

Abstract
Apolipoprotein E (apoE) and the low density lipoprotein receptor (LDLr) are well recognized determinants of atherosclerosis. In addition to hepatocytes, where both are highly expressed and contribute to plasma lipoprotein clearance, they are expressed in vascular cells and macrophages. In this study, we examined the effects of human apoE isoforms and LDLr levels in atherogenic pathways in primary macrophages ex vivo and atherosclerosis development after bone marrow transfer in vivo using mice expressing human apoE isoforms and different levels of LDLr expression. Increases in LDLr expression significantly increased cholesterol delivery into macrophages in culture, and the effects were more prominent with lipoproteins containing apoE4 than those containing apoE3. Conversely, increased LDLr expression reduced cholesterol efflux in macrophages expressing apoE4 but not in macrophages expressing apoE3. Furthermore, apoE3 protected VLDL from oxidation in vitro more than did apoE4. In LDLr-deficient mice expressing the human apoE4 isoform, Apoe4/4 Ldlr−/−, the replacement of bone marrow cells with those expressing LDLr increased atherosclerotic lesions in a dose-dependent manner compared with mice transplanted with cells having no LDLr. In contrast, atherosclerosis in Apoe3/3 Ldlr−/− mice, expressing the human apoE3 isoform, did not differ by the levels of macrophage LDLr expression. Our results demonstrate that apoE4, but not apoE3, in macrophages enhances atherosclerotic plaque development in mice in an LDLr-dependent manner and suggests that this interaction may contribute to the association of apoE4 with an increased cardiovascular risk in humans.
Elevated levels of cholesterol in plasma, carried by remnant particles of very low density lipoproteins (VLDL)3 and low density lipoproteins (LDL), are well known risk factors for atherosclerosis. The bulk of these atherogenic lipoproteins are cleared from the circulation by the LDL receptor (LDLr) in the liver with apolipoprotein E (apoE) and apoB100 as ligands. There are three common apoE isoforms in humans: apoE2, apoE3, and apoE4, each having distinct influences on both lipid metabolism and atherogenesis in humans (1). Possession of at least one copy of the APOE*2 allele is associated with higher plasma apoE and triglyceride but lower LDL cholesterol levels and atherosclerosis risk when compared with APOE*3 homozygotes (1). In contrast, the presence of at least one APOE*4 allele is associated with lower plasma apoE, increased LDL cholesterol, and a greater risk of coronary artery disease than APOE*3 homozygotes. This association is thought to be mainly due to differences in lipoprotein clearance and is counterintuitive, considering the LDLr affinity of apoE2 is lower while the affinity of apoE4 is slightly higher than apoE3 (2, 3).
To gain insights into the mechanisms underlying the relationship between atherosclerosis risk and apoE isoform in humans, we previously made mice expressing human apoE2, apoE3, or apoE4 in place of the endogenous mouse apoE (3–5). Notably, the atherosclerosis risk associated with the resulting mice was different from that in humans; mice with apoE2 had increased plasma lipids and developed atherosclerosis, whereas mice with apoE3 and apoE4 were normolipidemic and resistant to atherosclerosis. To further test whether the apoE isoform-dependent atherosclerosis risk is affected by the LDLr expression, we also developed mice in which the endogenous mouse Ldlr gene was replaced with a gene (hLdlr) coding for human LDLr. The transcriptional regulation of the hLdlr gene was normal, but the steady state levels of its mRNA in the liver were elevated, because the transcripts carry a more stable 3′-untranslated region sequence than normal. Somewhat unexpectedly, we found that, when this hLdlr allele was introduced into mice expressing human apoE isoforms, physiologic overexpression of the LDLr was protective in mice with apoE2 but caused severe atherosclerosis in mice with apoE4, recapitulating the associations between apoE isoforms and atherosclerosis risk seen in humans (6,7). These data suggest that the LDLr apoE interaction is central to the increased atherosclerosis risk associated with apoE4.
A substantial portion of the atherosclerosis risk associated with apoE4 is probably due to its hepatic metabolism by the LDLr and the resultant changes to plasma lipids. However, there is ample in vitro evidence that the interaction of apoE with the LDLr in the liver may not be entirely responsible for the risk of coronary artery disease associated with apoE4 in humans (8–10). Both ApoE and the LDLr are expressed in many cell types and are thought to play roles in the atherosclerotic process beyond their role in lipoprotein clearance (11). The effects on atherogenesis of apoE isoforms and levels of LDLr in extrahepatic tissues (e.g. in macrophages) are difficult to study in vivo, because their combined effects on plasma lipids are confounding.
