

Significance
Understanding host–enteric virus interactions has been limited by the inability to culture nontransformed small intestinal epithelial cells and to infect animal models with human viruses. We report epithelial responses in human small intestinal enteroid cultures from different individuals following infection with human rotavirus (HRV), a model enteric pathogen. RNA-sequencing and functional assays revealed type III IFN as the dominant transcriptional response that activates interferon-stimulated genes, but antagonism of the IFN response negates restriction of HRV replication. Exogenously added IFNs reduce HRV replication, with type I IFN being most effective. This highlights a paradox between the strong type III transcriptional response and the weaker functional role of type III IFN in human enteric viral restriction in human small intestinal cultures.
Keywords: enteric virus, interferon, human enteroids, human rotavirus
Abstract
The intestinal epithelium can limit enteric pathogens by producing antiviral cytokines, such as IFNs. Type I IFN (IFN-α/β) and type III IFN (IFN-λ) function at the epithelial level, and their respective efficacies depend on the specific pathogen and site of infection. However, the roles of type I and type III IFN in restricting human enteric viruses are poorly characterized as a result of the difficulties in cultivating these viruses in vitro and directly obtaining control and infected small intestinal human tissue. We infected nontransformed human intestinal enteroid cultures from multiple individuals with human rotavirus (HRV) and assessed the host epithelial response by using RNA-sequencing and functional assays. The dominant transcriptional pathway induced by HRV infection is a type III IFN-regulated response. Early after HRV infection, low levels of type III IFN protein activate IFN-stimulated genes. However, this endogenous response does not restrict HRV replication because replication-competent HRV antagonizes the type III IFN response at pre- and posttranscriptional levels. In contrast, exogenous IFN treatment restricts HRV replication, with type I IFN being more potent than type III IFN, suggesting that extraepithelial sources of type I IFN may be the critical IFN for limiting enteric virus replication in the human intestine.
The human small intestinal epithelium is the primary site of infection and replication for many gastrointestinal pathogens. However, fundamental knowledge about intestinal epithelial cell–pathogen interactions in humans is limited as a result of the impracticality of studying these cells in vivo. Modeling these interactions in vitro is difficult because many human enteric pathogens fail to replicate or replicate poorly in cancer cell lines derived primarily from the human colon (1–4). Human intestinal enteroids (HIEs) represent a new in vitro model of the human small intestinal epithelium; this model was developed following advances in stem cell biology, and it recapitulates many of the biological and physiological properties of the human small intestine in vivo (5, 6). Unlike other in vitro models, HIEs are easily established from nontransformed small intestinal tissue, can be maintained on a long-term basis, and contain a stem cell niche and the diversity of intestinal epithelial cell types (enterocytes, goblet, enteroendocrine, and Paneth cells) present in vivo (6, 7). HIEs allow for comparisons of intestinal host responses across genetically diverse individuals (8) and reproduce many of the in vivo pathophysiological properties of infection by a human enteric viral pathogen, human rotavirus (HRV) (9).
HRVs are the major etiology of severe diarrhea in children under the age of 5 y worldwide, resulting in an estimated 215,000 deaths annually (10). HRVs replicate to high titers in humans but replicate poorly or not at all in small animal models (11, 12). This host restriction of HRV in animal models is based on properties of host and virus and is not unique to HRVs; it is exhibited by other human enteric viruses such as human noroviruses and adenoviruses (12–14). In addition, HRV generally replicates to lower titers than animal rotaviruses in transformed cell lines (2). Therefore, most knowledge of rotavirus–host interactions comes from studies that analyzed animal rotavirus replication in animal models or in transformed cell lines (15–18). Induction of the adaptive immune system and secretion of IgA are critical factors in protecting the host against rotavirus infection based on in vivo models of animal rotavirus infection (19, 20). However, in a naïve host, the adaptive immune system is not fully activated until several days after primary infection (21, 22). Epithelial cells mount an early innate immune response to infection, and this immediate antiviral response is often crucial in limiting viral replication and ensuring tissue viability until the adaptive immune response is activated (22–25).
IFNs are potent antiviral cytokines that function in an autocrine and paracrine fashion following the detection of specific pathogen-associated molecular patterns (PAMPs) (26). Currently, three classes of IFNs have been identified: type I, type II, and type III IFN (27, 28). Type II IFN (IFN-γ) is mainly produced by immune cells, whereas type I (IFN-α/β) and type III IFN (IFN-λ) are produced by immune and epithelial cells and thus are relevant to host–pathogen interactions at epithelial surfaces (27). The type I IFN receptor is expressed on most cell types, whereas the type III IFN receptor is primarily restricted to epithelial cells (29). Type I and III IFNs are generally induced by similar stimuli (30). Stimulation of an IFN receptor leads to the induction of a variety of IFN-stimulated genes (ISGs), and ISG-encoded proteins are effector molecules of the antiviral state (31). One unanswered question is whether unique type I and III IFN expression profiles are produced by the human intestinal epithelium in response to different stimuli or viral pathogens.
Based on studies in mice, the innate response to rotavirus infection is complex and virus strain-specific; homologous (murine) rotavirus replication is insensitive to IFNs whereas type I and III IFNs restrict heterologous (nonmurine) rhesus rotavirus (RRV) replication (17, 32, 33). Host restriction is hypothesized to be the basis for attenuation of an HRV vaccine widely used today, RotaTeq, which is derived from a heterologous bovine strain (34). Although a wealth of knowledge has been gained from mouse studies with nonhuman enteric viruses, these models do not account for evolutionary differences in host and pathogen. Subtle but possibly important differences exist between human and mouse type I and III IFN systems and between human and mouse rotaviruses (18, 35–38), necessitating functional studies of interactions between human host cells and human enteric pathogens.
HIEs represent a new model to study epithelial cell innate immune responses to human enteric pathogens. In this study, we used HIEs to investigate the host response to live and inactivated HRVs, the dsRNA structural analog polyinosinic:polycytidylic acid [poly(I:C)], and exogenous IFNs. Transcriptional responses to HRV infection were examined in cultures established from different individuals. We demonstrate that viral infection induces a conserved type III IFN-regulated ISG transcriptional response in HIEs from different individuals. However, this type III IFN transcriptional response is not translated into a robust protein response and does not restrict viral growth. In contrast, exogenously added IFNs restrict HRV replication, with type I IFN being more effective than type III IFN. HIEs are a valuable model to define human-specific epithelial cell responses to stimuli such as human enteric pathogens and have revealed a difference in our understanding of the role of type I and III IFN as innate mediators in human intestinal epithelial cells.
Results
HIEs Demonstrate Minimal Intraculture Variability by Microarray Analysis, and the Epithelial Transcriptional Response to HRV Infection Is Reproducible Across HIEs from Different People.
HIEs are novel, physiologically active, nontransformed cultures that are increasingly used to study human pathogen–host interactions (5, 8, 9, 39, 40). We first evaluated the variability in the transcriptional response to HRV infection in HIEs by analyzing transcriptomes from one jejunal HIE culture by microarray analysis. Independent HIE cultures (individual j11) were mock-inoculated or inoculated with HRV (strain Ito) at a high multiplicity of infection (MOI). Three technical replicates for each condition were analyzed at 6 and 10 h post infection (hpi), as HRV-infected cells lyse by 14 hpi (9). The percentage of infected cells was 51.4 ± 14.7% (mean ± SD of triplicate cultures) as assessed by flow cytometry, demonstrating that the HRV-infected cultures contain infected cells and uninfected bystander cells. Principal component analysis showed that the first principal component (time point in hpi) explained 42% of the variance in the global gene expression profiles, indicating that time point is an important covariate. The second principal component explained 22% of the variance and discriminated the expression profiles based on infection status (Fig. 1A). Although time and infection status influenced HIE transcriptional profiles, the intraculture transcriptional profiles for a specific combination of time and infection status clustered together.
Fig. 1.

Transcriptional response to HRV infection in HIEs assessed by using whole-genome microarray chips. (A) HIE cultures from one individual (j11) were mock- or HRV-inoculated (MOI of 20). At 6 and 10 hpi, gene expression profiling was performed on 12 independent samples (three technical replicates for each of the four conditions). The x and y axes depict the first and second principal components, which differentiate time point (explanatory variable for 42% of variance) and infection status (explanatory variable for 22% of variance), respectively. Mock- (purple) and HRV-inoculated (orange) samples are plotted at 6 hpi (filled squares) and 10 hpi (open circles). (B) HIEs from three individuals (j2, j3, and j11) were mock- or HRV-inoculated at an MOI of 20. At 6 and 10 hpi, gene expression profiling was performed on HIEs from each patient. Clustering is based on Spearman rank correlation. Each individual's (green) HIE samples are shown as mock- (purple) or HRV-inoculated (orange). (C) Spearman rank correlation (listed within each square) of the infection-induced gene expression fold changes of 20,948 transcripts was calculated for all sample pairs. Intensity of red indicates correlation strength. (D) Venn diagrams depict the overlap among each individual’s top 100 up-regulated genes at 6 and 10 hpi.
We next evaluated interculture variability in the HIE response to HRV infection by microarray analysis. Jejunal HIEs from three individuals (j2, j3, and j11) were mock- or Ito-HRV–infected at a high MOI. The percentages of HRV-infected cells were 52.9% (j2), 43.9% (j3), and 47.7% (j11). Hierarchical clustering of global gene expression profiles accounting for time point revealed that HIEs first clustered by each individual and then by infection status (Fig. 1B), indicating significant baseline variability between HIEs derived from different individuals. To assess if the response to HRV infection was conserved between HIEs from the three different people, we calculated gene expression fold changes with HRV infection across more than 20,000 transcripts from each individual. We observed a significant correlation between HRV-induced transcriptional changes in the three individuals’ HIEs; the lowest correlations of 0.18 at 6 hpi (between j2 and j11) and 0.31 at 10 hpi (between j3 and j11) correspond to P values <2.2 × 10−16 (Fig. 1C). Notably, 18 genes at 6 hpi and 28 genes at 10 hpi ranked in the top 100 up-regulated genes in each of the three HIEs’ transcriptional profiles (P < 10−5 for both time points, permutation-based approach; Materials and Methods and Fig. 1D). Further analysis of these genes revealed that 16 of 18 and 15 of 28 were ISGs at 6 and 10 hpi, respectively (Fig. S1A). Thus, despite a baseline variability at the transcriptional level between HIEs from different individuals, all three mounted a conserved ISG transcriptional response to HRV infection.
Fig. S1.

Further analysis of microarray and RNA-seq results. (A) Depicted are the 18 and 28 overlapping genes induced by HRV infection and identified by microarray analysis at 6 and 10 hpi in Fig. 1D, respectively. (B) Heat-map representation of RNA-seq gene expression data from mock- and HRV-inoculated samples shows differentially expressed genes (FDR < 0.05) during HRV infection. (C) Pathway analysis was performed on all genes identified by RNA-seq with a significant fold change (FDR < 0.05, |log2 fold change| ≥ 1). The top three induced pathways are shown. (D) The 63 genes that were analyzed in C are listed in order of fold change with infection. Blue asterisks in D represent common genes that were also identified by microarray at 6 hpi. In A and D, gene symbols in red indicate ISGs and gene symbols in purple indicate IFNs. Gene symbols in black are neither ISGs nor IFNs. Ensembl gene IDs are shown if no gene symbol is available. (E) Empirical cumulative distribution plot shows log2 fold change (x-axis) and fold change percentile (y-axis), with genes demarcated as ISGs (red) or non-ISGs (blue).
