

Significance
Alzheimer’s disease (AD) is the most common form of dementia, but the cause of AD remains poorly understood. Using highly purified recombinant γ-secretase, we examined the effect of 138 AD-derived presenilin-1 (PS1) mutations on the production of β-amyloid peptides (Aβ42 and Aβ40). These 138 mutations cover virtually all AD-targeted amino acids in PS1. Our results reveal no significant correlation between the Aβ42/Aβ40 ratio produced by a γ-secretase variant with a specific PS1 mutation and the mean age at onset of patients carrying this mutation. The comprehensive characterization of pathogenic PS1 mutations serves as a valuable resource for the analysis of γ-secretase activities and AD pathogenesis.
Keywords: Alzheimer’s disease, γ-secretase, Aβ peptides, cleavage activity, amyloid hypothesis
Abstract
A hallmark of Alzheimer’s disease (AD) is the aggregation of β-amyloid peptides (Aβ) into amyloid plaques in patient brain. Cleavage of amyloid precursor protein (APP) by the intramembrane protease γ-secretase produces Aβ of varying lengths, of which longer peptides such as Aβ42 are thought to be more harmful. Increased ratios of longer Aβs over shorter ones, exemplified by the ratio of Aβ42 over Aβ40, may lead to formation of amyloid plaques and consequent development of AD. In this study, we analyzed 138 reported mutations in human presenilin-1 (PS1) by individually reconstituting the mutant PS1 proteins into anterior-pharynx–defective protein 1 (APH-1)aL–containing γ-secretases and examining their abilities to produce Aβ42 and Aβ40 in vitro. About 90% of these mutations lead to reduced production of Aβ42 and Aβ40. Notably, 10% of these mutations result in decreased Aβ42/Aβ40 ratios. There is no statistically significant correlation between the Aβ42/Aβ40 ratio produced by a γ-secretase variant containing a specific PS1 mutation and the mean age at onset of patients from whom the mutation was isolated.
The first case of Alzheimer’s disease (AD) was reported more than 100 y ago, and the brain of the deceased patient contained characteristic senile plaques (1). These plaques were found to be amyloid in nature by electron microscopy (2), and analysis of amino acid sequence revealed the amyloid to be derived from amyloid precursor protein (APP) (3). APP is first cleaved by β-secretase to produce a 99-residue transmembrane fragment C99, which then undergoes additional cleavages by γ-secretase to generate a series of amyloidogenic β-amyloid peptides (Aβ) with 39–43 amino acids (4, 5). The slightly longer Aβ peptides, particularly Aβ42 and Aβ43, are thought to be more prone to aggregation than the shorter ones such as Aβ40 (6–8). Indeed, the amyloid plaques contain mostly longer Aβ peptides such as Aβ42 (9).
Familial cases of AD were found to be genetically linked to missense mutations in APP (10, 11). These observations, together with prior knowledge on amyloid plaques, prompted proposition of the amyloid hypothesis, which regards the formation of β-amyloid plaques as the cause of AD through apoptosis induction of neuronal cells (12). Consistent with this hypothesis, Aβ42 was found to induce cell death (13, 14). Human presenilin-1 (PS1) was cloned and found to be targeted by missense mutations in early-onset familial AD (EOFAD) (15). Remarkably, these EOFAD-linked PS1 mutants led to elevated molar ratios of Aβ42 over Aβ40 both in cell lines and in the brains of transgenic animals (16), and the plasma levels of Aβ42 and Aβ43 in FAD patients with PS1 or APP mutations were significantly increased compared with those of healthy individuals (17). These observations led to refinement of the amyloid hypothesis, which identifies increased ratios of longer Aβ peptides over shorter ones as a key early event in AD development (18). The increased molar ratio of Aβ42 over Aβ40 is widely used as a surrogate indicator for the pathogenic effect of presenilin and APP mutations.
Deficiency of PS1 in neuronal cultures caused a fivefold drop in Aβ production (19). Two transmembrane aspartate residues in PS1 were found to be essential for the endoproteolytic maturation of PS1 and the cleavage of C99, linking PS1 to γ-secretase (20). The γ-secretase activity was copurified with a PS1-containing complex (21); photoactivated inhibitors of γ-secretase specifically labeled the active site of PS1, identifying PS1 as a component of γ-secretase (22). The other three components of γ-secretase—presenilin enhancer protein 2 (PEN-2), anterior-pharynx–defective protein 1 (APH-1), and Nicastrin (NCT)—were subsequently identified (23, 24). Because formation of amyloid plaques is a hallmark of AD, γ-secretase is naturally linked to AD. Specifically, the increased molar ratio of Aβ42 over Aβ40 as a potential causal factor of AD is attributed to altered proteolytic activity of γ-secretase.
Despite its popularity, the amyloid hypothesis is yet to be experimentally proven and is challenged by unexplained observations (25, 26). Because PS1 is required for Aβ generation, it was initially suggested that AD-derived mutations might reflect a gain of function (19). However, FAD-derived mutations appeared to decrease γ-secretase activity in vitro and in cells (27–31). Enzymatic inhibition of γ-secretase in the clinic failed to alleviate the symptoms of AD patients (32). Thus, the overall protease activity of γ-secretase is no longer considered a causal factor for AD. The altered ratio of Aβ peptides, most frequently referred to as the increased ratios of Aβ42 or Aβ43 over Aβ40, is thought to induce formation of the β-amyloid plaque and consequently lead to AD. Unfortunately, the appearance and amount of β-amyloid plaques correlate poorly with the development and severity of AD, and normal individuals may contain β-amyloid plaques in the brain (33, 34). PS1 mutations have also been found in frontotemporal dementia, which exhibits the same functional defect but lacks amyloid pathology (35–37). Finally, if γ-secretase activity underlies AD genesis, one would expect to find disease mutations in the other three components of γ-secretase which presumably modulate its protease activity. No AD-associated mutation has been reported in PEN-2, APH-1, or NCT.
In contrast to the amyloid hypothesis, the presenilin hypothesis posits that the loss of normal function by the mutant PS1 causes AD through dominant negative effect on the wild-type allele (38, 39). This hypothesis provides a satisfactory explanation to the puzzling observation that AD-derived mutations are exclusively missense in nature but not nonsense or frameshift. It also potentially explains why heterozygous mouse with one mutant PSEN1 allele, but not PSEN1+/− mouse with one allele deleted, develops memory impairment and neurodegeneration (40). At present, whether AD is caused by loss of function in presenilin (the presenilin hypothesis) or qualitative changes in Aβ production (the amyloid hypothesis) remains controversial (27, 38).
Another important indicator of AD development is the mean age at onset (AAO) for patients with a shared mutation in PS1, PS2, or APP. A more deleterious mutation is likely to be reflected by a lower AAO. Analysis of small subsets of AD-derived mutations supported a strong correlation: the higher the Aβ42/Aβ40 ratio, the lower the AAO (41, 42). In contrast, a different set of PS1 mutations only suggested a weak correlation (43). Notably, each of these conclusions was drawn from a limited number of mutations, lacking statistical significance.
In this study, we report a systematic analysis of 138 AD-derived PS1 mutations, which include all reported amino acids that have been targeted for mutations in AD patients by January 2016. Each of the 138 PS1 mutations was recapitulated in an APH-1aL–containing γ-secretase variant, which was purified to homogeneity and analyzed for its ability to produce Aβ42 and Aβ40. These results reveal no statistically significant correlation between the combined production of Aβ42 and Aβ40 by a γ-secretase variant and the mean AAO for patients with the corresponding mutation, or between the Aβ42/Aβ40 ratio and the mean AAO.
Results
Preliminary Analysis of 10 PS1 Mutations.
To examine the relationship between Aβ42/Aβ40 ratio and the mean AAO, we performed a pilot experiment on 10 representative PS1 mutations, which affect six residues on four transmembrane helices (TMs) and four amino acids from the inter-TM loops. Each of these PS1 variants was reconstituted into γ-secretase for overexpression, purification, and assessment of proteolytic activity using APP-C99 as the substrate (Fig. 1A). APH-1aL, which is widely expressed in both neuronal and glia-enriched regions in the brain (44), was used in all γ-secretase variants. To ensure detection sensitivity, we used a monoclonal antibody-based, fluorescence-detecting AlphaLISA assay.
Fig. 1.

