

Abstract
Aim
The expression level of DNA repair-related genes and their association with breast cancer status among participants of the New York site of the Breast Cancer Family Registry was investigated.
Materials and Methods
RNA from mononuclear cells in 194 sister sets (n=475 women) were assayed for ATM, BRCA1, MSH2, MUTYH and XPC gene expression levels and analyzed using generalized estimating equations (GEE).
Results
Individuals with decreased ATM and MSH2 expression had significantly higher odds for breast cancer compared to individuals with higher levels of expression (odds ratio (OR)=1.1, 95% confidence interval (CI)=1.02, 1.18) and (OR=1.90, 95% CI=1.21, 2.97), respectively. Upon stratifying the GEE model, reductions in ATM and MSH2 expression levels was heightened among women with an extended family history (FH) of breast cancer.
Conclusion
Reduced expression of ATM and MSH2 compromises DNA repair capacity and, thereby, increases breast cancer prevalence.
Keywords: Breast cancer, DNA repair, gene expression, family history
Based on current rates, one in eight women will develop breast cancer over their lifetime (1). The presence of familial aggregation of breast cancer is one of the major known risk factors, accounting for roughly 20% of all breast cancer cases and resulting in an additional two to three-fold increase in risk among individuals stemming from families in which multiple members present with the disease (2). In addition to a heightened risk of breast cancer, familial breast cancer cases typically present at a younger age and are more likely to develop bilateral breast cancer. There are also implications for histological presentation of the disease and risk for additional cancer-subtypes, as observed by the triple-negative phenotype and increased risk for ovarian cancer among familial cases due to BRCA1 mutations (3).
The indication of a distinct disease profile for familial breast cancer highlights the need to understand the factors driving familial presentation of the disease to better inform means of prevention and treatment relevant to this population. The role of compromised DNA repair capacity as a component driving familial aggregation has been demonstrated by the fact that the major predisposition genes BRCA1 and BRCA2 are now known to be involved in double-strand break repair (DSBR) (4). In addition to these genes, a handful of other less penetrant susceptibility genes have been identified, including ATM, a gene also involved in DSBR. Women who have inherited mutations in either BRCA1 or BRCA2 have up to an 80% likelihood of developing breast cancer by age 75, while women who are carriers of mutations in ATM are at a 2-fold increased risk of breast cancer compared to non-carriers (5, 6). However, these mutations only account for a proportion of all familial cases, likely due to the severity of the defect associated with complete loss of function in these genes, indicating additional factors remain to be identified.
Familial-based case-control studies have also demonstrated associations with breast cancer due to reductions in overall DSBR pathway activity, as well as nucleotide excision repair (NER) capacity, as indicated by the phenotypic assessment of repair (7, 8). The subtle modulation of repair activity observed in these studies may be met by means other than complete loss of function mutations, including epigenetic and post-transcriptional regulation of gene activity.
As gene expression levels capture regulation of gene activity, we assessed the association between breast cancer status and the expression levels of 5 genes, ATM, BRCA1, MSH2, MUTYH and XPC, known to be involved in various DNA repair pathways in peripheral blood mononuclear cells (PBMCs) from sister-sets enrolled in the New York site of the Breast Cancer Family Registry (NY-BCFR).
Materials and Methods
Study population
The study participants consisted of 194 sister-sets (n=475) enrolled at the New York site of the Breast Cancer Family Registry (BCFR) with RNA available from viable PBMCs. Participants had to meet one or more of the following eligibility criteria: (i) A female relative who had been diagnosed with either breast or ovarian cancer prior to the age of 45; (ii) a female relative who had been diagnosed with breast and ovarian cancer at any age; (iii) two or more female relatives who had been diagnosed with breast or ovarian cancer after the age of 45; (iv) a male relative diagnosed with breast cancer at any age; (v) a known carrier of BRCA1 or 2 mutation. Demographic characteristics, epidemiologic risk factors, family history of cancer and food frequency information were determined through questionnaires. Blood was collected at the time of recruitment, on average 5 years after diagnosis for cases (9).
DNA and RNA isolation
PBMCs previously isolated via a Ficoll gradient from 45 ml of blood collected with ACD as an anticoagulant and frozen at −140°C were thawed, pelleted and washed with phosphate buffered saline (PBS). DNA and RNA were subsequently extracted using an All-prep DNA/RNA 96 kit (Qiagen, Valencia, CA, USA). Purified RNA and DNA were quantified using a Nanodrop 2000 (Thermo Scientific, Pittsburgh, PA, USA).