RNA-Sequencing Analysis of HIEs Reveals IFN Signaling Is the Dominant Pathway Induced by HRV Infection.
We next performed an RNA-sequencing (RNA-seq) experiment on mock- and HRV-infected HIE cultures. RNA-seq more accurately measures transcriptional activity than microarrays, particularly of low-expressing genes (41). Because minimal transcriptional variability was observed between samples from the same individual (Fig. 1A), one mock- and one HRV-infected culture of HIEs from two individuals (j2 and j11) were sequenced at 6 hpi. Approximately 25% of the RNA-seq reads in the two infected samples mapped to the rotavirus genome, confirming that the infection was robust (Fig. 2A).
Fig. 2.

Transcriptional response to HRV infection assessed by RNA-seq. HIEs from two individuals (j2 and j11) were mock- or HRV-inoculated (MOI of 20). At 6 hpi, total RNA was extracted and RNA-seq was performed on rRNA-reduced samples. (A) Bar plot depicts relative abundance of reads mapped to the human and rotavirus genomes. (B) Scatter plot depicts mean baseline expression estimates (mock-inoculated samples, 6 hpi) of transcripts from the microarray (x-axis) and RNA-seq (y axis) experiments. (C) Volcano plot depicts the results of the differential gene expression analysis. The x and y axes correspond to the log2 fold change and −log10 q-value (FDR), respectively. (D) Scatter plot depicts basal expression levels in mock-inoculated samples (x-axis) and the log2 fold change (y axis) with infection. ISGs are colored in blue. Gene symbols in C and D indicate specific ISGs (red) or IFNs (purple).
Comparison of microarray and RNA-seq experiments showed significant correlation between baseline gene expression estimates (Spearman correlation R = 0.67, P < 2.2 × 10−16). Transcripts present at levels >8 in the microarray dataset (referred to here as medium- and high-expressing genes) correlated relatively well with transcript levels in the RNA-seq dataset (Fig. 2B). However, there was a wide distribution of transcript levels in the RNA-seq dataset for genes with expression <8 in the microarray dataset, confirming that RNA-seq improved the quantification and resolution of transcript levels for low-expressing genes.
To identify genes involved in the HIE response to infection, differential expression analysis was performed using HIE origin (j2 or j11) and infection status as the explanatory variables. We identified a total of 230 genes differentially expressed by infection status [false discovery rate (FDR) < 0.05], 172 were up-regulated and 58 were down-regulated (Fig. 2C and Fig. S1B). Ingenuity Pathway Analysis of these 230 genes revealed that the pathways most affected by HRV infection were IFN signaling (P = 9.8 × 10−21) and activation of IRF by cytosolic pattern recognition receptors (P = 2.7 × 10−12).
Examination of those genes that significantly changed with infection (FDR < 0.05) and also had an absolute log2 fold change ≥1 revealed 63 genes, all of which were up-regulated. Analysis of these 63 genes again demonstrated that the top induced pathway during HRV infection was IFN signaling (P = 9.6 × 10−16; Fig. S1C). Of these 63 genes, 55 were ISGs and three were IFNs; all three belonged to the type III IFN family (Fig. S1D). Type I (IFNB1) and type III IFN (IFNL1) had similarly low basal expression levels in the mock-inoculated HIEs (Fig. 2D, purple symbols), but only type III IFN (IFNL1, IFNL2, IFNL3) was significantly up-regulated with infection (Fig. 2 C and D). We identified baseline expression levels of ISGs (total, n = 295) in the mock-inoculated HIE cultures. Gene-set enrichment analysis showed that ISGs were significantly up-regulated in HRV-infected cultures (P < 2.2 × 10−16, Kolmogorov–Smirnov test; Fig. S1E). Overall, IFN-ISG signaling is the dominant pathway induced at the transcriptional level in HRV-infected HIEs.
The ISG Response to HRV Infection of HIEs Is Regulated by Signaling Through the Type III IFN Receptor.
Infection-induced gene expression changes were verified by quantitative RT-PCR (RT-qPCR). Transcript levels of multiple IFNs and ISGs were compared between mock- and HRV-infected jejunal HIE cultures at 6 hpi. The type III IFNs were highly up-regulated in infected cultures from six individuals, with 2–3(log10) increases in transcript levels of IFNL1 and IFNL2, whereas type I IFNs were only slightly up-regulated (<1–10-fold; Fig. 3A). ISGs constituted the majority of the top up-regulated genes. Six ISGs (IFI44L, IFIT1, RSAD2, MX1, OAS2, and HERC5) with varying basal expression levels (Fig. 2D, red symbols) were assessed for their fold up-regulation by RT-qPCR. Up-regulation in the infected cultures ranged from 2.7-fold (MX1) to 35.3-fold (IFIT1; Fig. 3B).
Fig. 3.

The IFN-mediated response to HRV infection in HIEs. Independent samples of HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were mock- or HRV-inoculated (MOI of 10). (A) The IFN (type I and III) and (B) ISG transcriptional responses were compared between HRV-inoculated and mock-inoculated cultures by RT-qPCR at 6 hpi. (C) Using HIEs from the same six individuals, HRV infection without any receptor-blocking antibodies (none) was compared with infection performed in the presence of blocking antibodies to the type III IFN receptor (anti-type III IFN), blocking antibodies to the type I IFN system (anti-type I IFN), and antibodies to both the type III IFN receptor and type I IFN system (anti-both IFNs). HIEs were inoculated with HRV and, after 1.5 hpi, the virus inoculum was washed off and HIEs were resuspended in medium with or without antibodies to the indicated IFN receptors. HIEs were harvested for transcriptional (RT-qPCR) analysis at 6 hpi. The fold up-regulation of each ISG in the control infections without receptor-blocking antibodies (shown in B) was normalized to 100% and then compared with the up-regulation of individual ISGs under each receptor-blocking condition. Data in A–C are of HIEs from six individuals and are a compilation of three independent experiments. Results are expressed as geometric mean ± 95% confidence interval (CI) of the geometric mean (n = 6 biological replicates) in A and as arithmetic mean ± SEM (n = 6 biological replicates) in B and C. Statistical analyses were performed by Mann–Whitney U test in A and C. (D) Expression levels of type III IFN (IFNL1), type I IFN (IFNB1), and the ISG IFI44L were assessed from mock- and HRV-inoculated HIEs (MOI of 4) from individual j3 at 3, 10, and 22 hpi. Each data point represents geometric mean ± 95% CI of the geometric mean of four technical replicates.
Based on the robust induction of the type III IFNs and minimal to no induction of type I IFN (IFNA2 and IFNB1) transcripts, we hypothesized that ISG induction was regulated by signaling through the type III IFN receptor. We tested this hypothesis by performing HRV infection in the presence of blocking antibodies to the type I or III IFN receptors. Basal expression levels of the six previously tested ISGs in mock-inoculated HIEs from six individuals were not significantly different with or without blocking antibody treatment (P > 0.05, Wilcoxon signed-rank test, n = 6 biological replicates), suggesting minimal to no baseline IFN regulation of these six ISGs in mock-inoculated HIEs. In addition, type III IFN (IFNL1 and IFNL2) transcriptional levels were not significantly different between HRV infections performed with or without type III IFN receptor-blocking antibody treatment (P > 0.05, Wilcoxon signed-rank test, n = 6 biological replicates), indicating that this antibody did not induce nor suppress IFNL transcriptional activity. However, HRV infection performed in the presence of blocking antibodies to the type III IFN receptor significantly reduced the induction of the six ISGs analyzed, ranging from a 66% reduction in MX1 transcript levels to an 81% reduction in RSAD2 transcript levels compared with HRV infection without receptor-blocking antibodies (Fig. 3C). Maximum (100%) up-regulation of each ISG corresponds to the fold inductions shown in Fig. 3B. HRV infection in the presence of a control antibody (isotype matched to the type III IFN receptor-blocking antibody) did not decrease ISG levels compared with infection without antibodies. Infection performed in the presence of blocking antibodies to type I and type III IFN receptors failed to further suppress ISG levels compared with type III IFN receptor blockade alone (Fig. 3C). These data suggest that signaling through the type III IFN receptor largely regulates ISG induction in response to HRV infection in HIEs.
Analysis of IFN-ISG transcriptional kinetics at three additional time points showed that IFN transcripts were present at 3 hpi and decreased by ∼1 log10 at 10 hpi, at which point their levels plateaued (Fig. 3D). In contrast, up-regulation of ISG transcripts (assessed by using IFI44L as a marker) was not observed at 3 hpi but was present by 10 hpi, demonstrating that the type III IFN response preceded the ISG response. Together, these data suggest that HRV infection of HIEs results in robust up-regulation of type III IFN, which in turn activates the type III IFN receptor and leads to the induction of ISGs.
HRV Replication in HIEs Is Not Restricted by Endogenously Produced IFN.
HRV infection of epithelial cells in the HIE model induced a type III IFN-regulated ISG response, and IFN receptor signaling appeared to start between 3 and 6 hpi, as ISGs were up-regulated at 6 hpi but not at 3 hpi (Fig. 3). To assess whether the endogenous host IFN response can restrict rotavirus replication, HRV replication was assessed in the presence of IFN-receptor blocking antibodies. We hypothesized that inhibiting the host IFN response by blocking IFN signaling would increase virus titer yields. However, blockade of neither the type I nor type III IFN receptors during multiple rounds of replication significantly increased viral yields by 60 hpi (P = 0.75, Kruskal–Wallis test; Fig. 4). In addition, neither replication of Ito-HRV nor RRV was enhanced by simultaneous blockade of type I and type III IFN receptors for 60 h (P > 0.05, Mann–Whitney U test). These data suggest that, although homologous HRV and heterologous RRV induce a type III IFN response at the epithelial level (Fig. S2), this host cell response does not restrict viral growth because rotavirus grows to similar titers regardless of whether the type III IFN response is neutralized.
Fig. 4.