Preliminary analysis of 10 AD-derived PS1 mutations. (A) A schematic diagram of the major workflow in this study. The WT human γ-secretase and 138 variants, each containing an AD-derived mutation in PS1, were individually overexpressed and purified. The peak fractions were incubated with APP-C99 and examined for the production of Aβ42 and Aβ40 using the AlphaLISA assay. (B) Effect of 10 AD-derived PS1 mutations on the cleavage activities of the corresponding γ-secretases. Shown here is the combined production of Aβ42 and Aβ40 peptides by the 10 γ-secretase variants. The activity of WT γ-secretase was normalized as 1. Each experiment was repeated three times, and the SD is shown. (C) Effect of 10 AD-derived PS1 mutations on the generation of Aβ42 and Aβ40. (D) All but two variants show increased molar ratios of Aβ42 over Aβ40 compared with WT γ-secretase. Only the variant A285V displays a slightly lower Aβ42/Aβ40 ratio, and S365A is nearly identical to WT γ-secretase. (E) The combined production of Aβ42 and Aβ40 by each of the 10 γ-secretase variants shows no obvious correlation with the mean AAO. (F) The Aβ42/Aβ40 ratio exhibits no obvious correlation with the mean AAO.
The combined production of Aβ42 and Aβ40 was decreased for 7 of the 10 variants and increased for three: V272A, S365A, and L424V (Fig. 1B). The variants S365A and L424V also increased production of Aβ40 compared with WT γ-secretase, whereas six variants, including S365A and L424V, increased Aβ42 production (Fig. 1C). Eight of these 10 variants exhibit increased ratios of Aβ42 over Aβ40 compared with WT γ-secretase (Fig. 1D). Among the other two variants, S365A displays the same Aβ42/Aβ40 ratio as WT γ-secretase, and A285V has a slightly lower ratio of 0.84 ± 0.08 (Table S1).
Table S1.
Summary of biochemical characterization of 138 γ-secretase variants with PS1 mutations
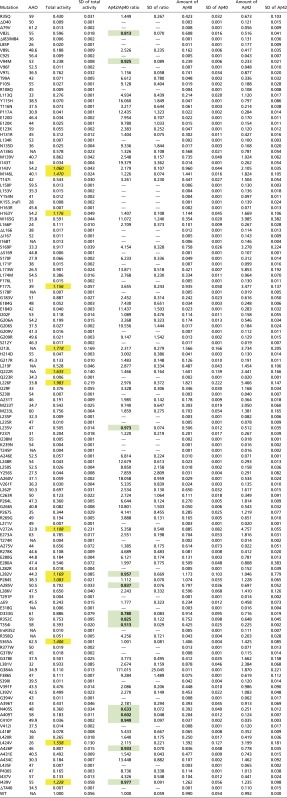 |
“Total activity” stands for the combined production of Aβ42 and Aβ40. The result of Aβ42 production, Aβ40 production, combined production of Aβ42 and Aβ40, and Aβ42/Aβ40 ratio for the WT γ -secretase is normalized as 1. NA, not available. The mutations with increased total production of Aβ40 and Aβ42 are italic with yellow shading. The mutations with decreased Aβ42 over Aβ40 ratios are bold with green shading.
The combined production of Aβ42 and Aβ40 by these 10 γ-secretase variants was plotted against the mean AAOs that correspond to the PS1 mutations, revealing no obvious correlation (Fig. 1E). For example, two variants with increased production are placed on two extreme ends of the AAO range, with L424V and S365A corresponding to AAOs of 26 and 62.5 y, respectively (45, 46). Next, the Aβ42/Aβ40 ratios were plotted against the mean AAOs, again revealing no obvious correlation (Fig. 1F). The mutation L424V yields an Aβ42/Aβ40 ratio of 2.12 ± 0.22, whereas the mutation F105I from patients with an AAO of 55 y corresponds to a ratio of 9.13 ± 0.40 (47).
Experimental Design and Selection of PS1 Mutations.
Our preliminary analysis suggested a lack of correlation between the Aβ42/Aβ40 ratio for a specific γ-secretase variant and the mean AAO of the patients from whom the corresponding PS1 mutation was isolated. To reach a statistically significant conclusion, we must include a much larger set of AD-derived mutations in PS1. By January 2016, there had been 228 reported mutations in PS1 that were derived from AD patients (www.alzforum.org). These mutations affect 121 amino acids in PS1, among which 58 were each mutated to two or more types of amino acids (Fig. S1A). To ensure a comprehensive, unbiased analysis of PS1, we included at least one mutation for each disease-targeted amino acid. For 13 PS1 residues that are mutated to multiple types of amino acids, we chose two or three mutations for investigation (Fig. S1B). Altogether, 138 distinct mutations were selected for in-depth analysis, covering all 121 AD-targeted residues in PS1. These 138 mutations include those that were subjected to preliminary analysis (Fig. 1).
Fig. S1.