Reverse transcription (RT) and gene expression
Sixty nanograms of RNA was reverse transcribed using a High Capacity cDNA reverse transcription kit (Life Technologies, Carlsbad, CA, USA). The reaction consisted of 10X RT buffer, 25X dNTP Mix (100 mM), 10X RT random primers and MultiScribe Reverse Transcriptase. An RT (−) control, in which the reaction mixture included a randomly selected RNA template and all RT reagents with the exception of the reverse transcription enzyme, was included as a means to assess the presence of genomic DNA contamination. The thermocycling conditions were as follows: 25°C for 10 min, 37°C for 120 min and 85°C for 5 min.
Expression of the target genes was assessed using pre-designed Taqman assays: Hs01112347_m1 (ATM), Hs01556194_m1 (BRCA1), Hs00953523_m1 (MSH2), Hs01014856_m1 (MUTYH), Hs00190295_m1 (XPC) (Life Technologies). The reaction mixture consisted of 2.5 ng cDNA template, 20X Gene expression assay and 2X Taqman Universal Master Mix. The thermocycling conditions included an initial incubation at 95°C for 10 min, followed by a 95°C for 15 s and 60°C for 1 min, repeated for 40 cycles. Level of fluorescence from targeted transcripts was determined using FAM-reporter labeled probes.
Samples were run in duplicate for each target and endogenous control assay. The selection of β-actin as endogenous control was based on demonstrating high and lowly variable expression in PBMCs in a prior pilot assessment of three commonly assessed housekeeping genes (ACTB, GAPDH and RPL0). Each assay was also conducted with cDNA derived from commercially purchased human total RNA (Life Technologies) as a calibrator sample, an RT (−) control and a non-template control. Relative expression was determined using the 2−ΔΔCt method.
Statistical methods
Pearson’s Chi-square tests were performed to assess the distribution of the study population according to selected measured variables. Known contributors to breast cancer onset that were included in the analyses comprised age at blood draw, smoking status (ever vs. never), body mass index (BMI) (≥25kg/m2 vs. <25 kg/m2, cut-off based on WHO definition of normal range), age at menarche (>13 vs. ≤13 years, based on median age at menarche among unaffected sisters), age at first parity (>30 vs. ≤30 years, nulliparous women were assigned age at blood draw) and extended family history (>1 vs. 1).
Generalized estimating equations (GEE) were run to assess the associations between repair gene expression levels and breast cancer status. Relevance to breast cancer status was determined by evaluating expression level as a continuous variable, with a change in odds of breast cancer assessed due to a 1 unit-fold decrease in expression level and evaluating expression level partitioned into tertiles with cut-offs based on controls. To adjust for potential confounding, additional known relevant factors for disease onset, including age at blood draw and smoking status, were included in the models. Effect modification was assessed to determine whether extended family history impacted the association between the lowest tertile of expression in the assayed genes and breast cancer status. All statistical analyses were conducted using SAS software 9.3 (SAS Institute, Cary, NC, USA). Figures were generated using the ggplot2 R package (10).
Results
Selection of candidate genes
We selected candidate genes spanning over four DNA repair pathways based on information available regarding repair-related findings in studies conducted within the NY-BCFR and otherwise reported in the literature. ATM and BRCA1 are known breast cancer susceptibility genes participating in DSBR. MSH2, a component of mismatch repair (MMR), and MUTYH, a component of base excision repair (BER), are targets for which single nucleotide polymorphisms (SNPs) were previously identified in association with breast cancer status in a study conducted within the registry (Kappil et al., manuscript in preparation). While overall reductions in NER capacity have been associated with breast cancer in studies conducted within the registry, no specific component of NER has been identified as contributing to the observed association (8, 11). Hence, we selected XPC as our component of interest in NER, as an association between reduced expression levels of XPC and lung cancer risk has been previously reported (12).
Study population characteristics
The distributions of various factors known to be relevant to breast cancer are shown in Table I. No significant differences between cases and controls were observed based on age at blood draw, BRCA1/2 mutation status, ethnicity, smoking status, BMI, age at menarche, parity or family history.
Table I.