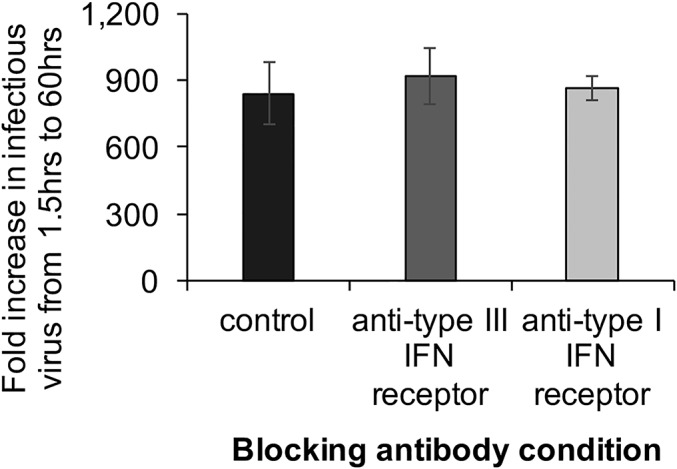
HRV growth in the presence of IFN receptor-blocking antibodies. HIEs (j2) were HRV-inoculated (MOI of 0.1). At 1.5 hpi, viral inoculum was washed off and the HIEs were resuspended in medium containing no IFN receptor-blocking antibodies (control) or antibodies to the type III IFN receptor or type I IFN system. Infectious virus titers [in focus forming units (FFU)] were assessed at 1.5 hpi and 60 hpi, and the fold increase (60 hpi/1.5 hpi) is shown. The raw increase in viral titer (60 hpi − 1.5 hpi) was 256,015 FFU (control), 279,695 FFU (anti-type III IFN), and 264,015 FFU (anti-type I IFN). Data from one representative experiment is presented as arithmetic mean ± SD of four technical replicates of HIEs from individual j2, and are representative of data from additional experiments performed with HIEs from five additional individuals (j3, j6, j8, j10, and j11) with four technical replicates each.
Fig. S2.

IFN induction in response to different RV strains. Independent samples of HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were mock- or RV-inoculated (MOI of 10) with an animal strain (i.e., RRV) or one of two human strains (Wa and Ito), and transcriptional responses at 6 hpi were assessed by RT-qPCR. Data are of HIEs from six individuals and are a compilation of two independent experiments. Results are expressed as geometric mean ± 95% CI of the geometric mean (n = 6 biological replicates). Statistical analyses are shown for levels of IFNL1 using the Wilcoxon signed-rank test.
Different Strains of Rotavirus and a dsRNA Analog Preferentially Induce IFNL over IFNB in HIEs.
The preceding experiments were primarily performed with the human strain Ito, genotype G3P[8]. Rotavirus infection of HIEs recapitulates the host-range restriction observed in vivo; human strains infect and replicate at higher levels than animal strains such as simian RRV in HIEs (9). To assess whether the type III IFN induction in response to Ito-HRV infection was strain-specific, HIEs were infected at the same MOI with an additional human strain (Wa, G1P[8]) and a simian strain (RRV; G3P[3]). Irrespective of virus strain, the pattern of IFN induction was similar; all three strains led to increased fold induction of IFNL1 compared with IFNB1 (Fig. S2). However, IFNL1 levels were significantly higher in HIEs infected with HRVs than with RRV, suggesting that type III IFN transcriptional intensity is strain-specific depending on the host susceptibility in HIEs.
Next, we evaluated if the preferential up-regulation of IFNL1 instead of IFNB1 with rotavirus infection was intestinal segment- or rotavirus-specific. Responses of 12 different individuals’ HIEs showed that IFNL1 induction in response to HRV infection was not significantly different across the three sections of the small intestine (P = 0.28, Kruskal–Wallis test), but IFNL1 fold up-regulation was ∼10–100-fold more than IFNB1 in each intestinal section (Fig. 5A) despite similar basal expression levels of the two IFNs in uninfected HIEs (Fig. 2D, purple symbols). We also tested the response to the dsRNA analog poly(I:C), which causes robust induction of the type I IFN system in transformed and nontransformed cell lines (42, 43). Unlike rotavirus or other viruses (24, 44), poly(I:C) does not antagonize IFN transcriptional responses. Similar to the response to rotavirus, all 12 HIEs treated with poly(I:C) exhibited significantly increased IFNL1 transcript fold up-regulation (range, 419–225,332 fold) compared with IFNB1 (range, 1–142 fold; Fig. 5B). These findings indicate that HIEs preferentially respond to rotavirus and dsRNA stimuli with type III IFN rather than type I IFN.
Fig. 5.

IFN transcriptional response by HIEs across the small intestine and cellular localization of dsRNA. Independent samples of four duodenal HIEs (d1, d4, d6, and d104), four jejunal HIEs (j2, j3, j6, and j11), and four ileal HIEs (i2, i103, i16, and i104) were inoculated with (A) HRV (MOI of 5) or (B) treated with poly(I:C) (30 μg/mL), and transcriptional responses were assessed at 6 hpi by RT-qPCR. Results are expressed as geometric mean ± 95% CI of the geometric mean (n = 4 biological replicates per intestinal segment, compiled from two independent experiments). Statistical analyses were performed by Mann–Whitney U test. (C) HIEs (j3) were mock- (Top) or HRV-inoculated (Bottom; MOI of 10). At 6 hpi, HIEs were fixed and stained for dsRNA (magenta), the rotavirus viroplasm protein NSP2 (green), and nuclei (DAPI, blue). (Scale bar: 10 μm.)
The specific rotavirus PAMP(s) recognized by cellular pathogen recognition receptors (PRRs) is unclear. Rotavirus replication was not thought to release free dsRNA genome into the cell (45). However, dsRNA has recently been detected independent of viroplasms in animal rotavirus-infected MA104 cells (46). dsRNA is a potent PAMP for many other viruses (47), and it is therefore pertinent that the dsRNA analog poly(I:C) and HRV induced similar IFN transcriptional profiles in HIEs. Staining HIEs for dsRNA and the viroplasm marker nonstructural protein (NSP2) revealed their colocalization in the cytoplasm of infected cells as well as dsRNA independent of viroplasms (Fig. 5C). These data demonstrate the presence of viroplasm-independent dsRNA in HRV-infected cells that could be the PAMP recognized by cellular PRRs.
A Majority of IFNL Transcript in HRV-Infected Cells Is Not Translated into Detectable Type III IFN Protein.
Blockade of the type III IFN receptor reduced ISG up-regulation (Fig. 3C), suggesting that type III IFN (IFN-λ) protein was secreted during infection and engaged its receptor. Therefore, either (i) HRV replicated despite high quantities of type III IFN protein or (ii) low levels of type III IFN protein produced during infection were sufficient to induce ISGs but insufficient to restrict viral growth. IFN protein production was evaluated in supernatants from mock-inoculated and HRV-infected HIE cultures by ELISA. Despite robust up-regulation of IFNL1 transcripts at 6 hpi (Fig. S3A), IFN-λ1 and IFN-λ2/3 proteins were below the limit of detection in the supernatant from infected HIEs at multiple time points (6–48 hpi) and under multiple different conditions, including from a homogenate of supernatant and cell lysate (Table S1). These data suggested that HRV infection induced type III IFN protein at levels below the limit of detection of standard ELISAs but sufficient to induce ISGs. This was confirmed by observing that exogenous treatment of HIEs with concentrations of IFN-λ1 protein below the limit of detection of standard ELISAs (<2 U/mL; Materials and Methods) induced ISGs (Fig. 6A) to similar levels as HRV (Fig. 3B). Rotavirus infection of transformed cells also induces the neutrophil chemoattractant IL-8 (48). High but similar levels of IL-8 were detected in HIE supernatants from mock- and HRV-infected samples previously tested for IFN (P > 0.05 at each time point, Mann–Whitney U test; Fig. S3B), showing that not all host proteins were as affected by HRV infection as type III IFN.
Fig. S3.

Effect of HRV-infection on IL-8. (A) HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were mock- or HRV-inoculated (MOI of 10), and transcript levels were assessed at 6 hpi by RT-qPCR. Results are expressed as geometric mean ± 95% CI of the geometric mean (n = 6 biological replicates). (B) The surrounding media from the same experiment described in A was analyzed for secreted IL-8 by ELISA. Results are expressed as mean ± SEM (n = 6 biological replicates). Data from one representative experiment of two independent experiments are shown.
Table S1.
Analysis of IFN protein from HIEs
| IFN sample analyzed | 6 hpi | 8 hpi | 12 hpi | 20 hpi | 24 hpi | 48 hpi |
| IFN-λ1 | ||||||
| Supernatant | − | − | − | − | − | |
| Concentrated supernatant | − | − | ||||
| Supernatant + anti-IL28RA | − | − | − | |||
| Cells + supernatant | − | − | ||||
| IFN-λ2/3 | ||||||
| Supernatant | − | − | ||||
| Supernatant + anti-IL28RA | − | − | ||||
| Cells + supernatant | − | − | ||||
| IFN-β1 | ||||||
| Supernatant | − | − | −* | − | − |
MOI > 10 FFU per cell for each experiment. −, below the limit of detection. “Concentrated supernatant” indicates supernatant from n = 5–6 samples concentrated into a single sample. “Supernatant + anti-IL28RA” indicates supernatant from HIEs incubated with anti-IL28RA antibody (25 μg/mL) during infection to block the type III IFN receptor. “Cells + supernatant” indicates sonicated homogenate of HIEs and surrounding media. IFN-λ1 ELISA sensitivity is 8 pg/mL. IFN-λ2/3 ELISA sensitivity is 15.6 pg/mL. and IFN-β1 ELISA sensitivity is 50 pg/mL.
Limit of detection is 2 pg/mL.
Fig. 6.

Detection of cytokines and the effect of HRV infection on host translation. (A) HIEs from four individuals (j2, j3, j6, and j11) were treated with increasing doses of IFN-λ1, and ISG expression was assessed after 3 h by RT-qPCR. Data are of HIEs from four individuals and are a compilation of two independent experiments. Results are expressed as geometric mean ± 95% CI of the geometric mean (n = 4 biological replicates). (B) HIEs (j3) were mock-inoculated, HRV-inoculated (MOI of 10), or treated with poly(I:C) (30 μg/mL). After 1.5 hpi, HIEs were washed and resuspended in new medium, which was analyzed at 8 hpi for secreted IFN-λ1 by ELISA. (C) HIEs (j2) were mock-inoculated, HRV-inoculated (MOI of 10), treated with poly(I:C) (100 μg/mL), or HRV-inoculated (MOI of 10) and treated 10 min later with poly(I:C) (100 μg/mL). After 1.5 hpi, HIEs were washed and resuspended in new medium, which was analyzed at 20 hpi for secreted IFN-λ1 by ELISA. In B and C, data from one representative experiment of three independent experiments is presented as arithmetic mean ± SD of three technical replicates. Statistical analyses in C were performed by Student’s t test.
Rotavirus antagonizes IFN production pretranscriptionally in a variety of cell lines (HT29, Caco-2, and 293T) through degradation of factors involved in IFN transcription (35, 49–51). We observed robust type III IFN transcription with HRV and poly(I:C) treatment (∼100–1,000-fold increases; Fig. 5), but protein was detected only from poly(I:C)-treated HIEs at 8 hpi (Fig. 6B). We next evaluated the possibility of rotavirus-mediated inhibition of IFNL1 translation in HIEs treated with HRV and/or poly(I:C). IFN-λ1 protein levels were below the limit of detection from mock- and HRV-infected HIEs, but were 224 pg/mL from poly(I:C)-treated HIEs at 20 hpi (Fig. 6C). The IFN-λ1 protein level significantly decreased to 41 pg/mL when poly(I:C)-treated HIEs were also infected with HRV. These data (Fig. 6 B and C) indicate a rotavirus-mediated posttranscriptional partial suppression of host IFNL1 mRNA translation. Of note, IFN-β1 protein from HIE supernatant was below the limit of detection by ELISA (sensitivity, 2 pg/mL) for each of the four conditions described in Fig. 6C at 20 hpi.