AD-derived mutations in PS1. (A) A topology diagram of PS1. The residues, each mutated to only one type of amino acid, are colored red. The residues, each mutated to two or more types of amino acid, are colored yellow. (B) A flowchart on the selection of 138 PS1 mutations for in-depth analysis. These 138 mutations include 129 missense mutations, seven truncations, and two insertions. All 121 AD-targeted amino acids are covered by these 138 mutations.
These 138 PS1 variants were individually cloned and coexpressed with the other three components of γ-secretase in mammalian cells (48). The 138 resulting γ-secretase variants, each containing a specific PS1 mutant and APH-1aL, were individually purified to homogeneity. All 138 variants were eluted from gel filtration with a nearly identical elution volume, suggesting that none of the AD-associated PS1 mutations disrupts the folding or overall structure of γ-secretase. Thus, these mutations likely affect the functional aspects of PS1 and γ-secretase.
Analysis by SDS/PAGE revealed five discrete bands for the WT γ-secretase (Fig. S2, lane 1-9) and for the vast majority of the variants (Figs. S2 and S3A, lanes 1-1 through 16-9). These five bands correspond to NCT (120–150 kDa), the N-terminal fragment of PS1 (PS1-NTF, 34 kDa), APH-1aL (29 kDa), the C-terminal fragment of PS1 (PS1-CTF, 19 kDa), and PEN-2 (12 kDa). The apparent size of PEN-2 is larger than the predicted molecular weight, in part due to presence of the FLAG affinity tag. The endoproteolytic processing of PS1 is incomplete for some γ-secretase variants exemplified by G209V, L235R, and E280G (Fig. S2, lanes 1-2, 1-5, and 1-6). Eight of these γ-secretase variants nearly abrogated endoproteolysis, as evidenced by the intact PS1 and missing NTF/CTF bands (Fig. S4A).
Fig. S2.

WT γ-secretase and variants used in this study. WT γ-secretase and variants are visualized on SDS/PAGE gels by Coomassie staining. For most variants, five polypeptide bands are clearly visible, representing Nicastrin (NCT), the N-terminal fragment (NTF) of PS1, APH-1aL, the C-terminal fragment (CTF) of PS1, and PEN-2. Each variant is identified by a specific PS1 mutation labeled at the top of the lane. Each lane is identified by two numbers, with the first representing the gel and the second specifying location within the gel. For example, WT γ-secretase is in lane 1-9 (gel 1, lane number 9).
Fig. S3.

γ-secretase and variants used in this study. (A) γ-secretase variants are visualized on SDS/PAGE gels by Coomassie staining. (B) Thirteen engineered γ-secretase variants used in this study are visualized on SDS/PAGE gels by Coomassie staining.
Fig. S4.

Eight γ-secretase variants abolish autoproteolysis. (A) These eight γ-secretase variants are visualized on a SDS/PAGE gel by Coomassie staining. In contrast to WT γ-secretase, these variants nearly abrogated endoproteolysis, as evidenced by the intact PS1 and missing NTF/CTF bands. (B) These eight variants exhibit severely compromised cleavage activities compared with WT γ-secretase. (C) Seven of the eight residues affected by these mutations map to the vicinity of the active site in PS1. Shown here are two perpendicular views of PS1, with the mutated residues colored red. The two catalytic aspartate residues are shown in spheres.
Generation of Aβ42 and Aβ40 by 138 γ-Secretase Variants.
The 138 γ-secretase variants were individually examined for their abilities to cleave APP-C99 into Aβ42 and Aβ40 using the AlphaLISA assay (Table S1). Compared with WT γ-secretase, only 10 variants increased production of Aβ40 (Fig. 2). The largest increase of 82% for Aβ40 production comes from the variant L226F, which replaces Leu226 by Phe in TM5 of PS1. All 10 variants with increased Aβ40 production, hereafter referred to as TOP10, yielded higher amounts of Aβ42 and Aβ40 (Fig. 3A). All 10 variants also enhanced production of Aβ42; except L282V and I439V, the extent of increased production for Aβ42 is greater than that for Aβ40 (Fig. 2). These eight variants exhibit increased ratios of Aβ42 over Aβ40 (Fig. 3B).
Fig. 2.

Effect of 138 AD-derived PS1 mutations on the abilities of the corresponding γ-secretase variants to generate Aβ42 and Aβ40. The amounts of Aβ42 and Aβ40 produced by WT γ-secretase are normalized. The relative amounts of Aβ42 and Aβ40 produced by these 138 γ-secretase variants are shown here. The vast majority of these variants exhibit reduced production of both Aβ42 and Aβ40. In particular, production of Aβ42 as well as Aβ40 was abrogated to background levels for 11 γ-secretase variants, including S178P, S212Y, Q223R, S230I, I238M, K239N, T245P, L271V, T274R, T291P, and E318G. Only 10 γ-secretase variants exhibit increased cleavage activities toward both Aβ42 and Aβ40 compared with WT γ-secretase.
Fig. 3.