Distribution of selected variables among sisters affected and unaffected with breast cancer in the Breast Cancer Family Registry (BCFR).
| Variable | No. cases (%) (n=219) |
No. controls (%) (n=271) |
p-Valuea |
|---|---|---|---|
| Age at blood draw (in years) | |||
| ≤50 | 127 (58.0) | 172 (63.5) | 0.22 |
| >50 | 92 (42.0) | 99 (36.5) | |
| BRCA1 (+) mutation | 12 (5.5) | 8 (3.0) | 0.16 |
| BRCA2 (+) mutation | 10 (4.6) | 5 (1.8) | 0.08 |
| Ethnicity | |||
| Non-Hispanic White | 117 (54.2) | 127 (47.0) | 0.27 |
| African American | 5 (2.3) | 9 (3.3) | |
| Other | 94 (43.5) | 134 (49.6) | |
| Smoking Status | |||
| Never | 129 (58.9) | 164 (60.5) | 0.72 |
| Ever | 90 (41.1) | 107 (39.5) | |
| BMI (kg/m2) | |||
| <25 | 106 (48.4) | 127 (47.4) | 0.82 |
| ≥25 | 113 (51.6) | 141 (52.6) | |
| Age at menarche (in years) | |||
| <13 | 101 (46.3) | 117 (43.2) | 0.49 |
| ≥13 | 117 (53.7) | 154 (56.8) | |
| Parity | |||
| Nulliparous | 39 (17.8) | 52 (19.2) | 0.70 |
| Parous | 180 (82.2) | 219 (80.8) | |
| Family History | |||
| 1 | 122 (55.7) | 172 (63.5) | 0.08 |
| >1 | 97 (44.3) | 99 (36.5) |
Pearson’s χ2-test; BMI, body mass index.
Analysis conducted using GEE indicated an inverse association between ATM expression level and breast cancer status with expression assessed as a continuous variable (odds ratio (OR)=1.10, 95% confidence interval (CI)=1.02, 1.18). Upon tertiling expression level, a significant association with breast cancer status was observed among women in the lowest tertile of expression for MSH2 (OR=1.90, 95%CI=1.21, 2.97). Adjusting for age at blood draw and smoking status did not appreciably alter the estimates from the crude model (Table II).
Table II.
Generalized estimating equations (GEE) analysis of the relationship between expression levels of various DNA repair genes and breast cancer risk among sisters discordant for breast cancer in the Breast Cancer Family Registry (BCFRa).
| OR (95% CI) | Adjusted OR (95% CI) | |
|---|---|---|
| ATM (n=172 sets) | ||
| Continuous | 1.10 (1.02,1.18) | 1.10 (1.02, 1.18) |
| T3 (>1.34) | 1.00 (referent) | 1.00 (referent) |
| T2 (>0.57−≤1.34) | 1.03 (0.66,1.61) | 1.02 (0.66, 1.59) |
| T1 (≤0.57) | 1.33 (0.90,1.96) | 1.35 (0.92, 1.99) |
| BRCA1 (n=114 sets) | ||
| Continuous | 1.43 (0.13, 16.04) | 1.52 (0.14, 16.93) |
| T3 (>0.05) | 1.00 (referent) | 1.00 (referent) |
| T2 (>0.02−≤0.05) | 1.21 (0.73, 2.03) | 1.23 (0.73, 2.06) |
| T1 (≤0.02) | 0.74 (0.43, 1.26) | 0.75 (0.44, 1.27) |
| MSH2 (n=155 sets) | ||
| Continuous | 2.09 (0.48, 9.03) | 2.17 (0.51, 9.24) |
| T3 (>0.09) | 1.00 (referent) | 1.00 (referent) |
| T2 (>0.03−≤0.09) | 0.93 (0.56, 1.53) | 0.92 (0.55, 1.54) |
| T1 (≤0.03) | 1.87 (1.19, 2.92) | 1.90 (1.21, 2.97) |
| MUTYH (n=134 sets) | ||
| Continuous | 0.99 (0.89, 1.09) | 0.99 (0.90, 1.09) |
| T3 (>1.02) | 1.00 (referent) | 1.00 (referent) |
| T2 (>0.39−≤1.02) | 0.96 (0.58, 1.59) | 0.97 (0.59, 1.59) |
| T1 (≤0.39) | 1.02 (0.69, 1.51) | 1.02 (0.68, 1.51) |
| XPC (n=178 sets) | ||
| Continuous | 1.01 (0.98, 1.03) | 1.01 (0.98, 1.03) |
| T3 (>3.30) | 1.00 (referent) | 1.00 (referent) |
| T2 (>1.05−≤3.30) | 0.90 (0.59, 1.38) | 0.90 (0.58, 1.38) |
| T1 (≤1.05) | 1.18 (0.81, 1.73) | 1.18 (0.81, 1.74) |
Model adjusted for age at blood draw and smoking status; OR, odds ratio; CI, confidence interval.