Rotavirus antagonism of the host response is primarily mediated through the nonstructural proteins NSP1 and NSP3, which are present only in cells infected with replication-competent virus (24). We assessed if viral antagonism of the IFN response in HIEs was a replication-dependent process by treating HIEs with purified, inactivated HRV particles that contain the dsRNA genome, structural proteins, and can enter cells but do not transcribe viral mRNAs or express nonstructural proteins (16, 52). Inactivated HRV induced ∼10-fold higher transcript levels of IFNL1 and IFNB1 compared with replication-competent virus early after infection (Fig. 7A), demonstrating that the robust IFNL1 transcriptional response induced by replication-competent HRV is partially antagonized at a pretranscriptional state by infectious virus. Furthermore, high levels of IFN-λ1 protein were detected from HIEs treated with inactivated (i.e., replication-incompetent) HRV (Fig. 7B), but only if infection was performed in the presence of antibody to the type III IFN receptor, suggesting that secreted IFN-λ1 protein rapidly bound its receptor and did not remain free in the supernatant for an extended duration. A similar technique of using receptor-blocking antibodies to enhance cytokine detection has been described for IL-4 (53). Thus, IFN transcriptional induction in HRV-infected HIEs is replication-independent, but pre- and posttranscriptional antagonism of the IFN response in HRV-infected HIEs is replication-dependent.
Fig. 7.

IFN response to inactivated HRV. HIEs (j3) were inoculated with replication-competent HRV (MOI of 10 FFU) or with an equivalent amount (in micrograms) of inactivated HRV particles. (A) Expression of type III IFN (IFNL1), type I IFN (IFNB1), and the ISG IFI44L was assessed at 6 hpi by RT-qPCR. Each data bar represents the geometric mean ± 95% CI of the geometric mean (n = 4 technical replicates). Statistical analyses were performed by Mann–Whitney U test. (B) HIEs were infected as described in A and, after 1 h, HIEs were washed and resuspended in medium containing type III IFN receptor-blocking antibody (25 μg/mL anti-IL28RA). The media was analyzed at 20 hpi for secreted IFN-λ1 by ELISA. Data from one representative experiment of two independent experiments is presented as arithmetic mean ± SD of three or four technical replicates.
Exogenous IFN Treatment Restricts Rotavirus Growth in HIEs.
Endogenously produced IFN did not restrict HRV growth in HIEs (Fig. 4). However, endogenous type III IFN restricts animal rotavirus replication in mice (54, 55). The mouse epithelium was thought to produce the type III IFN in vivo, although immune cells can also produce type III IFN (54, 56, 57). In contrast, murine type I IFN predominantly originates from lamina propria immune cells during rotavirus infection (58). We simulated the presence of surrounding lamina propria immune cells by adding IFN before infection to compare the potential of exogenous type I and type III IFN to restrict rotavirus growth in HIEs.
Anti-rotavirus efficacy of type I IFN vs. type III IFN was compared by pretreating HIEs for 24 h with escalating doses of IFN-λ1 or IFN-β1 ranging from 100 to 10,000 U/mL. IFN was subsequently washed off, and the HIEs were infected with HRV at a low MOI to allow for multiple rounds of replication. IFN-λ1 and IFN-β1 inhibited HRV replication, but inhibition by IFN-λ1 was weaker than IFN-β1 at all concentrations (Fig. 8A). Maximal inhibition of HRV replication by IFN-λ1 (79%) was achieved at 1,000 U/mL of IFN-λ1. In contrast, each 10-fold increase in IFN-β1 concentration reduced viral replication by ∼10-fold, with replication 90% inhibited with 100 U/mL of IFN-β1 and completely inhibited with 10,000 U/mL IFN-β1 (Fig. 8A).
Fig. 8.

Pretreatment of HIE cultures with exogenously administered type I and type III IFN. (A) HIEs (j3) were pretreated with escalating doses of type III IFN (IFN-λ1) or type I IFN (IFN-β1) for 24 h, after which HIEs were washed to remove IFN and then inoculated with HRV (MOI of 0.1). The increase in infectious virus was calculated by subtracting the titer at 1.5 hpi from the titer at 24 hpi for each condition and depicted in the panel as the percentage of viral growth compared with the control infection (no IFN pretreatment). The control infection represents 100% viral growth. The raw increase in viral titer (24–1.5 hpi) for the control (no IFN pretreatment) sample was 35,275 FFU (geometric mean of three replicates). (B) HIEs (j3) were pretreated with 1,000 U/mL of the indicated IFN for 24 h, following which the same procedures were performed as described in A. The raw increase in viral titer (24–1.5 hpi) for the control (no IFN pretreatment) sample was 15,741 FFU (geometric mean of four replicates). (C) HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were pretreated with 1,000 U/mL of IFN-λ1 or IFN-β1 for 6 or 24 h, washed, and HRV-inoculated (MOI of 0.1). (D) HIEs from five individuals (j2, j3, j8, j10, and j11) were pretreated with the indicated combinations of IL-22 (100 ng/mL) and IFN-λ1 for 24 h, after which they were HRV-inoculated (MOI of 0.1). In C and D, the percentage of viral growth was determined as described for A. In A–D, the efficacy [1 − (% viral growth)] of each IFN pretreatment is displayed at the top. In A and B, data from one representative experiment of two or three independent experiments is presented as arithmetic mean ± SEM of three or four technical replicates. In C and D, data are of HIEs from five or six individuals and are a compilation of two (C) and four (D) independent experiments; results are expressed as arithmetic mean ± SEM (n = 5–6 biological replicates). (E) HIEs (j2) were HRV-inoculated (MOI of 0.1) and, after 1.5 hpi, HIEs were washed and resuspended in medium containing pancreatin without IFN (control) or with 1,000 U/mL of the indicated IFN. The amount of infectious virus under each condition was assessed at 1.5, 24, and 48 hpi. Each data point represents arithmetic mean ± SEM of six technical replicates. Data from one representative experiment of three independent experiments is shown. Statistical analyses in A–E were performed by Mann–Whitney U test.
Our studies focused on IFN-λ1 and IFN-β1 because we did not find differences in transcriptional up-regulation between IFNL1-3 and because IFNB1 was the only type I IFN that was even slightly up-regulated (Fig. 3A). However, heterogeneity in the antiviral properties of different type I and type III IFN variants has been reported with respect to other viruses (59, 60). To compare efficacies of alternative type I and type III IFNs, HIEs were pretreated with IFN-λ3 (type III IFN) or IFN-α2 (type I IFN) and compared with their analogs, IFN-λ1 and IFN-β1, respectively. There was no significant difference between the two type III IFNs; both reduced viral replication by ∼80% (Fig. 8B). However, pretreatment with IFN-β1 was significantly more protective than IFN-α2, indicating heterogeneity in the anti-rotavirus efficacy of the different type I IFNs.
The HRV replication cycle in HIEs is rapid, with new progeny virus present by 7 hpi (9). To evaluate the time needed for IFN to establish an antiviral state, HIEs were treated for 6 or 24 h with IFN-λ1 or IFN-β1 before HRV infection. Following 6 h of pretreatment, IFN-λ1 and IFN-β1 reduced viral replication by 62% and 93%, respectively (Fig. 8C). After 24 h of pretreatment, the antiviral efficacy of IFN-λ1 increased an additional 26% (88% protection), whereas the antiviral efficacy of IFN-β1 was only slightly enhanced to 99% after 24 h of pretreatment. Thus, exogenous IFN-β1 induced a faster and more effective anti-rotavirus state in epithelial cells than IFN-λ1.
The protective effects of IFN-λ in acute rotavirus infection in mice are enhanced by IL-22, which also cured chronic murine rotavirus infection (54, 61). In the HIE model, pretreatment with IL-22 decreased HRV replication by 54% (Fig. 8D). However, IL-22 did not significantly enhance the efficacy of low-dose (100 U/mL) or high-dose (1,000 U/mL) IFN-λ1 under the conditions tested. In conjunction with the results from Fig. 8A, these data demonstrate that the anti-HRV efficacy of IFN-λ saturates between 80% and 90% in HIEs, regardless of whether higher doses or an additional cytokine (IL-22) are used.
IFNs are typically present at very low basal levels before infection in vivo and are induced following detection of microbial PAMPs during infection (62). To simulate this condition, HIEs were first infected with HRV and, 1.5 h later, exogenous IFN-λ1 or IFN-β1 was added to the maintenance media for the duration of the infection. At 24 and 48 hpi, viral growth in IFN-λ1- or IFN-β1–treated HIEs was reduced by ∼50–55% (IFN-λ1) and 79% (IFN-β1) compared with that in untreated HIEs (Fig. 8E). Viral growth was not further restricted by treating HIEs during infection with a combination of 1,000 U/mL IFN-λ1 and 1,000 U/mL IFN-β1 compared with 1,000 U/mL IFN-β1 alone.
Discussion
HIEs provide a new model to evaluate the human small intestinal epithelial response to infection and assess human host effects by generating HIEs from multiple individuals. We found that the basal human epithelial transcriptome differs significantly between HIEs from different individuals, and this person effect was more pronounced than the response to enteric viral infection. These findings highlight that transcriptional data from single individual sources, such as cell lines, may have limited generalizability as they cannot capture the distinct transcriptional signatures exhibited by different individual sources and across small intestinal segments (63). This inherent variability captured in the HIE model provides a unique opportunity to study similarities and differences in susceptibility to enteric infection among individuals.
Despite a significant baseline person effect at the transcriptional level, all 14 lines of HIEs tested responded to HRV infection with a single predominant and conserved type III IFN-regulated ISG pathway. The lack of a type I IFN response in these cells is consistent with the knowledge that murine type I IFN is produced by immune cells and not intestinal epithelial cells following murine rotavirus infection (58). The innate type III IFN response in HIEs was preserved irrespective of rotavirus strain or replication competency or following treatment with the dsRNA analog poly(I:C), a potent PAMP. The rotavirus PAMP(s) that drives the type III IFN response is unknown, but rotavirus dsRNA is a potential candidate, as we and others (46) detected viroplasm-free dsRNA in the cytoplasm of rotavirus-infected cells. dsRNA [mimicked by poly(I:C)] can induce type I and III IFN and is a key intermediate in the replication cycle of many viruses, including other major human enteric viral pathogens (13, 43, 47, 64, 65). Thus, it is possible that human intestinal epithelial cells are programmed to respond to viral dsRNA with type III rather than type I IFN.