Effect of 138 AD-derived PS1 mutations on the abilities of the corresponding γ-secretase variants to modulate the Aβ42/Aβ40 ratios. (A) Effect of 138 AD-derived PS1 mutations on the combined production of Aβ42 and Aβ40 by the corresponding γ-secretase variants. The vast majority of the mutations negatively affect the combined production of Aβ42 and Aβ40. Only 13 variants exhibit increased total cleavage activity compared with WT γ-secretase. (B) Effect of 138 AD-derived PS1 mutations on the abilities of the corresponding γ-secretase variants to modulate the Aβ42/Aβ40 ratios. The vast majority of these mutations lead to increased Aβ42/Aβ40 ratios. Only 13 variants exhibit decreased Aβ42/Aβ40 ratios compared with WT γ-secretase.
Compared with WT γ-secretase, 34 variants increased production of Aβ42 (Fig. 2). All other 104 variants produced lower levels of both Aβ42 and Aβ40. Of these 104 variants, 67 exhibit a severely compromised ability to produce Aβ40, which is defined by loss of at least 95% activity compared with WT γ-secretase. By the same criteria, only 14 variants exhibit a severely compromised ability to produce Aβ42, and they represent a subset of the 67 variants with crippled Aβ40 generation (Fig. 2). These results demonstrate that the vast majority of AD-derived PS1 mutations negatively affect the cleavage activities of the resulting γ-secretase variants in vitro. In addition, a PS1 mutation is more likely to decrease the production of Aβ40 than Aβ42 or to increase the generation of Aβ42 than Aβ40. Either scenario would result in an increased ratio of Aβ42 over Aβ40.
Our conclusion is corroborated by analysis of the total cleavage activities toward Aβ42 and Aβ40 (Fig. 3A). Because the molar quantity of Aβ40 generated by WT γ-secretase exceeds that of Aβ42 by ∼10-fold, the combined amount of Aβ42 and Aβ40 is dominated by Aβ40 production. Consequently, although 34 variants increased Aβ42 production, only 13 variants exhibit a higher level of combined Aβ42 and Aβ40 production compared with WT γ-secretase (Fig. 3A). In addition to TOP10, these 13 variants include I143V, F177L, and V272A, each of which has a WT level of Aβ40 but markedly elevated Aβ42 production. Among these 13 variants, 6 have activities that are within 20% range of the WT value, and the most active variant, L226F, has an activity that is ∼98% higher than WT γ-secretase (Fig. 3A). In sharp contrast, 101 variants each display a combined production of Aβ42 and Aβ40 that is less than 50% of that by WT γ-secretase. Of these 101 variants, 57 exhibit a severely compromised ability, with about 42 variants abrogating generation of Aβ42 and Aβ40 altogether. The Aβ42/Aβ40 ratios of these 42 γ-secretase variants cannot be calculated because they only produce background levels of Aβ42 and Aβ40.
Our conclusion is also confirmed by quantification of the Aβ42/Aβ40 molar ratios by the remaining 96 γ-secretase variants (Fig. 3B). Only 13 variants exhibit decreased Aβ42/Aβ40 ratios. Among the 13 variants, 9 have Aβ42/Aβ40 ratios that are within 20% of the normalized WT value, and only 4 variants (D333G, T354I, N405S, and A409T) exhibit ratios that are significantly less than the WT value. The lowest Aβ42/Aβ40 ratio of 0.51 ± 0.03 is associated with the PS1 mutation T354I, and all four corresponding mutations in PS1 affect residues in the CTF. In contrast to the moderate reduction of Aβ42/Aβ40 ratios, 83 variants exhibit higher ratios, of which 64 are at least 100% higher and 31 are at least fourfold higher (Fig. 3B). These 31 variants are hereafter referred to as RATIO31. The highest Aβ42/Aβ40 ratio of 171 ± 25 is associated with the PS1 mutation G384A.
Aβ42/Aβ40 Ratios in Liposome- and Cell-Based Assays.
Our proteolytic cleavage assay was performed on each of the 138 γ-secretase variants in the presence of low amounts of phospholipids and 0.5% zwitterionic detergent CHAPSO. Such mild conditions are expected to maintain the structural integrity as well as biochemical property for most membrane proteins and enzymes. Nevertheless, to rule out the possibility of potentially disruptive effects from the detergents, we performed the cleavage assays in liposomes and in cells. Liposomes were prepared using polar lipid extracts from pig brain, and both WT γ-secretase and 11 variants were individually reconstituted into these liposomes in the presence of the substrate APP-C99 (Fig. 4A). These liposomes, both before and after γ-secretase reconstitution, remained intact as revealed by electron microscopy (Fig. S5).
Fig. 4.

The Aβ42/Aβ40 ratios generated in liposome- and cell-based cleavage assays are similar to those obtained in detergent micelle-based assays. (A) A schematic diagram of the liposome-based cleavage assay. The WT and mutant γ-secretases were individually reconstituted into the liposomes together with APP-C99 and examined for their abilities to generate Aβ42 and Aβ40 using the AlphaLISA assay. (B) The Aβ42/Aβ40 ratios generated in the liposome-based cleavage assays are similar to those obtained in detergent micelle-based assays. Results from 11 representative γ-secretase variants are shown here. (C) A schematic diagram of the cell-based cleavage assay. The CRISPR/Cas9 technology was used to generate PS1−/− N2a cells. The pCAG vectors, each encoding WT or a distinct γ-secretase variant, were individually transfected into N2a cells for assessment of production of Aβ42 and Aβ40. (D) The Aβ42/Aβ40 ratios generated in the cell-based cleavage assays are similar to those obtained in detergent micelle-based assays.
Fig. S5.

Negatively stained electron microscopy images of the (Left) empty liposomes and (Right) γ-secretase-loaded proteoliposomes. These liposomes are mostly intact.
Following overnight incubation of these proteoliposomes at 37 °C, the cleavage products were analyzed by the AlphaLISA assay (Fig. 4A). Compared with the normalized value of WT γ-secretase, the overall results for the 11 variants are quite similar to those obtained using the detergent CHAPSO micelles (Fig. 4B). The ratios of Aβ42/Aβ40 are similar (within 30% difference of each other) between detergent micelle- and liposome-based assays for eight variants, F105I, A246E, L248R, V272A, A285V, S365A, L381V, and L424V. The other three variants exhibit some differences between the two assay conditions, but the trend of the Aβ42/Aβ40 ratios remains the same between the two assay conditions. For example, compared with WT γ-secretase, the variant I229F has a higher Aβ42/Aβ40 ratio under both assay conditions, although the value measured in the liposome-based assay is ∼30% higher than that in detergent micelle (Fig. 4B).
Next, we examined whether similar Aβ42/Aβ40 ratios would be maintained in cell-based assays. Pilot experiments using the mouse neuronal N2a cells revealed significant background cleavage from endogenous γ-secretase. To eliminate the background, we successfully applied the CRISPR/Cas9 technology to knock out the PSEN1 genes in N2a cells (Fig. 4C and Fig. S6). Each of the 11 PS1 variants and the APP-C99 substrate were coexpressed in the PSEN1−/− N2a cells, and the production of Aβ42 and Aβ40 was monitored by the AlphaLISA assay. Compared with WT γ-secretase, the cell-based results are generally comparable to those under detergent micelles (Fig. 4D). The ratios of Aβ42/Aβ40 are similar (within 30% difference of each other) between detergent micelle- and cell-based assays for six variants F105I, A246E, L248R, P264L, L381V, and L424V. The other five variants exhibit some differences between the two assay conditions, but the trend remains the same for two variants. Compared with WT γ-secretase, the variants I229F and V272A each has a higher Aβ42/Aβ40 ratio under both assay conditions, although in each case the value measured in the cell-based assay is more than 30% higher than that in detergent micelle (Fig. 4D). Of the remaining three variants, S365A has a WT-level Aβ42/Aβ40 ratio in detergent micelle and a 35% higher ratio in the cell-based assay.
Fig. S6.