To further characterize the association between breast cancer status and the expression of DNA repair genes, we stratified our population based on extent of family history, with individuals stemming from families with more than one diagnosed case defined as having an extended family history. Among women with an extended family history of breast cancer, breast cancer prevalence was heightened due to reduced expression levels of ATM (OR=1.98, 95%CI=1.07, 3.68) and MSH2 (OR=2.19, 95%CI=1.02, 4.68) compared to women without an extended history of breast cancer (Figure 1).
Figure 1.
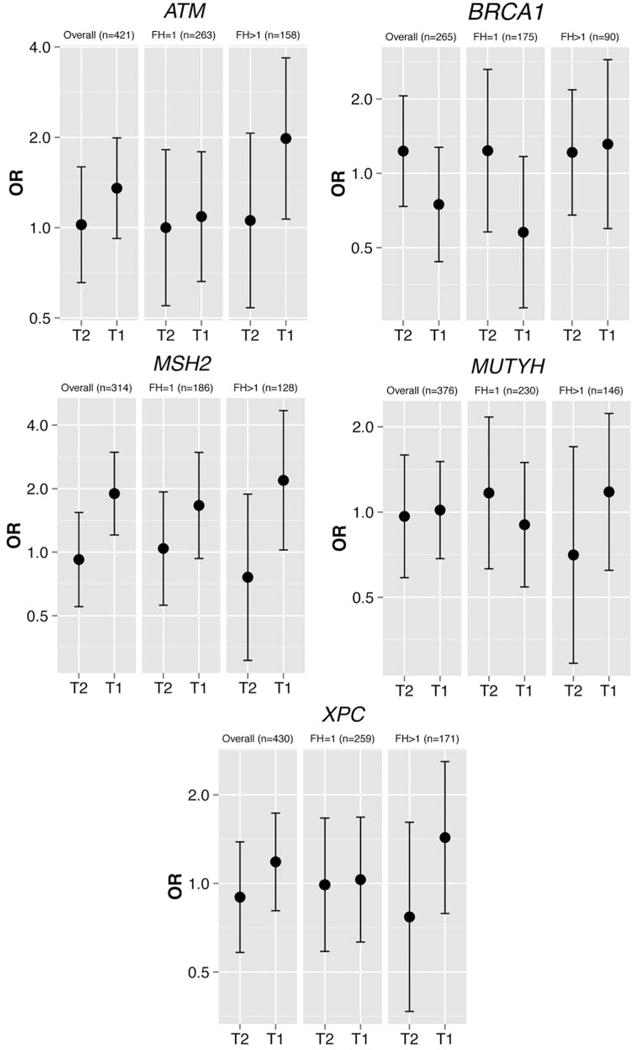
Generalized estimating equations (GEE) analysis of effect modification due to family history on the association of DNA repair gene and breast cancer adjusted for age at blood draw and smoking status in the Breast Cancer Family Registry (BCFR). *T1, Tertile 1; T2, tertile 2; FH, family history; OR, odds ratio.
Discussion
In the current study, we demonstrated an association between reduced ATM and MSH2 expression levels and breast cancer status. Similar to the major breast cancer predisposition genes, BRCA1 and BRCA2, ATM is also involved in the double-strand break repair pathway as a sensor of double-strand break damage. As a member of the P13K related protein kinases, the inactive serine-threonine kinase dimer phosphorylates into active monomers in the presence of double-strand breaks and transduces the signal to recruit downstream components of the pathway. Individuals with inherited mutations resulting in the inactivation of this kinase activity present with ataxia telangiectasia (AT), a disorder whose clinical features include a predisposition to cancer, with breast cancer leading as the predominant sub-type among women.
While reports focusing on the impact of individual, more highly penetrant variants in ATM on breast cancer status have been conflicting; a mutational screen of ATM in a familial-based breast cancer case-control study demonstrated a significantly higher count of variations among cases than among controls (13). However, as ATM spans 62 exons, such mutational screens seem infeasible to adopt in large-scale studies. mRNA transcript levels provide an alternative means of comprehensively capturing functional activity and several studies have shown reduced expression levels of ATM in breast tumors compared to normal tissue. To our knowledge, only one other breast cancer case-control study has reported on ATM expression levels in blood. Contrary to our findings, the study was not able to distinguish differences in expression levels based on case-control status (14). However, this may be related to the small sample size of the study population. Additionally, two studies have linked the epigenetic dysregulation of ATM with breast cancer susceptibility. Hypermethylation of an ATM intragenic region in peripheral blood was observed in bilateral breast cancer cases compared to controls (15). In a subsequent study, hypermethylation of this region was also detectable in pre-diagnostic blood samples of cases compared controls, indicating that the dysregulation of this gene is part of the carcinogenesis process rather than a response to cancer status or treatment (16).