Herein, we address the effect of LDLr expression level and human apoE3 and apoE4 isoforms on lipoprotein uptake and efflux in macrophages isolated from mice having human apoE isoforms and different levels of LDLr expression. We also examined in vivo whether the interactions between apoE and LDLr in macrophages, independent of global LDLr expression, affect atherosclerosis after bone marrow transfer (BMT) in mice expressing human apoE3 or apoE4 and lacking LDLr (3ko or 4ko) (12). We found that the expression of LDLr in macrophages directly correlated with the extent of atherosclerosis in mice with human apoE4. In contrast, macrophage LDLr expression did not affect atherosclerosis in mice expressing apoE3. These results indicate that apoE4 exerts adverse effects on bone marrow-derived cells in the vessel wall in an LDLr-dependent manner and may contribute to its pathogenesis.
Experimental Procedures
Mice
All the mutant mouse strains used in this work were individually backcrossed at least 6 generations to C57BL/6 genetic background before intercrossing. Mice heterozygous for a targeted replacement of the mouse Ldlr gene with the stabilized human Ldlr minigene (Apoe+/+Ldlrh/+) (13) were bred to mice homozygous for replacement of the mouse apoE gene with either the human APOE*3 or APOE*4 allele (Apoe3/3Ldlr+/+ and Apoe4/4Ldlr+/+) (3,5). The littermates generated by crossing Apoe3/3Ldlr+/+ (3m) with Apoe3/3Ldlrh/+ (3h) and Apoe4/4Ldlr+/+ (4m) with Apoe4/4Ldlrh/+ (4h), respectively, were used in experiments as donors of bone marrow cells. Mice with human APOE*3 and lacking LDLr (3ko) and mice with human APOE*4 and lacking LDLR (4ko) were generated by crossing either Apoe3/3 or Apoe4/4 mice with Ldlr−/− mice (6) and maintained as Apoe3/3 Ldlr−/− (3ko) or Apoe4/4 Ldlr−/− (4ko), respectively (see Table 1 for genotypes and nomenclature). Mice were fed either normal mouse chow containing 4.5% (w/w) fat and 0.022% (w/w) cholesterol (Prolab RMH 3000; Agway Inc.) or a high fat Western-type (HFW) diet containing 21% (w/w) fat and 0.2% (w/w) cholesterol (TD88137; Teklad). The animals were handled under protocols approved by the Institutional Animal Care and Use Committees of the University of North Carolina (Chapel Hill, NC).
Table 1. Genotypes of mice.
| Mice | Genotype | Description |
|---|---|---|
| 3m | Apoe3/3, Ldlr+/+ | Human apoE3 |
| 3h | Apoe3/3, Ldlrh/+ | Human apoE3 and increased LDLR |
| 3ko | Apoe3/3, Ldlr−/− | Human apoE3 and no LDLR |
| 4m | Apoe4/4, Ldlr+/+ | Human apoE4 |
| 4h | Apoe4/4, Ldlrh/+ | Human apoE4 and increased LDLR |
| 4ko | Apoe4/4, Ldlr−/− | Human apoE4 and no LDLR |
Peritoneal Macrophage Isolation
Macrophages (MPM) were obtained from the peritoneal cavity of mice 4 days after intraperitoneal injection of 1 ml of 4% (w/v) thioglycolate (BD Biosciences). The cells obtained were either directly used for gene expression analyses (below) or washed with Ham's nutrient mixture F-10 medium (F-10), spun at 1000 × g for 5 min, and plated in 12-well plates at a density of 6 × 105 cells/well in F-10 medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100μg/ml streptomycin, and 2 mm l-glutamine. Cells were washed 2 h later to remove nonadherent cells. Cells were cultured in medium without fetal bovine serum for 24 h prior to experiments.