PSEN1 deletion in N2a cells. (A) DNA sequencing results around the targeted deletion region of the coding sequences of PSEN1. Sequencing was performed on genomic DNA derived from WT N2a cells. The dinucleotide CG, present in WT PSEN1, has been designed for deletion. (B) Sequencing of genomic DNA derived from a mixture of PSEN1+/+, PSEN1+/−, and PSEN1−/− N2a cells. (C) Sequencing of genomic DNA derived from the PSEN1−/− N2a cells. (D) The production of Aβ40 is drastically reduced in PSEN1−/− N2a cells.
These results demonstrate that the Aβ42/Aβ40 ratios derived from liposome- and cell-based assays are qualitatively similar to those obtained under detergent micelles. Notably, none of these three conditions would faithfully recapitulate the cleavage environment in patient brain. Both liposome- and cell-based assays may suffer from poor reproducibility and uncertainties associated with lipid composition and/or cell growth status. For convenience and reproducibility, we had chosen the detergent micelle-based assay to assess WT γ-secretase and the 138 variants.
Lack of Correlation Between Aβ42/Aβ40 Ratios and AAOs.
We plotted the observed ratios of Aβ42 over Aβ40 for the 138 γ-secretase variants against the mean AAOs for patients from whom these PS1 mutations were derived (Fig. 5A). Strikingly, there is no obvious correlation between the Aβ42/Aβ40 ratios and the AAOs. A forced fit of these data points into a straight line produced a correlation coefficient of 0.038 and a P value of 0.06, which are statistically insignificant (Fig. S7A). The PS1 mutation L166P corresponds to a very low AAO of 24 y (49), and the corresponding γ-secretase variant exhibits a relatively low Aβ42/Aβ40 ratio of 2.71 ± 0.37. In contrast, the mutation E273A was derived from patients with a mean AAO of 63 y (50), and the corresponding γ-secretase variant has an Aβ42/Aβ40 ratio of 2.55 ± 0.20, similar to that for L166P. The mutation G384A, corresponding to a mean AAO of 39 y (51), has the highest Aβ42/Aβ40 ratio of 171 ± 25 (Fig. 5A). The cleavage activities of these 138 variants, as reflected by combined production of Aβ42 and Aβ40, also display no correlation with the mean AAOs (Fig. 5B).
Fig. 5.

There is no statistically significant correlation between the Aβ42/Aβ40 ratios produced by γ-secretase variants and the mean AAOs. (A) A plot of the Aβ42/Aβ40 ratio versus the mean AAO. Altogether, 90 data points are shown here. For 42 γ-secretase variants, the Aβ42/Aβ40 ratio cannot be computed due to background levels of Aβ42 and/or Aβ40. For six PS1 mutations, there are no reported AAOs. (B) A plot of the total cleavage activity of γ-secretase variant versus the mean AAO. Altogether, 127 data points are shown here. For 11 PS1 mutations, there are no reported AAOs. (C) Higher ratios of Aβ42/Aβ40 correlate with lower AAOs from a group of nine PS1 mutations. The correlation coefficient is 0.99, with a P value of less than 0.001. (D) Lower ratios of Aβ42/Aβ40 correlate with lower AAOs from a group of nine PS1 mutations. The correlation coefficient is 0.92, with a P value of less than 0.001.
Fig. S7.

Analysis of potential correlation between the Aβ42/Aβ40 ratios and the mean AAOs. (A) A forced fit of all data points into a straight line yields a statistically insignificant correlation coefficient of 0.038 and a P value of 0.06. (B) A plot of Aβ42/Aβ40 ratio versus mean AAO for the TOP10 variants. The TOP10 variants exhibit increased production of both Aβ42 and Aβ40. (C) A plot of Aβ42/Aβ40 ratio versus mean AAO for the RATIO31 variants. The RATIO31 variants exhibit the 31 highest ratios of Aβ42/Aβ40.
Because these 138 variants exhibit vastly different enzymatic activities, we wondered whether select subsets of these variants might exhibit meaningful correlation between Aβ42/Aβ40 ratios and mean AAOs. We plotted Aβ42/Aβ40 ratios and mean AAOs for the TOP10 variants, which exhibit increased production for both Aβ42 and Aβ40. The scattered distribution of this plot reveals no statistically significant correlation (Fig. S7B). A similar conclusion was reached for the RATIO31 variants, which exhibit the 31 highest Aβ42/Aβ40 ratios among all variants (Fig. S7C). Previous studies suggested potential correlations between AAO and the Aβ42/Aβ40 ratio on the basis of a small subset of FAD-derived mutations (41–43). We examined the same mutations and obtained similar results despite subtle shift of exact numbers (Fig. S8). Obviously, these correlations lack statistical significance.
Fig. S8.

Comparison with published studies on potential correlations between AAO and the Aβ42/Aβ40 ratio. (A) Five PS1 mutations appear to exhibit a positive correlation between the Aβ42/Aβ40 ratios and AAOs. This is mainly contributed by two mutations, A246E and L250S. These five PS1 mutations were investigated in a published study (43), where a weak correlation between AAO and the Aβ42/Aβ40 ratio was observed. (B) Six PS1 mutations appear to exhibit a negative correlation between the Aβ42/Aβ40 ratios and AAOs. These five PS1 mutations were investigated in a published study (41), where a perfectly negative correlation between AAO and the Aβ42/Aβ40 ratio was noted.
Our systematic analyses of nearly all reported missense mutations from PS1 using the in vitro assay shows no statistically significant correlation between AAOs and Aβ42/Aβ40 ratios. Because these 138 mutations include all AD-targeted residues in PS1, the statistical power embedded in our brute force approach cannot be understated. In fact, conclusions derived from a subset of the data are often erroneous. For example, a subset of nine PS1 mutations give a perfect correlation (R2 = 0.99, P value < 0.001) for the conclusion that higher ratios of Aβ42/Aβ40 correspond to lower AAOs, which is consistent with the amyloid hypothesis (Fig. 5C). Unfortunately, a subset of another nine PS1 mutations also yields an excellent correlation (R2 = 0.92, P value < 0.001) for the opposite conclusion that higher ratios of Aβ42/Aβ40 correspond to higher AAOs (Fig. 5D).
Effect of Random PS1 Mutations on Aβ42/Aβ40 Ratios.
Why do most FAD-derived PS1 mutations result in increased Aβ42/Aβ40 ratios by the corresponding γ-secretase variants? Results of the in vitro cleavage assays strongly suggest that γ-secretase may have evolved to optimize the Aβ42/Aβ40 ratio to a low value; namely, any PS1 mutation is likely to reduce Aβ40 production more than Aβ42 or to increase Aβ42 production more than Aβ40. If this assumption were true, random mutations of PS1 would be expected to mostly lead to increased Aβ42/Aβ40 ratios. To examine this assumption, we randomly chose 13 amino acids for missense mutation. These 13 residues are located in eight TMs of PS1 (Fig. 6A); the vast majority of these 13 mutations involve replacement of conserved amino acids such as L150V and L182V. The 13 corresponding γ-secretase variants were individually purified to homogeneity (Fig. S3B) and subjected to the detergent micelle-based, APP-C99 cleavage assays.
Fig. 6.

