA provided funding to M.M. and H.A.-P. under grant number 2009-03579. The U.S. Department of Defense provided funding to M.M. under grant W81XWH-08-1-0720.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00740-16.
REFERENCES
- 1.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O'Brien SJ, Jones TF, Fazil A, Hoekstra RM, International Collaboration on Enteric Disease “Burden of Illness” Studies . 2010. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis 50:882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 2.Crump JA, Mintz ED. 2010. Global trends in typhoid and paratyphoid fever. Clin Infect Dis 50:241–246. doi: 10.1086/649541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon MA. 2011. Invasive nontyphoidal Salmonella disease: epidemiology, pathogenesis and diagnosis. Curr Opin Infect Dis 24:484–489. doi: 10.1097/QCO.0b013e32834a9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilchrist JJ, MacLennan CA, Hill AVS. 2015. Genetic susceptibility to invasive Salmonella disease. Nat Rev Immunol 15:452–463. doi: 10.1038/nri3858. [DOI] [PubMed] [Google Scholar]
- 5.Kirk MD, Pires SM, Black RE, Caipo M, Crump JA, Devleesschauwer B, Dopfer D, Fazil A, Fischer-Walker CL, Hald T, Hall AJ, Keddy KH, Lake RJ, Lanata CF, Torgerson PR, Havelaar AH, Angulo FJ. 2015. World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med 12:e1001940. doi: 10.1371/journal.pmed.1001940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pegues DA, Miller SI. 2015. Salmonella species, p 2559–2568. In Bennett JE, Dolin R, Blaser MJ (ed), Mandell, Douglas, and Bennett's principles and practice of infectious diseases, 8th ed Elsevier, Philadelphia, PA. [Google Scholar]
- 7.Keestra-Gounder AM, Tsolis RM, Baumler AJ. 2015. Now you see me, now you don't: the interaction of Salmonella with innate immune receptors. Nat Rev Microbiol 13:206–216. doi: 10.1038/nrmicro3428. [DOI] [PubMed] [Google Scholar]
- 8.Dougan G, John V, Palmer S, Mastroeni P. 2011. Immunity to salmonellosis. Immunol Rev 240:196–210. doi: 10.1111/j.1600-065X.2010.00999.x. [DOI] [PubMed] [Google Scholar]
- 9.McSorley SJ. 2014. Immunity to intestinal pathogens: lessons learned from Salmonella. Immunol Rev 260:168–182. doi: 10.1111/imr.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsolis RM, Xavier MN, Santos RL, Baumler AJ. 2011. How to become a top model: impact of animal experimentation on human Salmonella disease research. Infect Immun 79:1806–1814. doi: 10.1128/IAI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedoui S, Kupz A, Wijburg OL, Walduck AK, Rescigno M, Strugnell RA. 2010. Different bacterial pathogens, different strategies, yet the aim is the same: evasion of intestinal dendritic cell recognition. J Immunol 184:2237–2242. doi: 10.4049/jimmunol.0902871. [DOI] [PubMed] [Google Scholar]
- 12.Bueno SM, Riquelme SA, Riedel CA, Kalergis AM. 2012. Mechanisms used by virulent Salmonella to impair dendritic cell function and evade adaptive immunity. Immunology 137:28–36. doi: 10.1111/j.1365-2567.2012.03614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenstein TK. 2001. Implications of Salmonella-induced nitric oxide (NO) for host defense and vaccines: NO, an antimicrobial, antitumor, immunosuppressive and immunoregulatory molecule. Microbes Infect 3:1223–1231. doi: 10.1016/S1286-4579(01)01482-4. [DOI] [PubMed] [Google Scholar]
- 14.Kullas AL, McClelland M, Yang HJ, Tam JW, Torres A, Porwollik S, Mena P, McPhee JB, Bogomolnaya L, Andrews-Polymenis H, van der Velden AWM. 2012. l-Asparaginase II produced by Salmonella Typhimurium inhibits T cell responses and mediates virulence. Cell Host Microbe 12:791–798. doi: 10.1016/j.chom.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torres A, Luke JD, Kullas AL, Kapilashrami K, Botbol Y, Koller A, Tonge PJ, Chen EI, Macian F, van der Velden AWM. 2016. Asparagine deprivation mediated by Salmonella asparaginase causes suppression of activation-induced T cell metabolic reprogramming. J Leukoc Biol 99:387–398. doi: 10.1189/jlb.4A0615-252R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porwollik S, Santiviago CA, Cheng P, Long F, Desai P, Fredlund J, Srikumar S, Silva CA, Chu WP, Chen X, Canals R, Reynolds MM, Bogomolnaya L, Shields C, Cui P, Guo JB, Zheng Y, Endicott-Yazdani T, Yang HJ, Maple A, Ragoza Y, Blondel CJ, Valenzuela C, Andrews-Polymenis H, McClelland M. 2014. Defined single-gene and multi-gene deletion mutant collections in Salmonella enterica sv Typhimurium. PLoS One 9:e99820. doi: 10.1371/journal.pone.0099820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatahet F, Boyd D, Beckwith J. 2014. Disulfide bond formation in prokaryotes: history, diversity and design. Biochim Biophys Acta 1844:1402–1414. doi: 10.1016/j.bbapap.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swain AL, Jaskolski M, Housset D, Rao JK, Wlodawer A. 1993. Crystal structure of Escherichia coli l-asparaginase, an enzyme used in cancer therapy. Proc Natl Acad Sci U S A 90:1474–1478. doi: 10.1073/pnas.90.4.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cedar H, Schwartz JH. 1967. Localization of the two-l-asparaginases in anaerobically grown Escherichia coli. J Biol Chem 242:3753–3755. [PubMed] [Google Scholar]