A preferential type III vs. type I IFN response to viral infection and dsRNA analogs does not appear to be common to all human epithelial cells. Whereas primary hepatocytes also respond to HBV/HCV and poly(I:C) predominantly with type III IFN (66, 67), primary lung epithelial cells respond to influenza virus infection and poly(I:C) with type I and III IFN (43, 68). Thus, the type of IFN induced in response to infection appears to be cell- and/or virus-specific rather than a pan-epithelial intrinsic response. The specific cellular factors that predispose induction of type III IFN or type I IFN are unclear because regulation of IFN is complex. Both types of IFN are induced by similar stimuli and generally rely on similar transcription factors, such as NF-κB and the IRF family (30), but preferential activation of type III IFN may be influenced by other transcription factors (69). In addition, peroxisomal content (70) and cell differentiation status (70) may significantly influence type III IFN induction. It is notable that the rotavirus capsid protein VP4 has a peroxisomal targeting motif (71) and that rotavirus infection of HIEs requires differentiated HIEs (9). Whether any of these factors influences the preferential type III IFN innate response of HIEs to HRV and poly(I:C) remains to be determined.
The predominant type III IFN response and its regulation of ISG induction observed in our studies with HIEs is consistent with previous studies reporting that murine intestinal epithelial cells from poly(I:C)-injected and murine rotavirus-infected mice exhibit equal or greater fold inductions of type III IFN than type I IFN transcripts (17, 55, 56, 58). This contrasts with results using transformed human colonic cell lines that respond to rotavirus infection and poly(I:C) with type I IFN-β at the transcript and protein levels, and type I IFN regulates ISG induction in these cells (16, 42, 72). Transformed cells often have significant deficits in their response to pathogens, including in IFN signaling pathways (73, 74). It seems likely that this critical difference in IFN responses to rotavirus and poly(I:C) between immortalized cell lines and nontransformed HIE cultures is partially attributed to their transformation status.
Despite robust up-regulation of type III IFN at the transcriptional level, rotaviral replication was not enhanced by neutralization of the type III IFN receptor in HIEs. This result was unexpected because enteric viral replication (rotavirus, reovirus, murine norovirus) is enhanced in mice lacking the type III IFN receptor (54–56, 75), even though it is notable that neutralization of the type III IFN response had a limited effect on reovirus replication (approximately twofold increase in viral transcripts) in a human colonic transformed cell line (70). The basis for the apparent discrepancy may be multifactorial and include differences in how the IFN system was blocked (neutralizing antibodies vs. receptor KOs), development of alternate compensatory pathways in IFN-KO mice, and/or differences in human and mouse type III IFN subtype expression (37, 38). Regardless of these differences, HRV infection inhibited the IFN response by HIEs. Most pathogenic viruses, including rotavirus, have developed ways to subvert IFN responses at multiple steps in their biologic pathways (24, 44). Rotavirus evades the host innate response primarily through its nonstructural proteins NSP1 and NSP3, and the structural protein VP3 has recently been shown to antagonize the cellular RNaseL antiviral response as well (33, 76). In several in vitro models, rotavirus NSP1 inhibits the host IFN response by preventing IFN from being transcribed (35, 51). Recent studies have shown that NSP1 acts as an adaptor that links the host Cullin–E3 ligase complex with β-transducin repeat-containing protein (β-TrCP), which is a necessary component for NF-κB–mediated induction of IFN transcripts (77, 78). Through this interaction with the Cullin–E3 ligase complex, NSP1 facilitates the proteasomal degradation of itself and β-TrCP (78). Using HIEs as a model, it was further shown that β-TrCP degradation by HRV was mitigated by inhibition of Cullin–RING ubiquitin ligases, demonstrating that HRV antagonizes IFN transcription by hijacking host proteins (78). Although we did not evaluate this in our studies, this mechanism may help explain the lower levels of IFN transcripts observed when HIEs were infected with replication-competent HRV compared with inactivated HRV, which does not produce NSP1. Although replication-competent HRV inhibited type III IFN transcription ∼10-fold compared with inactivated HRV, type III IFN transcripts were still robustly induced in replication-competent HRV-infected HIEs (100-1,000 fold) and are present at high levels in intestinal epithelial cells from rotavirus-infected mice (54, 55). Together, these data suggest that the transcriptional regulatory pathways upstream of IFN induction remain at least partially intact in these models. However, rotavirus can also inhibit host responses by decreasing translation of host mRNA through multiple mechanisms primarily via the protein NSP3 (24, 79–81). Although these mechanisms do not selectively antagonize IFN, production of IFN protein is likely reduced when there is a global decrease in host translation. Thus, translational inhibition is a potent method for HRV to antagonize the type III IFN antiviral response because IFN-λ protein was significantly decreased in HIEs infected with replication-competent HRV compared with inactivated HRV. Because very low levels of IFN-λ protein are required to elicit the ISG responses observed in HRV-infected HIEs, our data suggest that minimal amounts of type III IFN protein were produced during HRV infection of HIEs: enough to induce ISGs but not enough to restrict viral growth. These results suggest there may be a threshold level of IFN production and perhaps ISG production needed to restrict viral growth. Overall, this represents the paradox of IFN responses in HIEs.
In contrast to HRV-infected HIEs, high levels of IFN-λ protein are detected in the small intestine from rotavirus-infected mice. These studies examined whole small intestinal homogenates; therefore, the cellular origin of the IFN-λ protein could not be determined (17, 54). Murine intestinal epithelial cells were proposed to be the major source of IFN-λ protein in these homogenates because IFNL transcript levels were approximately four to five fold higher in intestinal epithelial cells than in hematopoietic cells (54). However, our data demonstrate that IFNL transcripts do not parallel protein levels in epithelial cells during rotavirus infection. Hematopoietic cells can produce IFN-λ protein in response to multiple viruses (57), and a recent study has demonstrated that steady-state IFN-λ in the intestine may actually originate from CD45+ hematopoietic cells rather than epithelial cells in uninfected mice (56). Taken together, the HIE and mouse data suggest that the predominant source of the IFN-λ protein in the small intestine in response to rotavirus infection may be of hematopoietic rather than epithelial origin.
Lamina propria hematopoietic cells are the primary source of type I IFN during rotavirus infection in mice, rather than intestinal epithelial cells (58). This is consistent with our data in which HIEs produced no detectable IFN-β protein in response to HRV, and blockade of the type I IFN system had minimal to no effect on transcription of the six ISGs assessed. These results suggest that the origin of type I IFN in humans, as in mice in vivo, is extraepithelial, such as from plasmacytoid dendritic cells (82). We simulated the effect of extraepithelial sources of IFN by exogenously treating HIEs with IFN. Treatment with type I IFN-β was significantly more effective than type III IFN at restricting HRV replication. This differs from reports in mice, in which exogenous treatment with IFN-λ is more protective against enteric viral replication than type I IFN (55, 56), and type I IFN is primarily considered to inhibit systemic spread of enteric viruses (17, 56, 75). This difference may be a result of the type I IFN subtype tested; previous studies in mice and HIEs used an IFN-α subtype rather than IFN-β for exogenous treatment (17, 55, 56, 83); however, IFN-α2 was significantly weaker than IFN-β in restricting HRV growth in HIEs. Heterogeneity in the host response to different type I IFNs has been previously characterized; IFN-β binds the type I IFN receptor with greater affinity than IFN-α2 (84), and other viral pathogens including LCMV and HCV have differing sensitivities to IFN-α and IFN-β (60). Thus, the choice of type I IFN tested may have a significant impact on the evaluation of IFN antiviral efficacy. Our data indicate that treatment with human type I IFN-β potently decreases local spread and replication of enteric viruses in the human intestinal epithelium, but whether extraepithelial production of type I IFN during infection in vivo is of sufficient quantity or interacts with epithelial cells to impact homologous virus replication remains a question. The ISGs required to limit HRV replication have yet to be identified, as individual ISG efficacy is virus-specific (31), but exogenous IFN-β likely induces these ISGs to higher levels and/or at a faster rate than IFN-λ in the human intestinal epithelium.
A current limitation of the HIE system is that it lacks the microbiome, and immune cells have yet to be incorporated with the epithelial cells to create a model that recapitulates the complex cross-talk that occurs in the small intestine (85, 86). Although we partially simulated the role of immune cells on epithelial cells by exogenously adding IFN to HIE cultures, we were not able to evaluate the effects of epithelial-produced IFN-λ on immune cells. Plasmacytoid dendritic cells respond to exogenous IFN-λ as well as produce large amounts of type III and type I IFN protein (30, 57). This raises the possibility of IFN-λ–mediated cross-talk between intestinal epithelial cells and intestinal plasmacytoid dendritic cells and leaves open the prospect that epithelial-derived IFN-λ can act on and even induce its own production from immune cells because IFN-λ is an ISG itself (87). As HIE platforms and cellular coculture techniques are further developed, it will be possible to dissect such microbiome–immune–epithelial interactions.
To better understand infectious disease pathogenesis, host–pathogen interactions are studied in model systems. However, these models can yield differing results depending on the choice of host organism and pathogen strain. This is particularly relevant for human infections because modeling human pathogens in vivo is often impractical. Advances in the organoid/enteroid and transcriptomic fields provide much needed tools to study host–pathogen interactions in the human intestine. Recently, RNA-seq of iPSC-derived human intestinal organoids treated with the human enteric pathogen Salmonella enterica serovar Typhimurium revealed new insights into how the epithelium responds to bacterial infection with a proinflammatory transcriptional profile (3). Like Salmonella Typhimurium, many human enteric viruses also lack robust models, and our studies demonstrate that HIEs are a useful model to study the human enteric virus HRV (9). In the present study, a complex host–pathogen interplay between the human intestinal epithelial type III IFN system and viral antagonism of this response is described. Although epithelial-derived type III IFN has been proposed as a critical defense against murine enteric viruses (54–56), our data are consistent with a recent report showing type III IFN does not significantly restrict homologous murine rotavirus infection (17). Even though the type III IFN system is activated in HIEs following HRV infection, the virus effectively evades this epithelial antiviral response. Whether other human enteric viruses similarly evade the type III IFN system remains to be determined. The recent cultivation of human noroviruses in HIEs establishes HIEs as a relevant model for studying host–pathogen interactions of other human enteric viruses in addition to HRV (4). The limited role of type III IFN in restricting HRV replication in the human epithelium raises the question of whether virus-induced type III IFN has undiscovered biologic functions in the mucosal epithelium that are not directly antiviral but may have alternative effects on bystander epithelial and/or nonepithelial cells. Given that HIEs are derived from nontransformed human small intestinal tissue and can be generated from many different individuals, they provide a preclinical model for assessing the host response to human enteric pathogens and an additional system to evaluate if findings in animal models can be translated directly to human tissue or if important differences in these systems exist.
Materials and Methods
Detailed methods and descriptions of cell lines, HIE cultures, viruses and viral infections, immunohistochemistry, cytokines, antibodies, ELISA kits, RNA extraction, RT-qPCR protocols, and statistical analyses are provided in SI Materials and Methods.
Microarray-Based Transcriptome Analysis.