γ-secretase variants with engineered PS1 mutations exhibit similar biochemical properties as the variants with AD-derived mutations. (A) Location of the 13 amino acids affected by engineered mutations. Except TM4, each of the other eight TMs contributes at least one residue for mutation. Shown here is a ribbon representation of PS1 structure. The residues targeted for mutation are colored magenta, and the two catalytic Asp residues are shown in spheres. (B) All 13 engineered mutations result in compromised protease activities of the corresponding γ-secretase variants for the generation of both Aβ42 and Aβ40. (C) All 13 engineered mutations lead to decreased levels of protease activity as judged by the combined production of Aβ42 and Aβ40. (D) Seven mutations cause increased ratios of Aβ42/Aβ40 compared with WT. Only three mutations (I140V, I253V, and I414V) have no significant effect on the Aβ42/Aβ40 ratios.
Strikingly, none of these 13 variants produced more Aβ40 or Aβ42 than WT γ-secretase (Fig. 6B). Six variants (P88S, V97F, L182V, S230A, L443V, and Y446H) nearly abrogated production of Aβ40 and diminished generation of Aβ42. Consequently, these six variants exhibit a severely compromised ability to generate the Aβ peptides (Fig. 6C). The Aβ40 production by three variants (P88S, V97F, and Y446H) is at the background level and thus disallows calculation of the Aβ42/Aβ40 ratios. In complete agreement with our assumption, the extent of reduction for Aβ40 is considerably more than that for Aβ42 for most of the 13 variants. Seven variants exhibit increased ratios of Aβ42/Aβ40 compared with WT γ-secretase, and three variants have a ratio that is similar to that by WT γ-secretase (Fig. 6D).
The patterns of Aβ42/Aβ40 production by these 13 randomly generated mutations in PS1 are reminiscent of those by 138 FAD-derived mutations. In both cases, the vast majority of the γ-secretase variants exhibit reduced production of Aβ42 and Aβ40 and increased ratios of Aβ42/Aβ40. A sizable fraction of the variants generate background levels of Aβ42 and/or Aβ40, disallowing calculation of Aβ ratios.
Discussion
Mutations in APP, PS1, and PS2 have been unequivocally linked to the development of AD, and most AD-derived mutations in PS1 result in increased ratios of Aβ42/Aβ40. Although the amyloid hypothesis is yet to be proved, it is supported by these observations and by the finding that oligomeric Aβ peptides induced synapse engulfment by microglia (52). A human monoclonal antibody, aducanumab, was shown to selectively target aggregated Aβ and reduce the amyloid plaque in patients with prodromal or mild AD. A dose-dependent slowing of clinical progression was observed in the phase 1b trial and awaits further confirmation by the ongoing phase 3 trials (53).
Notably, nearly 90% of FAD mutations occur in presenilin, but no mutations have been identified in the other three subunits of γ-secretase. Through knock-in studies, presenilin is shown to play an important role in synaptic plasticity (54–57). Such observations support the presenilin hypothesis, where presenilin mutations are considered to be closely associated with neurodegeneration by impairing the γ-secretase–dependent and –independent PS functions. PS1 may function, either alone or in complex with other proteins, as an ion channel to regulate calcium flux (58–61). Examination of FAD-derived mutations in PS1 identified two mutational hotspots, each located in the center of a four-TM bundle (62). Whether this observation is linked to the function of PS1 awaits further investigation.
The mean AAO is an indicator of disease severity and progression, with a lower AAO suggestive of a more deleterious mutation in PS1 and thus more debilitating disease. If increased ratios of longer Aβ peptides over shorter ones is a key early event in AD development, then one might be able to find a statistically significant correlation between such ratios and AAO. This study is aimed at investigating the effect of PS1 mutations on γ-secretase activity and examining a potential correlation between the Aβ42/Aβ40 ratio and disease progression as represented by AAO. To rule out bias from subsets of PS1 mutations, we collected a large number of reported PS1 mutations and individually generated the corresponding γ-secretase variants. Except 13 mutations, all others led to decreased production of Aβ42 and Aβ40 compared with WT γ-secretase. The vast majority of PS1 mutations lead to increased ratios of Aβ42/Aβ40. The Aβ42/Aβ40 ratios by these γ-secretase variants, each containing a specific PS1 mutation and APH-1aL, show no statistically significant correlation with the mean AAOs for the corresponding mutations.
In this study, APH-1aL was used to reconstitute γ-secretase, and consequently, the general conclusions reported in our study are restricted to APH-1aL–containing γ-secretase. To examine the validity of such conclusions under other settings, we generated 10 γ-secretase variants, each reconstituted with APH-1b and a specific PS1 mutation, and individually examined their abilities to produce Aβ42 and Aβ40. The combined production of Aβ42 and Aβ40 by these APH-1b–containing γ-secretase variants displays a similar trend to that by the APH-1aL–containing γ-secretase (Fig. S9A). No significant correlation was found between the combined production of Aβ42 and Aβ40 and the AAO (Fig. S9B). Except one PS1 mutation (F105I), the APH-1b–containing γ-secretases generally exhibit higher Aβ42/Aβ40 ratios than APH-1aL–containing γ-secretases (Fig. S9C). The trend of Aβ42/Aβ40 ratio changes is quite similar for these two classes of γ-secretases, suggesting general applicability of such preliminary conclusion. No significant correlation between the Aβ42/Aβ40 ratios and AAOs is observed (Fig. S9D).
Fig. S9.