- 20.Gouzy A, Larrouy-Maumus G, Bottai D, Levillain F, Dumas A, Wallach JB, Caire-Brandli I, de Chastellier C, Wu TD, Poincloux R, Brosch R, Guerquin-Kern JL, Schnappinger D, de Carvalho LPS, Poquet Y, Neyrolles O. 2014. Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PLoS Pathog 10:e1003928. doi: 10.1371/journal.ppat.1003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gesbert G, Ramond E, Rigard M, Frapy E, Dupuis M, Dubail I, Barel M, Henry T, Meibom K, Charbit A. 2014. Asparagine assimilation is critical for intracellular replication and dissemination of Francisella. Cell Microbiol 16:434–449. doi: 10.1111/cmi.12227. [DOI] [PubMed] [Google Scholar]
- 22.Popp J, Noster J, Busch K, Kehl A, zur Hellen G, Hensel M. 2015. Role of host cell-derived amino acids in nutrition of intracellular Salmonella enterica. Infect Immun 83:4466–4475. doi: 10.1128/IAI.00624-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jelsbak L, Hartman H, Schroll C, Rosenkrantz JT, Lemire S, Wallrodt I, Thomsen LE, Poolman M, Kilstrup M, Jensen PR, Olsen JE. 2014. Identification of metabolic pathways essential for fitness of Salmonella Typhimurium in vivo. PLoS One 9:e101869. doi: 10.1371/journal.pone.0101869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali MM, Newsom DL, Gonzalez JF, Sabag-Daigle A, Stahl C, Steidley B, Dubena J, Dyszel JL, Smith JN, Dieye Y, Arsenescu R, Boyaka PN, Krakowka S, Romeo T, Behrman EJ, White P, Ahmer BMM. 2014. Fructose-asparagine is a primary nutrient during growth of Salmonella in the inflamed intestine. PLoS Pathog 10:e1004209. doi: 10.1371/journal.ppat.1004209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibayama K, Takeuchi H, Wachino J, Mori S, Arakawa Y. 2011. Biochemical and pathophysiological characterization of Helicobacter pylori asparaginase. Microbiol Immunol 55:408–417. doi: 10.1111/j.1348-0421.2011.00333.x. [DOI] [PubMed] [Google Scholar]
- 26.Hofreuter D, Novik V, Galan JE. 2008. Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host Microbe 4:425–433. doi: 10.1016/j.chom.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Philips JA, Ernst JD. 2012. Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7:353–384. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- 28.Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. 2015. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev 264:74–87. doi: 10.1111/imr.12274. [DOI] [PubMed] [Google Scholar]
- 29.Velin D, Straubinger K, Gerhard M. 2016. Inflammation, immunity, and vaccines for Helicobacter pylori infection. Helicobacter 21:26–29. doi: 10.1111/hel.12336. [DOI] [PubMed] [Google Scholar]
- 30.Janssen R, Krogfelt KA, Cawthraw SA, van Pelt W, Wagenaar JA, Owen RJ. 2008. Host-pathogen interactions in Campylobacter infections: the host perspective. Clin Microbiol Rev 21:505–518. doi: 10.1128/CMR.00055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowley SC, Elkins KL. 2011. Immunity to Francisella. Front Microbiol 2:26. doi: 10.3389/fmicb.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shrivastava A, Khan AA, Khurshid M, Kalam MA, Jain SK, Singhal PK. 2016. Recent developments in l-asparaginase discovery and its potential as anticancer agent. Crit Rev Oncol Hematol 100:1–10. doi: 10.1016/j.critrevonc.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 33.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 34.Stojiljkovic I, Baumler AJ, Heffron F. 1995. Ethanolamine utilization in Salmonella Typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol 177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santiviago CA, Reynolds MM, Porwollik S, Choi SH, Long F, Andrews-Polymenis HL, McClelland M. 2009. Analysis of pools of targeted Salmonella deletion mutants identifies novel genes affecting fitness during competitive infection in mice. PLoS Pathog 5:e1000477. doi: 10.1371/journal.ppat.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of FLP-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 38.Quan S, Hiniker A, Collet J-F, Bardwell JCA. 2013. Isolation of bacteria envelope proteins, p 359–366. In Delcour HA. (ed), Bacterial cell surfaces: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 39.Schagger H. 2006. Tricine-SDS-PAGE. Nat Protoc 1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 40.Pujol C, Bliska JB. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun 71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.