HumanHT-12 v4 Expression BeadChips (Illumina) were used for microarray analysis. Transcriptome analysis was performed by using R statistical software (88). Two microarray experiments were performed: the first experiment included samples from a single individual’s HIEs (j11) and the second with samples of HIE cultures from three individuals (j2, j3, and j11). In the first and second microarray experiments, 20,948 genes were defined as expressed and used in subsequent analysis. Expression values for these 20,948 genes were quantile normalized by using the normalize.quantiles() function of the preprocessCore R library. Analysis of the microarray-acquired data is described in SI Materials and Methods.
RNA-seq–Based Transcriptome Analysis.
Total RNA was prepared by using the Illumina TruSeq Stranded RNA Sample preparation protocol. Paired-end sequencing (100 bp) was performed by using the IlluminaHiSeq 2500 machine. A total of 103,002,651, 101,422,021, 90,511,554, and 84,412,802 reads were sequenced for samples j11-uninfected, j2-uninfected, j11-infected, and j2-infected, respectively. Analysis of the RNA-seq data are described in SI Materials and Methods.
SI Materials and Methods
Cell Lines and HIE Cultures.
African green monkey kidney (MA104) cells were grown in Dulbecco’s modified Eagle medium (Invitrogen and Sigma-Aldrich) supplemented with 10% (vol/vol) FBS (Invitrogen). Three-dimensional cultures of HIEs were generated, cultured, and infected with rotavirus as detailed by Saxena et al. (9). Each culture/sample of HIEs from an individual person contains many (∼50–100) single 3D HIEs of varying size. Calculations of MOI are based on the total number of cells within a sample of HIEs rather than the number of individual HIE units (10). Three different types of media were used to wash (complete medium without growth factors [CMGF(−)]), proliferate [complete medium with growth factors (CMGF+)], and differentiate/maintain (differentiation medium) the HIEs, which are detailed by Saxena et al. (9). The specific HIEs used in each experiment are referred to by the intestinal segment of origin followed by a number. Culture codes beginning with “d” correspond to duodenal HIEs, “j” to jejunal HIEs, and “i” to ileal HIEs.
Viruses and Viral Infections.
RRV and HRV strains Wa and Ito were grown in MA104 cells in the presence of 2 μg/mL of trypsin (Worthington Biochemical) and 10 mM CaCl2, titered by fluorescent focus assay (FFA) (37) and expressed in FFU. MOI was calculated according to the formula (infectious virus based on FFA titers in MA104 cells)/(total number of cells), and MOI is represented as FFU per cell. The number of cells in a sample of HIEs was quantified with a Coulter particle counter (10). The inoculum for infections performed at an MOI of <0.5 FFU per cell was a lysate of rotavirus-infected MA104 cells, and uninfected MA104 lysate was used for the mock inoculation. The inoculum for infections performed at an MOI of >0.5 FFU per cell consisted of purified triple-layered particles (TLPs) of rotavirus, and an equal volume of TNC (10 mM Tris·HCl, 140 mM NaCl, 10 mM CaCl2, pH 7.4) buffer was used as the mock inoculum. TLPs were purified from viral lysate by using a CsCl gradient (89) followed by centrifugation (2 h, 35,000 rpm; SW 41 Ti rotor) through a 20% sucrose cushion in TNC buffer to remove excess cesium and then dialysis in TNC buffer using a Slide-A-Lyzer cassette (molecular weight cutoff, 3.5 kDa; Thermo Fisher Scientific) to remove residual sucrose.
To make inactivated rotavirus particles, an aliquot of purified Ito-HRV TLPs was treated with 40 μg/mL of psoralen-AMT (cat. no. A4330; Sigma-Aldrich) and UV-irradiated for 2 h (17). The inactivated-HRV produced no fluorescent foci on MA104 cells in an FFA, demonstrating replication incompetence of the inactivated viral preparation. HIEs were infected with equal amounts (in micrograms) of Ito-HRV and inactivated HRV based on the protein concentration of each viral preparation, which was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific).
Viral infections, assessment of the percentage of infected cells by flow cytometry, and quantification of viral replication in 3D HIEs were performed as described by Saxena et al. (9). HIEs were differentiated for 3–4 d before infection. For infections performed at an MOI <0.5, viral inoculum was washed off after 1–1.5 hpi. The HIE maintenance media applied after washing off nonadsorbed virus contained 0.5 mg/mL of porcine pancreatin (Sigma-Aldrich) to allow for multiple rounds of replication. For infections performed at higher MOIs, viral inoculum was washed off after 1–1.5 hpi but did not include pancreatin in the subsequent maintenance (i.e., differentiation) media. HRV indicates Ito-HRV unless specifically stated otherwise, and all rotavirus preparations were treated with 10 μg/mL of trypsin (Worthington) for 30 min at 37 °C before use in infections to activate the VP4 spike protein (89).
Because the FFA to quantify viral titers was performed with MA104 cells to measure the amount of infectious virus in the HIEs and surrounding media, we evaluated the possibility that IFN in the media could inhibit rotavirus infection of MA104 cells during viral titration. Stock aliquots of Ito-HRV were incubated with no IFN (control) or 1,000 U/mL of IFN-λ1 or IFN-β1. MA104 cells were then inoculated with control (Ito-HRV) aliquots or IFN-treated Ito-HRV aliquots for 1 h, washed twice, incubated overnight in DMEM, and fixed in cold methanol for 10 min. Viral titers were obtained by FFA as described (37). The viral titers were equivalent in control and IFN-spiked aliquots, demonstrating that our results in Fig. 8 were not confounded by titration of the HIE-produced rotavirus particles in the presence of IFN on MA104 cells.
Immunohistochemistry.
Immunofluorescent staining and confocal microscopy were performed on formalin-fixed, paraffin-embedded sections (3 μm) of 3D HIEs (10). To detect dsRNA, the mouse anti-dsRNA J2 antibody that detects dsRNA structures greater than 40 bp was used (1:100; Scions) (48, 90). The rotavirus viroplasmic marker NSP2 was detected by using a guinea-pig anti-NSP2 antibody at a concentration of 1:500 (91). Secondary antibodies were used at a concentration of 1:500 (Rockland Immunochemicals).
Cytokines, Agonists, Antibodies, and ELISA Kits.
All human IFNs were obtained from PBL Assay Science and stored according to the manufacturer’s instructions. IFN concentrations were based on standardized international units, which were quantified by the manufacturer by using a cytopathic effect inhibition assay in which the EC50 of encephalomyocarditis virus infection on A549 cells is ∼1 U/mL of IFN, regardless of IFN subtype.
Human IL-22 was quantified by and purchased from PeproTech. HIEs were treated with high molecular weight poly(I:C) HMW (InvivoGen) by direct application of poly(I:C) suspended in CMGF(−) onto HIEs.
The type III IFN receptor was blocked with a polyclonal sheep antibody (25 μg/mL) against the IL28RA subunit (cat. no. AF5260, lot CCXR01; R&D Systems). To our knowledge, use of this antibody for blocking purposes has not been characterized or described before. Therefore, we characterized the antibody for this new indication in Fig. S4. A mixture of antibodies was used to block the type I IFN system: polyclonal rabbit anti-human IFN-α serum (6.88 × 102 NU/mL; cat. no. 31130–1, lot 5896; PBL), protein A purified polyclonal rabbit anti-human IFN-β (4.3 × 102 NU/mL; cat. no. 31405–1, lot 5277; PBL), and a monoclonal mouse anti-IFNAR2 antibody (10 μg/mL; clone MMHAR-2, lot 5989; PBL). All three of these anti-type I IFN antibodies were used together to block signaling through the type I IFN receptor; no experiment used only one or two of the anti-type I IFN antibodies.
Fig. S4.

Effect of receptor-blocking antibodies on rotavirus infectivity and induction of OAS2 transcripts. (A) Stock aliquots of Ito-HRV were incubated with no antibody (control), the anti-type III IFN receptor-blocking antibody, or the anti-type I IFN system antibody mixture for 2 h. To assess the titer of HRV under these conditions, MA104 cells were infected with control Ito-HRV aliquots or antibody-treated Ito-HRV aliquots for 1 h, washed twice, and incubated overnight in DMEM for 18 h. Viral titer was subsequently obtained by FFA. (B) HIEs (j11) suspended in differentiation medium containing 0.5 mg/mL pancreatin were treated with or without blocking antibodies to the type III IFN receptor (anti-IL28RA) or the type I IFN system (anti-type 1 IFN mixture). After 1 h, 100 U/mL of IFN-λ1 or IFN-β1 was added to the HIEs. After 26 h, total RNA was extracted, and the transcriptional response was compared with HIE cultures that were neither antibody-treated nor IFN-treated. Transcript levels of the ISG OAS2 were first normalized to GAPDH levels, and the fold increase was calculated by using the 2−ΔΔCt method. Displayed above the bars is the percent reduction in OAS2 levels between the two groups being compared. Data in A and B are presented as geometric mean ± 95% CI of the geometric mean of three technical replicates. (C) HIEs (j3) were treated with or without blocking antibodies to the type III IFN receptor (anti-IL28RA) or the type I IFN system (anti-type 1 IFN mixture). After 2 h, HIEs were treated with 100 U/mL of IFN-λ1 or IFN-β1 for 24 h. After 24 h, HIEs were washed to remove the antibodies and IFN and inoculated with Ito-HRV (MOI of 0.1). The control samples consisted of HIEs which did not receive antibodies nor IFN before Ito-HRV infection. The increase in infectious virus was calculated by subtracting the titer at 1.5 hpi in the control infection from the titer at 24 hpi in each of the five test groups. Data from one representative experiment of two independent experiments is presented as arithmetic mean ± SEM of four technical replicates. Statistical analyses were performed by Mann–Whitney U test.
We verified that blocking antibodies [anti-type III IFN receptor (anti-IL28RA) and anti-type I IFN system (anti-IFNAR2, anti–IFN-β, anti–IFN-α)] did not contain rotavirus-neutralizing antibodies. Stock aliquots of Ito-HRV were incubated with the blocking antibodies, followed by titration on MA104 cells. There was no significant change in viral titer compared with the control aliquots, indicating that the blocking antibodies were not contaminated with antibodies that influence the assessment of rotavirus infectivity (Fig. S4A). Because the maintenance media during HRV growth experiments contains pancreatin, we also evaluated the stability of the receptor-blocking antibodies in the presence of pancreatin. HIEs were incubated with antibodies to the type I IFN or type III IFN receptors in the presence of 0.5 mg/mL pancreatin. After 1 h, 100 U/mL of IFN-λ1 or 100 U/mL of IFN-β1 was added to the HIEs, which were maintained in pancreatin-containing media for the duration of the experiment. After 26 h of IFN treatment, the levels of the ISG OAS2 were evaluated in HIEs treated with IFN alone or with IFN and receptor-blocking antibodies. Compared with untreated HIEs, OAS2 was up-regulated in IFN-λ1–treated and IFN-β1–treated HIEs. However, when HIEs were incubated with IFN receptor-blocking antibodies before IFN treatment, OAS2 levels did not increase relative to untreated HIEs, indicating that the antibodies had the ability to block the effect of IFN even when the surrounding media contained pancreatin (Fig. S4B). In addition, we confirmed that the antibodies blocked the inhibitory effect of IFN-λ1 and IFN-β1 on rotavirus replication; viral growth was equivalent in control HIEs compared with HIEs that were pretreated with exogenous IFN in the presence of IFN receptor-blocking antibodies before infection (Fig. S4C).