In our study, the association between ATM expression level and breast cancer was modified by family history. This is in line with findings from a breast cancer case-control study where a higher mutation frequency in ATM among women with breast cancer was observed with increasing extent of family history (17). Furthermore, as in this study, other studies implicating ATM deficiency with breast cancer status have primarily been restricted to study populations selected for cases with a family history of breast cancer, while population-based case-control studies have failed to demonstrate similar associations. This further suggests that this marker may be of particular relevance to women with a family history of breast cancer and may, therefore, have implications for targeted genetic counseling.
While ATM is a known breast cancer susceptibility gene, a role for a deficiency in MSH2, or any other constituents of the mismatch repair pathway, has not been commonly reported in association with breast cancer risk. MSH2, acts in conjunction with either MSH6 to recognize small loops and insertions or MSH3 to recognize single base mismatches in the mismatch repair pathway. While microsatellite instability, a marker of MMR phenotype, has been observed in breast tumor tissues, mismatch repair has been much more clearly implicated in colorectal cancer and other Lynch syndrome affiliated outcomes. However, more recently, several groups have been calling for the inclusion of breast cancer among the cancer-subtypes that constitute Lynch syndrome. In support of this claim, a recently conducted prospective study within the Colon Cancer Family Registry has shown a nearly four-fold increase in risk of breast cancer over the general population among unaffected carriers of MMR gene mutations, while no increase in risk was observed among their non-carrier relatives (18). In addition to the increase in breast cancer incidence observed due to overall MMR deficiency, upon stratifying by MMR genotype, several studies have shown a heightened incidence among individuals with mutations specifically in MSH2 (19, 20).
In summary, we report on associations between familial breast cancer and reductions in the expression levels of ATM and MSH2. Given a true relationship between these genes and breast cancer status, the ability to detect meaningful differences in blood, as in our study, indicates a potential utility as a non-invasive marker of breast cancer status. These findings also hint at the etiology underlying familial instances of breast carcinogenesis as the identification of targets in differing DNA repair pathways indicates that a general compromise in the ability to rectify DNA damage is relevant in the familial presentation of the disease. Furthermore, findings from this study may also have prognostic implications given the known sensitivity to ionizing radiation observed among AT individuals, possibly resulting in a greater likelihood of subsequent disease among ATM deficient breast cancer cases due to treatment of the primary tumor.
The major strength of this study lies in the matched sister-set study design, ensuring comparability between cases and controls. However, the implications of the findings from the current study are also hindered by several inherent limitations. Primarily, the retrospective nature of the study precludes us from ruling out that our observations may be driven by reverse causality. This issue is of special concern in this population, as biospecimens were often collected following onset of treatment. Additionally, in our study, the markers of interest were assessed in PBMCs. This biospecimen serves as an attractive resource for biomarker development since it is easily procurable and composed of some long-lived blood-cell types. However, as PBMCs consist of a mixture of blood cell-types, it is unclear whether our findings may have been impacted by confounding due to cell-type composition. Due to the limitations in inferences that can be drawn from this study, additional studies will have to be conducted to validate these findings within a prospective setting. Given the importance of these genes in determining breast cancer status, factors influencing the varying levels of expression of these genes also remain to be determined. Finally, given an ultimate motivation to better define risk groups, further clarification on the implication for prognosis among women who present with deficiencies in these genes will need to be addressed.
Acknowledgments
This work was supported by a grant from the Breast Cancer Research Foundation and NIH grants P30ES009089, R01CA159868 and P30CA013696. The BCFR was supported by grant UM1 CA164920 from the USA National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products or organizations imply endorsement by the USA Government or the BCFR.