Lipoprotein Uptake
LDL was isolated from human plasma as described (7). Mouse VLDL fractions enriched with human apoE3 or apoE4 were isolated from pooled plasma of 3ko or 4ko mice, respectively, by ultracentrifugation at d < 1.006. Lipoproteins were labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI C18; Molecular Probes, Inc., Eugene, OR), as described by Stephan and Yurachek (14). Macrophages in culture were washed and incubated in F-10 medium without fetal bovine serum for 24 h, followed by incubation with medium containing 1 μg/ml of DiI-labeled human LDL or mouse VLDL. After 2 h, cells were washed with fresh medium, and fluorescence was observed with an IX70 inverted microscope (Olympus) equipped with a filter set for Texas red (exciter 560/55, dichroic 595, emitter 645/75; Chroma Technology Corp.). Fluorescence pixel intensity of each cell was recorded with a SPOT RT Slider digital camera and analyzed with SPOT version 4.0.9 (Diagnostic Instruments) and ImageJ 1.33u software (National Institutes of Health).
Cholesterol Efflux
MPM were cultured 24 h in F-10 medium without fetal bovine serum. Cells were radiolabeled by incubating with 2 μCi/ml [3H]cholesterol (PerkinElmer Life Sciences) for 24 h. They were then washed and further incubated for 24 h with F-10 medium with or without 10 μg/ml of apolipoprotein A-I (apoA-I) protein (Sigma). Following incubation, medium was removed, spun at 12,500 × g for 15 min, and assayed for radioactivity. Cells were washed with ice-cold PBS, and lipids were extracted with isopropyl alcohol for 4 h and assayed for radioactivity. Radiolabel in the medium and the cellular isopropyl alcohol extract was measured, and percentage effluxed was calculated as the ratio of radioactivity in the medium divided by the total (cells + medium). To analyze efflux of cholesterol from cholesterol-loaded foam cells, MPM were incubated with acetylated human LDL (AcLDL) and 2 μCi/ml [3H]cholesterol for 24 h, and efflux was measured after 16 h. LDL (density 1.019–1.063 g/ml) was acetylated by acetic anhydride (15).
Western Blot
To measure ApoE protein content, MPM in culture were washed and cultured in lipid-free F-10 medium. After 24 h, the amount of apoE protein associated with cells and in the medium was measured using Western blot with an antibody against human apoE (Calbiochem). Protein loading was controlled by the amount of tubulin using anti-tubulin antibody (Sigma), and gel to gel control for densitometric analysis was done with a standard amount of apoE.
Oxidative Stress
VLDL (1 μg of protein) from the 3ko or 4ko mice was incubated with 10 μm CuSO4 as described (16). Oxidation and the increase in the formation of conjugated dienes were measured by recording their absorbance at 234 nm every 20 min for 3 h. The lag time was determined graphically. Amounts of thiobarbituric acid-reactive substances (TBARS) in plasma, VLDL, and cultured medium were assayed as described (16). MPM were cultured with VLDL isolated from apoE-deficient mice (50 μg/ml of F-10 medium) for 24 h. Oxidized VLDL was prepared by incubating VLDL in μm CuSO4 for 24 h.
Gene Expression
RNA was extracted from MPM using RNAeasy kit (Qiagen, Valencia, CA) according to the manufacturer's protocol, either directly from peritoneal cells of mice that underwent BMT or cultured MPM from mice fed the HFW diet. Cultured macrophage cells were incubated with F-10 medium or F-10 medium plus 50 μg/ml apoE-deficient VLDL for 16 h. Real time RT-PCR amplifications (8) were performed in a 96-well plate in the ABI Prism 7700 sequence detector (PE Biosystems) in a total volume of 30 μl, which included 10 μl of RNA sample from the ABI Prism 6700 plus 20 μl of a reaction mixture. Each RT-PCR amplification was performed in duplicate: 30 min at 48 °C for the RT reaction and then 10 min at 94 °C, followed by a total of 40 temperature cycles (15 s at 94 °C and 1 min at 60 °C). To determine total Ldlr mRNA levels, a primer probe system specific for murine exon 1, which is also present in mice targeted for the hLdlr, was used (13).
Bone Marrow Transfer
Bone marrow cells were collected from the femurs and tibias of donor male mice by flushing with F-10 medium. Recipient female mice, 6–8 weeks of age, were lethally irradiated (9.5 grays) and injected with 2 × 106 bone marrow cells in 0.2 ml of medium through tail veins. For 2 weeks after transplantation, mice were given drinking water acidified to pH 2.0 by HCl and containing 100 mg/liter neomycin. Mice were fed an HFW diet for 3 months.
Plasma Lipids, Lipoprotein Analysis
Plasma was isolated, and total cholesterol (free and esterified) and triglycerides were measured as described previously (6). Lipoprotein distribution in pooled plasma samples (100 μl) was analyzed by fast protein liquid chromatography using a Superose 6 HR10/30 column (Amersham Biosciences).