The amounts of HIE-secreted IFN-λ1, IFN-λ2/3, and IL-8 were quantified by using ELISA kits from eBioscience according to the manufacturer’s protocol. Both the VeriKine Human IFN-β ELISA kit (PBL) and the VeriKine-HS Human IFN-β ELISA kit (PBL) were used to assess for IFN-β secretion. Values below the limit of detection by ELISA were represented as one half of the limit of detection. The limits of detection of IFN-λ1, IFN-λ2/3, and IFN-β by ELISA were 8 pg/mL, 15.6 pg/mL, and 50 pg/mL (1.2 pg/mL for the VeriKine-HS Human IFN-β ELISA), respectively. Because of the high sensitivity (2 pg/mL) of the IL-8 ELISA, the cell culture supernatant was initially diluted 1:20 for testing. Because the IFN-λ1 ELISA (eBioscience) quantified IFN-λ1 amount by weight (in picograms) and the IFN-λ1 (PBL) used for HIE treatment was quantified by International Units, a standard curve was generated to assess the limit of detection by ELISA of PBL-obtained IFN-λ1 (units). The IFN-λ1 ELISA limit of detection of 8 pg/mL by weight corresponded to a limit of detection of 2–4 U/mL of IFN-λ1.
Before assessing HIE supernatants for protein content by ELISA, HIEs were centrifuged at 400 × g for 3 min to pellet the cells from the supernatant. The supernatant was then collected and placed directly onto the ELISA plate. To quantify protein content from concentrated media (Table S1), supernatant was combined from multiple samples and was concentrated through an Amicon Ultra-4 filter unit with a 10-kDa cutoff per the manufacturer’s instructions (Merck Millipore).
RNA Extraction and RT-qPCR Analysis.
Immediately before RNA extraction, HIEs were centrifuged at 400 × g for 3–5 min, and the supernatant was discarded. Total RNA was extracted from pelleted HIEs by using the QIAshredder kit and RNeasy Mini Kit followed by treatment with RNase-free DNase (Qiagen) for all experiments, including microarray and RNA-seq experiments, except for the experiments described in Figs. S3A and S4B, in which the Direct-zol RNA MiniPrep Kit was used (Zymo Research). Transcriptional analyses performed in Figs. 3–8 were determined by RT-qPCR.
RT-qPCR was performed (10) by using TaqMan primer-probe mixes (Thermo Fisher Scientific) with the following accession identification numbers: Hs99999905_m1 (glyceraldehyde-3-phosphate dehydrogenase, GAPDH), Hs00601677_g1 (IFN-λ1, IFNL1), Hs00820125_g1 (IFN-λ2, IFNL2), Hs00265051_s1 (IFN-α2, IFNA2), Hs01077958_s1 (IFN-β1, IFNB1), Hs00174103_m1 (IL-8, IL8), Hs00915292_m1 (IFN-induced protein 44-like, IFI44L), Hs01911452_s1 (IFN-induced protein with tetratricopeptide repeats 1, IFIT1), Hs00942643_m1 (2′-5′-oligoadenylate synthetase 2, OAS2), Hs00180943_m1 (HECT and RLD domain containing E3 ubiquitin protein ligase 5, HERC5), Hs00369813_m1 (radical S-adenosyl methionine domain-containing 2, RSAD2), and Hs00895608_m1 (MX dynamin-like GTPase 1, MX1). For all transcriptional fold changes determined by RT-qPCR, total RNA was extracted from infected HIE cultures and compared with matched uninfected (mock-inoculated) HIE cultures. Expression levels were first normalized to GAPDH levels and analyzed by using StepOne version 2.1 software (Applied Biosystems). The fold increase was calculated by application of the 2−ΔΔCt method as previously described (97).
Microarray-Based Transcriptome Analysis.
Principal component analysis was performed on global gene expression data of the first microarray experiment by using the princomp() function of the stats R library. Clustering analysis was performed on global gene expression data of the second microarray experiment. To account for time point as a covariate, gene expression values were corrected by regressing out the time point status for each gene. Next, Spearman correlation of gene expression values was calculated between all possible sample pairs. Complete linkage clustering was performed on 1 − Spearman coefficient by using the hclust() function of the stats R library.
Correspondence between the global transcriptional responses to infection across HIEs from different individuals was assessed. Gene-expression fold changes between infected and uninfected cultures were calculated. Next, Spearman rank correlation of the fold changes was calculated between the three HIEs from different individuals.
To assess the significance of gene overlap across the 100 most up-regulated genes, we used a permutation-based approach. In 100,000 permutations, we randomly selected 100 genes from the 20,948 expressed genes three times and counted how many genes were chosen all three times for each permutation.
RNA-seq–Based Transcriptome Analysis.
Total RNA was extracted, and RNA-seq was performed on rRNA-reduced samples of RNA. Reads acquired from RNA-seq were aligned to the human hg19 and hg38 reference genomes by using STAR (version 2.3.1) (92). Totals of 87%, 86%, 68%, and 68% of reads for samples j11-uninfected, j2-uninfected, j11-infected, and j2-infected aligned uniquely to the human genome, respectively. The reads from this RNA-seq experiment for each of the four samples were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under accession no. GSE90796. To identify rotavirus RNA sequences, the resulting unmapped reads were mapped against a preassembled database of all rotavirus genomes from GenBank using Bowtie 2 (93) with default settings.
For analysis of gene expression, read summarization was performed by using the featureCounts function of the Subread package (94) and the Ensembl gene annotation gtf file with genome-build-accession identifier NCBI:GCA_000001405.14. Genes with less than one read per 10 million sequenced reads in two or more samples were excluded from subsequent analysis. Gene-expression estimates were normalized for gene length and sequencing depth by using the DESeq2 normalization method (95). Correspondence between gene expression estimates derived from microarrays and RNA-seq was assessed. Based on the gene symbols, 13,696 genes were profiled in the microarrays and RNA-seq. Mean expression levels of these 13,696 genes were calculated across the three uninfected samples from the second microarray experiment and the two uninfected RNA-seq samples independently. Next, Spearman rank correlation was used to evaluate correspondence between mean expression levels derived from microarrays and RNA-seq. Differential expression analysis was conducted by using R statistical software in conjunction with the DESeq2 (version 1.2.10) package (95). In short, samples were normalized for sequencing depth by using the standard DESeq2 function estimateSizeFactors. The DESeq2 modeling framework is based on the negative binomial distribution and uses the DESeq2 normalized gene counts as input. In our model, we accounted for a person effect and used infection status as the explanatory variable.
To identify ISGs in the RNA-seq dataset, the sequenced gene set was compared with the open-access Interferome version 2.01 database of IFN-regulated genes (96). Pathway analysis was performed on genes identified by RNA-seq using Ingenuity Pathway Analysis software (Qiagen).
Statistical Analyses.
Statistical analyses on data (not including microarray and RNA-seq results) were performed with Excel (Microsoft) and GraphPad Prism (GraphPad) software. For data with samples sizes ≥4, a nonparametric test such as the Mann–Whitney U test or Wilcoxon signed-rank test was used. For data with samples sizes of three, an unpaired Student’s t test was performed. Results were considered significant when the P value or FDR was <0.05. For graphs plotted on a logarithmic axis, the data are represented as the geometric mean ± 95% confidence interval (CI) of the geometric mean. For all other graphs, the data are represented as the arithmetic mean ± SD or SEM. Data are depicted as a single representative experiment from multiple repeat independent experiments or as a compilation figure of multiple independent experiments. Technical replicates refer to experimental replicates of HIEs from the same individual, and biological replicates refer to experimental replicates of HIEs from different individuals.
Acknowledgments
We thank Joel M. Sederstrom, Dr. Lisa D. White, Mylinh Bernardi, and Daniela Dias Xavier for their expert assistance, as well as Manasi Gadkari for expert assistance with performing microarray experiments. We also thank Dr. Harry Greenberg for helpful discussions and comments during preparation of this manuscript. This work was supported by National Institutes of Health (NIH) Grants U19-AI116497 and R01 AI080656 (to M.K.E.), U18-TR000552 (to M.D.), R21-AI117220 (to M.E.C.), and Howard Hughes Medical Institute Grant 570076890 (to K.S.). This project also was supported by Advanced Technology Core Laboratories at Baylor College of Medicine. This included core support from the Integrated Microscopy Core at Baylor College of Medicine with funding from Grants P30 DK-56338 (to H. El-Serag, Principal Investigator), P30 CA125123 (to C. K. Osborne, Principal Investigator), CPRIT RP150578 (to P. Davies, Principal Investigator), the Dan L. Duncan Cancer Center and the John S. Dunn Gulf Coast Consortium for Chemical Genomics; the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the Grants P30 AI036211 (to J. Butel, Principal Investigator), P30 CA125123 (to C. K. Osborne, Principal Investigator), and S10 RR024574 (to E. Lumpkin, Principal Investigator); and the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from the Grants P30 DK56338 (to H. El-Serag, Principal Investigator) and P30 CA125123 (to C. K. Osborne, Principal Investigator).
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE90796).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1615422114/-/DCSupplemental.
References
- 1.Duizer E, et al. Laboratory efforts to cultivate noroviruses. J Gen Virol. 2004;85(pt 1):79–87. doi: 10.1099/vir.0.19478-0. [DOI] [PubMed] [Google Scholar]
- 2.Kitamoto N, Ramig RF, Matson DO, Estes MK. Comparative growth of different rotavirus strains in differentiated cells (MA104, HepG2, and CaCo-2) Virology. 1991;184(2):729–737. doi: 10.1016/0042-6822(91)90443-f. [DOI] [PubMed] [Google Scholar]
- 3.Forbester JL, et al. Interaction of Salmonella enterica serovar typhimurium with intestinal organoids derived from human induced pluripotent stem cells. Infect Immun. 2015;83(7):2926–2934. doi: 10.1128/IAI.00161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ettayebi K, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016;353(6306):1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foulke-Abel J, et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014;239(9):1124–1134. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 7.Fuller MK, et al. Intestinal stem cells remain viable after prolonged tissue storage. Cell Tissue Res. 2013;354(2):441–450. doi: 10.1007/s00441-013-1674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanDussen KL, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64(6):911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxena K, et al. Human intestinal enteroids: A new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol. 2016;90(1):43–56. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD. World Health Organization–Coordinated Global Rotavirus Surveillance Network Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis. 2016;62(suppl 2):S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PubMed] [Google Scholar]
- 11.Ciarlet M, Estes MK, Barone C, Ramig RF, Conner ME. Analysis of host range restriction determinants in the rabbit model: Comparison of homologous and heterologous rotavirus infections. J Virol. 1998;72(3):2341–2351. doi: 10.1128/jvi.72.3.2341-2351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramig RF. The effects of host age, virus dose, and virus strain on heterologous rotavirus infection of suckling mice. Microb Pathog. 1988;4(3):189–202. doi: 10.1016/0882-4010(88)90069-1. [DOI] [PubMed] [Google Scholar]
- 13.Green K. 2013. Caliciviridae: The Noroviruses (Lippincott Williams & Wilkins, Philadelphia), 6th Ed.