Footnotes
Conflicts of Interest
The Authors declare that they have no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Singletary SE. Rating the risk factors for breast cancer. Ann Surg. 2003;237:474–482. doi: 10.1097/01.SLA.0000059969.64262.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lalloo F, Evans DG. Familial breast cancer. Clin Genet. 2012;82:105–114. doi: 10.1111/j.1399-0004.2012.01859.x. [DOI] [PubMed] [Google Scholar]
- 4.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 5.Rahman N, Stratton MR. The genetics of breast cancer susceptibility. Annu Rev Genet. 1998;32:95–121. doi: 10.1146/annurev.genet.32.1.95. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25:5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- 7.Machella N, Terry MB, Zipprich J, Gurvich I, Liao Y, Senie RT, Kennedy DO, Santella RM. Double-strand breaks repair in lymphoblastoid cell lines from sisters discordant for breast cancer from the New York site of the BCFR. Carcinogenesis. 2008;29:1367–1372. doi: 10.1093/carcin/bgn140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy DO, Agrawal M, Shen J, Terry MB, Zhang FF, Senie RT, Motykiewicz G, Santella RM. DNA repair capacity of lymphoblastoid cell lines from sisters discordant for breast cancer. J Natl Cancer Inst. 2005;97:127–132. doi: 10.1093/jnci/dji013. [DOI] [PubMed] [Google Scholar]
- 9.John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, Ziogas A, Andrulis IL, Anton-Culver H, Boyd N, Buys SS, Daly MB, O’Malley FP, Santella RM, Southey MC, Venne VL, Venter DJ, West DW, Whittemore AS, Seminara D. The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6:R375–389. doi: 10.1186/bcr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wickham H. ggplot2: Elegant graphics for data analysis. New York: Springer; 2009. Retrieved from http://had.co.nz/ggplot2/book. [Google Scholar]
- 11.Shen J, Desai M, Agrawal M, Kennedy DO, Senie RT, Santella RM, Terry MB. Polymorphisms in nucleotide excision repair genes and DNA repair capacity phenotype in sisters discordant for breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1614–1619. doi: 10.1158/1055-9965.EPI-06-0218. [DOI] [PubMed] [Google Scholar]
- 12.Wu YH, Tsai Chang JH, Cheng YW, Wu TC, Chen CY, Lee H. Xeroderma pigmentosum group C gene expression is predominantly regulated by promoter hypermethylation and contributes to p53 mutation in lung cancers. Oncogene. 2007;26:4761–4773. doi: 10.1038/sj.onc.1210284. [DOI] [PubMed] [Google Scholar]
- 13.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 14.Foroughizadeh M, Mozdarani H, Majidzadeh AK, Kaviani A. Variation of ATM Gene Expression in Peripheral Blood Cells of Sporadic Breast Carcinomas in Iranian Patients. Avicenna J Med Biotechnol. 2012;4:95–101. [PMC free article] [PubMed] [Google Scholar]
- 15.Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, Fletcher O, Dos Santos Silva I, Peto J, Boshoff C, Narod S, Petronis A. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet. 2009;18:1332–1342. doi: 10.1093/hmg/ddp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan K, Garcia-Closas M, Orr N, Fletcher O, Jones M, Ashworth A, Swerdlow A. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012;72:2304–2313. doi: 10.1158/0008-5472.CAN-11-3157. [DOI] [PubMed] [Google Scholar]
- 17.Teraoka SN, Malone KE, Doody DR, Suter NM, Ostrander EA, Daling JR, Concannon P. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001;92:479–487. doi: 10.1002/1097-0142(20010801)92:3<479::aid-cncr1346>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 18.Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, Macrae FA, Goldblatt J, Southey MC, Arnold J, Thibodeau SN, Gunawardena SR, Bapat B, Baron JA, Casey G, Gallinger S, Le Marchand L, Newcomb PA, Haile RW, Hopper JL, Jenkins MA. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–964. doi: 10.1200/JCO.2011.39.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buerki N, Gautier L, Kovac M, Marra G, Buser M, Mueller H, Heinimann K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer. 2012;51:83–91. doi: 10.1002/gcc.20935. [DOI] [PubMed] [Google Scholar]
- 20.Win AK, Lindor NM, Young JP, Macrae FA, Young GP, Williamson E, Parry S, Goldblatt J, Lipton L, Winship I, Leggett B, Tucker KM, Giles GG, Buchanan DD, Clendenning M, Rosty C, Arnold J, Levine AJ, Haile RW, Gallinger S, Le Marchand L, Newcomb PA, Hopper JL, Jenkins MA. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst. 2012;104:1363–1372. doi: 10.1093/jnci/djs351. [DOI] [PMC free article] [PubMed] [Google Scholar]