Comparison of Aβ42 and Aβ40 production by 10 γ-secretase variants containing either APH-1aL or APH-1b. (A) A plot of the total amount of Aβ42 and Aβ40 generated by γ-secretase variants containing either APH-1aL or APH-1b. (B) A plot of the total amount of Aβ42 and Aβ40 versus mean AAO for the APH-1b–containing γ-secretase variants. (C) The ratios of Aβ42 over Aβ40 produced by these γ-secretase variants. (D) A plot of the Aβ42/Aβ40 ratio versus mean AAO for the APH-1b–containing γ-secretase variants.
We acknowledge the caveat that our experimental conditions, where homogeneous γ-secretase is used in an artificial in vitro setting, are quite different from the complex circumstances in patient brain, where PSEN1 is heterozygous and potential dominant negative effect may be at work (39). Another complicating factor is the in vivo existence of six distinct γ-secretase complexes formed by different forms of presenilin (PS1 and PS2) and APH-1 (APH-1aL, APH-1aS, and APH-1b), which are localized to different regions of the brain (44, 63). Different Aβ profiles can be generated by the distinct γ-secretase complexes (64). FAD mutations in PS1 may shift its localization and increase the intracellular longer Aβ peptides (65).
In addition, the cleavage of APP-C99 by γ-secretase involves an endopeptidase activity, which generates Aβ48 or Aβ49, and a subsequent carboxypeptidase activity, which produces Aβ45/Aβ42 from Aβ48 and Aβ46/Aβ43/Aβ40 from Aβ49. In our study, we only assessed the production of Aβ42 and Aβ40, which is presumably affected by both endopeptidase and carboxypeptidase activities. Our study does not differentiate between these two activities, but it has been shown that some PS1 mutations have differential effects on these two activities. Some mutations had little effect on the endopeptidase activity but exhibited altered carboxypeptidase activity (66). Five AD-derived PS1 mutations led to elevated Aβ42/Aβ40 ratios using synthetic, longer Aβ peptides, reflecting altered carboxypeptidase activity of γ-secretase (67).
Although our data appear to contradict the amyloid hypothesis, the caveats listed above preclude a definitive conclusion. Notably, we cannot rule out the possibility that AD occurs through several different mechanisms and the amyloid hypothesis may constitute one of these mechanisms. Similarly, the presenilin hypothesis may account for other mechanism(s). In fact, AD, characterized by a pattern of cognitive and functional decline, is a very complex disease, with Aβ accumulating years ahead of any detectable symptom yet triggering a series of cellular and physiological responses. Thus, it is possible that there is no simple or single cause for this complex disease. Regardless of these different scenarios, our results involving characterization of 138 PS1 mutations represent a valuable resource for future investigation of γ-secretase and AD development.
Materials and Methods
Protein Preparation.
Generation of all FAD-derived PS1 mutants and expression and purification of the resulting γ-secretase variants were described as reported (48). The purified γ-secretase was applied to Superose-6 column (GE Healthcare) in 0.1% digitonin and Hepes buffer. The peak fractions were concentrated for activity assays.
Detergent-Based Activity Assays.
Purified 4 μg WT or mutant γ-secretase was mixed with 4 μg APP-C99 substrate in 0.5% CHAPSO, 50 mM Hepes, pH 7.0, 0.1% phosphatidylcholine, and 0.025% phosphatidylethanolamine and incubated in 37 °C for 16 h. Detection of Aβ production by the AlphaLISA assay was performed as described (68).
Liposome-Based Activity Assay.
Porcine brain polar lipid extract (Avanti) was dried by nitrogen gas after being dissolved in a mixture of chloroform and methanol (3:1 vol/vol) to 50 mg⋅mL−1 and then resuspended to 20 mg⋅mL−1 in 25 mM Hepes, pH 7.4, 150 mM NaCl. The lipids were frozen in liquid nitrogen and thawed at room temperature gently for eight cycles followed by extrusion through 0.4 μm filter to yield liposomes. CHAPSO was added to a final concentration at 1% (wt/vol) at 4 °C for 30 min. Purified γ-secretase and APP-C99 were added to the lipids and incubated at 4 °C for 30 min. Detergents were removed by two-step incubation with 400 mg⋅mL−1 Bio-Beads SM2 (Bio-Rad) after which the proteoliposomes were frozen and thawed for five cycles. The proteoliposomes were applied to centrifugation at 100,000 × g for 40 min at 4 °C and washed twice by ice-cold lysis buffer to remove free γ-secretase and substrate. The proteoliposomes were resuspended in lysis buffer to 20 mg⋅mL−1. Integrity of the proteoliposomes was checked by negative staining electron microscopy. The proteoliposomes were incubated at 37 °C for 16 h followed by detection of Aβ40 and Aβ42 using the AlphaLISA assay.
Generation of PSEN1 Knockout Cell Line.
The adherent N2a cell line was passaged and maintained in DMEM (Life Technologies) supplemented with 10% (vol/vol) FBS and MEM NEAA (GIBCO) at 37 °C under 5% (vol/vol) CO2 and 75% relative humidity. SgRNAs targeting mouse PSEN1 gene were designed in silico on a website (crispr.mit.edu), and several candidates were chosen. For generation of the complete knockout cell line, the annealing product of two sgRNA-containing primers (forward: 5′-caccgGCCATGCGGTCCATTCGGGG-3′; reverse: 5′-aaacCCCCGAATGGACCGCATGGCc-3′) was inserted into pSpCas9 vector digested with BbsI. Then, 0.5 μg plasmids were transiently transfected using neofect into ∼70% confluent cells in a 24-well plate. Twenty-four h after transfection, puromycin was added to a final concentration of 2 μg⋅mL−1 for selection. Finite serial dilution method was used to separate a single cell colony in 96-well plate. The cells alive were harvested, and their genome was extracted using tissue gDNA kit (Biomiga). The knockout efficiency was examined using a PCR-based method.
Cell-Based Activity Assay.
Using 0.5 μg neofect with 15-min incubation, 0.25 μg plasmid with a FAD-derived PS1 mutant and 0.25 μg plasmid containing the substrate APP-C99 were premixed and transfected into ∼70% confluent cells in a 24-well plate. Transfected cells were cultured for 60 h. Then the cell medium was harvested, and 2 μL was detected by the AlphaLISA assay as described above.
Acknowledgments
This work was supported by funds from the Ministry of Science and Technology (2014ZX09507003006 to Y.S.) and the National Natural Science Foundation of China (31130002 and 31321062 to Y.S.). L.S. is supported by a postdoctoral fellowship of the Tsinghua–Peking Joint Center for Life Sciences.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 629.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1618657114/-/DCSupplemental.
References
- 1.Alzheimer A. About a peculiar disease of the cerebral cortex. Centralblatt für Nervenheilkunde Psychiatrie. 1907;30:177–179. [Google Scholar]
- 2.Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122(3):1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 4.Estus S, et al. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992;255(5045):726–728. doi: 10.1126/science.1738846. [DOI] [PubMed] [Google Scholar]
- 5.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255(5045):728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 6.Burdick D, et al. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem. 1992;267(1):546–554. [PubMed] [Google Scholar]
- 7.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki N, et al. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 9.Roher AE, et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993;268(5):3072–3083. [PubMed] [Google Scholar]
- 10.Chartier-Harlin MC, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353(6347):844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 11.Mullan M, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1(5):345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 12.Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 13.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J Neurosci. 1993;13(4):1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gschwind M, Huber G. Apoptotic cell death induced by beta-amyloid 1-42 peptide is cell type dependent. J Neurochem. 1995;65(1):292–300. doi: 10.1046/j.1471-4159.1995.65010292.x. [DOI] [PubMed] [Google Scholar]