- 14.Jogler C, et al. Replication properties of human adenovirus in vivo and in cultures of primary cells from different animal species. J Virol. 2006;80(7):3549–3558. doi: 10.1128/JVI.80.7.3549-3558.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuadras MA, Feigelstock DA, An S, Greenberg HB. Gene expression pattern in Caco-2 cells following rotavirus infection. J Virol. 2002;76(9):4467–4482. doi: 10.1128/JVI.76.9.4467-4482.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frias AH, et al. Intestinal epithelia activate anti-viral signaling via intracellular sensing of rotavirus structural components. Mucosal Immunol. 2010;3(6):622–632. doi: 10.1038/mi.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin JD, et al. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 2016;12(4):e1005600. doi: 10.1371/journal.ppat.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J Virol. 2009;83(20):10322–10335. doi: 10.1128/JVI.01186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blutt SE, Miller AD, Salmon SL, Metzger DW, Conner ME. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. 2012;5(6):712–719. doi: 10.1038/mi.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franco MA, Greenberg HB. Immunity to rotavirus infection in mice. J Infect Dis. 1999;179(suppl 3):S466–S469. doi: 10.1086/314805. [DOI] [PubMed] [Google Scholar]
- 21.Burns JW, et al. Analyses of homologous rotavirus infection in the mouse model. Virology. 1995;207(1):143–153. doi: 10.1006/viro.1995.1060. [DOI] [PubMed] [Google Scholar]
- 22.Murphy K, Travers P, Walport M, Janeway C. Janeway’s Immunobiology. 7th Ed Garland Science; New York: 2008. [Google Scholar]
- 23.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7(2):131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 24.Arnold MM, Sen A, Greenberg HB, Patton JT. The battle between rotavirus and its host for control of the interferon signaling pathway. PLoS Pathog. 2013;9(1):e1003064. doi: 10.1371/journal.ppat.1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holloway G, Coulson BS. Innate cellular responses to rotavirus infection. J Gen Virol. 2013;94(pt 6):1151–1160. doi: 10.1099/vir.0.051276-0. [DOI] [PubMed] [Google Scholar]
- 26.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotenko SV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4(1):69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 29.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4(3):e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotenko SV. IFN-λs. CurrOpinImmunol. 2011;23(5):583–590. doi: 10.1016/j.coi.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoggins JW. Interferon-stimulated genes: Roles in viral pathogenesis. Curr Opin Virol. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng N, et al. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J Virol. 2008;82(15):7578–7590. doi: 10.1128/JVI.00391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez S, Sanchez-Tacuba L, Moreno J, Arias CF. Rotavirus strategies against the innate antiviral system. Annu Rev Virol. 2016;3(1):591–609. doi: 10.1146/annurev-virology-110615-042152. [DOI] [PubMed] [Google Scholar]
- 34.Velasquez DE, Parashar UD, Jiang B. Strain diversity plays no major role in the varying efficacy of rotavirus vaccines: An overview. Infect Genet Evol. 2014;28:561–571. doi: 10.1016/j.meegid.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Arnold MM, Patton JT. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J Virol. 2011;85(5):1970–1979. doi: 10.1128/JVI.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciarlet M, Estes MK. Human and most animal rotavirus strains do not require the presence of sialic acid on the cell surface for efficient infectivity. J Gen Virol. 1999;80(pt 4):943–948. doi: 10.1099/0022-1317-80-4-943. [DOI] [PubMed] [Google Scholar]
- 37.Hermant P, et al. Human but not mouse hepatocytes respond to interferon-lambda in vivo. PLoS One. 2014;9(1):e87906. doi: 10.1371/journal.pone.0087906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lasfar A, et al. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 2006;66(8):4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- 39.Zachos NC, et al. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem. 2016;291(8):3759–3766. doi: 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.In J, et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol. 2016;2(1):48–62.e3. doi: 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat Biotechnol. 2014;32(9):926–932. doi: 10.1038/nbt.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirata Y, Broquet AH, Menchén L, Kagnoff MF. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J Immunol. 2007;179(8):5425–5432. doi: 10.4049/jimmunol.179.8.5425. [DOI] [PubMed] [Google Scholar]
- 43.Ioannidis I, Ye F, McNally B, Willette M, Flaño E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol. 2013;87(6):3261–3270. doi: 10.1128/JVI.01956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015;36(3):124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawton JA, Estes MK, Prasad BV. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat Struct Biol. 1997;4(2):118–121. doi: 10.1038/nsb0297-118. [DOI] [PubMed] [Google Scholar]
- 46.Rojas M, Arias CF, López S. Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J Virol. 2010;84(20):10457–10466. doi: 10.1128/JVI.00625-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80(10):5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheth R, et al. Rotavirus stimulates IL-8 secretion from cultured epithelial cells. Virology. 1996;221(2):251–259. doi: 10.1006/viro.1996.0374. [DOI] [PubMed] [Google Scholar]
- 49.Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. 2007;81(9):4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J Virol. 2002;76(18):9545–9550. doi: 10.1128/JVI.76.18.9545-9550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold MM. The rotavirus interferon antagonist NSP1: Many targets, many questions. J Virol. 2016;90(11):5212–5215. doi: 10.1128/JVI.03068-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groene WS, Shaw RD. Psoralen preparation of antigenically intact noninfectious rotavirus particles. J Virol Methods. 1992;38(1):93–102. doi: 10.1016/0166-0934(92)90172-a. [DOI] [PubMed] [Google Scholar]
- 53.Bullens DM, Kasran A, Peng X, Lorré K, Ceuppens JL. Effects of anti-IL-4 receptor monoclonal antibody on in vitro T cell cytokine levels: IL-4 production by T cells from non-atopic donors. Clin Exp Immunol. 1998;113(3):320–326. doi: 10.1046/j.1365-2249.1998.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hernández PP, et al. Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. 2015;16(7):698–707. doi: 10.1038/ni.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pott J, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci USA. 2011;108(19):7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015;11(4):e1004782. doi: 10.1371/journal.ppat.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin Z, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol. 2012;189(6):2735–2745. doi: 10.4049/jimmunol.1102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sen A, et al. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc Natl Acad Sci USA. 2012;109(50):20667–20672. doi: 10.1073/pnas.1212188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg Microbes Infect. 2014;3(7):e51. doi: 10.1038/emi.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ng CT, Mendoza JL, Garcia KC, Oldstone MB. Alpha and beta type 1 interferon signaling: Passage for diverse biologic outcomes. Cell. 2016;164(3):349–352. doi: 10.1016/j.cell.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang B, et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science. 2014;346(6211):861–865. doi: 10.1126/science.1256999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 63.Middendorp S, et al. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells. 2014;32(5):1083–1091. doi: 10.1002/stem.1655. [DOI] [PubMed] [Google Scholar]
- 64.Wold W, Ison M. Adenoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1732–1767. [Google Scholar]
- 65.Mendez E, Arias C. Astroviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 609–628. [Google Scholar]
- 66.Park H, et al. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology. 2012;56(6):2060–2070. doi: 10.1002/hep.25897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sato S, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42(1):123–132. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 68.Crotta S, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013;9(11):e1003773. doi: 10.1371/journal.ppat.1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune functions at barrier surfaces and beyond. Immunity. 2015;43(1):15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Odendall C, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15(8):717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohan KV, Som I, Atreya CD. Identification of a type 1 peroxisomal targeting signal in a viral protein and demonstration of its targeting to the organelle. J Virol. 2002;76(5):2543–2547. doi: 10.1128/jvi.76.5.2543-2547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frias AH, Jones RM, Fifadara NH, Vijay-Kumar M, Gewirtz AT. Rotavirus-induced IFN-β promotes anti-viral signaling and apoptosis that modulate viral replication in intestinal epithelial cells. Innate Immun. 2012;18(2):294–306. doi: 10.1177/1753425911401930. [DOI] [PubMed] [Google Scholar]
- 73.Christian SL, et al. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One. 2012;7(9):e44267. doi: 10.1371/journal.pone.0044267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Critchley-Thorne RJ, et al. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci USA. 2009;106(22):9010–9015. doi: 10.1073/pnas.0901329106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nice TJ, et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347(6219):269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sánchez-Tacuba L, Rojas M, Arias CF, López S. Rotavirus controls activation of the 2′-5′-oligoadenylatesynthetase/RNase L pathway using at least two distinct mechanisms. J Virol. 2015;89(23):12145–12153. doi: 10.1128/JVI.01874-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lutz LM, Pace CR, Arnold MM. Rotavirus NSP1 associates with components of the Cullin RING ligase family of E3 ubiquitin ligases. J Virol. 2016;90(13):6036–6048. doi: 10.1128/JVI.00704-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding S, et al. Comparative proteomics reveals strain-specific β-TrCP degradation via rotavirus NSP1 hijacking a host Cullin-3-Rbx1 complex. PLoS Pathog. 2016;12(10):e1005929. doi: 10.1371/journal.ppat.1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gratia M, et al. Rotavirus NSP3 is a translational surrogate of the poly(A) binding protein-poly(A) complex. J Virol. 2015;89(17):8773–8782. doi: 10.1128/JVI.01402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Montero H, Rojas M, Arias CF, López S. Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J Virol. 2008;82(3):1496–1504. doi: 10.1128/JVI.01779-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubio RM, Mora SI, Romero P, Arias CF, López S. Rotavirus prevents the expression of host responses by blocking the nucleocytoplasmic transport of polyadenylated mRNAs. J Virol. 2013;87(11):6336–6345. doi: 10.1128/JVI.00361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB. Rotavirus structural proteins and dsRNA are required for the human primary plasmacytoid dendritic cell IFNalpha response. PLoS Pathog. 2010;6(6):e1000931. doi: 10.1371/journal.ppat.1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yin Y, et al. Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antiviral Res. 2015;123:120–131. doi: 10.1016/j.antiviral.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 84.Jaitin DA, et al. Inquiring into the differential action of interferons (IFNs): An IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006;26(5):1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baldridge MT, et al. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science. 2015;347(6219):266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shulzhenko N, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17(12):1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ank N, et al. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80(9):4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna: 2013. [Google Scholar]
- 89.Crawford SE, et al. Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol. 2001;75(13):6052–6061. doi: 10.1128/JVI.75.13.6052-6061.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schönborn J, et al. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991;19(11):2993–3000. doi: 10.1093/nar/19.11.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Criglar JM, et al. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol. 2014;88(2):786–798. doi: 10.1128/JVI.03022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dobin A, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liao Y, Smyth GK, Shi W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 95.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rusinova I, et al. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41(database issue):D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]