- 15.Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 16.Borchelt DR, et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 17.Scheuner D, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 18.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Strooper B, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 20.Wolfe MS, et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398(6727):513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 21.Li YM, et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA. 2000;97(11):6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li YM, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405(6787):689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 23.Francis R, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002;3(1):85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 24.Yu G, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407(6800):48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 25.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18(6):794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 26.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chávez-Gutiérrez L, et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31(10):2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentahir M, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006;96(3):732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 29.Shimojo M, Sahara N, Murayama M, Ichinose H, Takashima A. Decreased Abeta secretion by cells expressing familial Alzheimer’s disease-linked mutant presenilin 1. Neurosci Res. 2007;57(3):446–453. doi: 10.1016/j.neures.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Cacquevel M, Aeschbach L, Houacine J, Fraering PC. Alzheimer’s disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified γ-secretase complexes. PLoS One. 2012;7(4):e35133. doi: 10.1371/journal.pone.0035133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilig EA, Xia W, Shen J, Kelleher RJ., 3rd A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J Biol Chem. 2010;285(29):22350–22359. doi: 10.1074/jbc.M110.116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doody RS, et al. Alzheimer’s Disease Cooperative Study Steering Committee Semagacestat Study Group A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 33.Nordberg A. Amyloid plaque imaging in vivo: Current achievement and future prospects. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S46–S50. doi: 10.1007/s00259-007-0700-2. [DOI] [PubMed] [Google Scholar]
- 34.Villemagne VL, et al. The ART of loss: Abeta imaging in the evaluation of Alzheimer’s disease and other dementias. Mol Neurobiol. 2008;38(1):1–15. doi: 10.1007/s12035-008-8019-y. [DOI] [PubMed] [Google Scholar]
- 35.Raux G, et al. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology. 2000;55(10):1577–1578. doi: 10.1212/wnl.55.10.1577. [DOI] [PubMed] [Google Scholar]
- 36.Tang-Wai D, et al. Familial frontotemporal dementia associated with a novel presenilin-1 mutation. Dement Geriatr Cogn Disord. 2002;14(1):13–21. doi: 10.1159/000058328. [DOI] [PubMed] [Google Scholar]
- 37.Dermaut B, et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55(5):617–626. doi: 10.1002/ana.20083. [DOI] [PubMed] [Google Scholar]
- 38.Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104(2):403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ., 3rd Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβ production. J Neurosci. 2013;33(28):11606–11617. doi: 10.1523/JNEUROSCI.0954-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen J, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89(4):629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 41.Duering M, Grimm MO, Grimm HS, Schröder J, Hartmann T. Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol Aging. 2005;26(6):785–788. doi: 10.1016/j.neurobiolaging.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Kumar-Singh S, et al. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27(7):686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 43.Mehta ND, et al. Increased Abeta42(43) from cell lines expressing presenilin 1 mutations. Ann Neurol. 1998;43(2):256–258. doi: 10.1002/ana.410430217. [DOI] [PubMed] [Google Scholar]
- 44.Serneels L, et al. gamma-Secretase heterogeneity in the Aph1 subunit: Relevance for Alzheimer’s disease. Science. 2009;324(5927):639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guerreiro RJ, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. doi: 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robles A, et al. Clinical picture of a patient with a novel PSEN1 mutation (L424V) Am J Alzheimers Dis Other Demen. 2009;24(1):40–45. doi: 10.1177/1533317508324272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raux G, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: An update. J Med Genet. 2005;42(10):793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu P, et al. Three-dimensional structure of human γ-secretase. Nature. 2014;512(7513):166–170. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moehlmann T, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci USA. 2002;99(12):8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamimura K, et al. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160(1):76–81. doi: 10.1016/s0022-510x(98)00219-6. [DOI] [PubMed] [Google Scholar]
- 51.Cruts M, et al. Molecular genetic analysis of familial early-onset Alzheimer’s disease linked to chromosome 14q24.3. Hum Mol Genet. 1995;4(12):2363–2371. doi: 10.1093/hmg/4.12.2363. [DOI] [PubMed] [Google Scholar]
- 52.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sevigny J, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 54.Saura CA, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 55.Guo Q, et al. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: Involvement of calcium and oxyradicals. J Neurosci. 1997;17(11):4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia D, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85(5):967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia D, Kelleher RJ, 3rd, Shen J. Loss of Aβ43 production caused by presenilin-1 mutations in the knockin mouse brain. Neuron. 2016;90(2):417–422. doi: 10.1016/j.neuron.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoo AS, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27(3):561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 59.Tu H, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126(5):981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alattia JR, et al. Highly efficient production of the Alzheimer’s γ-secretase integral membrane protease complex by a multi-gene stable integration approach. Biotechnol Bioeng. 2013;110(7):1995–2005. doi: 10.1002/bit.24851. [DOI] [PubMed] [Google Scholar]
- 61.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275(24):18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 62.Bai XC, et al. An atomic structure of human γ-secretase. Nature. 2015;525(7568):212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai MT, et al. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J Biol Chem. 2003;278(25):22475–22481. doi: 10.1074/jbc.M300974200. [DOI] [PubMed] [Google Scholar]
- 64.Acx H, et al. Signature amyloid β profiles are produced by different γ-secretase complexes. J Biol Chem. 2014;289(7):4346–4355. doi: 10.1074/jbc.M113.530907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sannerud R, et al. Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell. 2016;166(1):193–208. doi: 10.1016/j.cell.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quintero-Monzon O, et al. Dissociation between the processivity and total activity of γ-secretase: Implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry. 2011;50(42):9023–9035. doi: 10.1021/bi2007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014;289(45):31043–31052. doi: 10.1074/jbc.M114.581165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun L, et al. Structural basis of human γ-secretase assembly. Proc Natl Acad Sci USA. 2015;112(19):6003–6008. doi: 10.1073/pnas.1506242112. [DOI] [PMC free article] [PubMed] [Google Scholar]