Evaluation of Atherosclerotic Lesions
BM recipient mice were fed an HFW diet for 12 weeks before sacrifice. Mice were perfused through the apex of the left ventricle with 4% paraformaldehyde under physiological pressures. Segments of the aortic sinus were embedded and sectioned, and the size of atherosclerotic plaques in aortic roots were scored as described previously (17).
Statistical Analysis
The statistical significance of genotype effects were analyzed by using analysis of variance (JMP software; SAS Inc., Cary, NC). Tukey-Kramer HSD was used for post hoc pairwise comparisons.
Results
Down-regulation of the LDLr Expression by Lipid in Macrophages
Our previous work (6, 13) showed that the 3′-untranslated region modification within the hLdlr allele made its transcripts in the liver more stable than the normal mouse allele, but the hLdlr allele retained the transcriptional repression in response to dietary cholesterol intake. To test whether the expression of the hLdlr allele is also normally regulated in macrophages by exogenous lipid, we isolated MPM from 3m, 3h, 4m, and 4h mice (see Table 1) and placed them in culture. Both 3h and 4h MPM cells cultured for 16 h without VLDL had higher LDLr expression than 3m and 4m MPM cells by ∼3–4–fold (Fig. 1, p ≤ 0.0001). The levels of Ldlr mRNA decreased in all MPM cells when they were incubated with apoE-deficient VLDL for 16 h, and the decrease was proportional whether cells had wild type or elevated LDLr. However, cells with apoE4 had greater proportional decreases in LDLr expression compared with cells with apoE3. Thus, the hLdlr gene in macrophages retains its normal feedback response to increased intracellular lipid and can be down-regulated by high levels of lipid, but its transcripts remain 2–3-fold higher than the wild type Ldlr mRNA.
Figure 1. Expression of the Ldlr gene in the primary culture of mouse peritoneal macrophages.
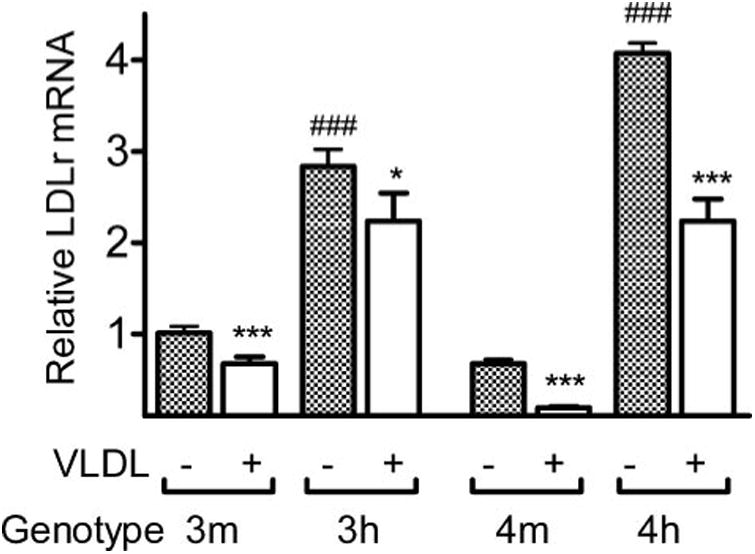
MPM from 3m, 3h, 4m, and 4ko mice were cultured in lipid free F-10 medium with or without 50 μg/ml VLDL isolated from apoE-deficient mice for 16 h. Total mRNA amount for the Ldlr gene was measured by RT-PCR, using the exon 1 sequence that is common between wild type mouse allele and hLdlr allele, normalized with the β-actin expression and plotted as expression levels relative to 3m equal to 1. The white bars indicate expression in cells cultured with VLDL, and black bars indicate without VLDL. *, p ≤ 0.05; ***, p ≤ 0.0005 for effect of VLDL. ###, p ≤ 0.0005 for the effect of LDLr with Tukey-Kramer HSD.
LDLr Expression and Lipoprotein Uptake in Macrophages
To test whether increased expression of the LDLr in macrophages increases uptake of lipoproteins we isolated MPM from 4ko, 4m, and 4h mice and incubated them with DiI-labeled human LDL in culture. After 2 h, intracellular fluorescent intensity in 4h macrophages was twice that of 4m macrophages and three times higher than 4ko cells (Fig. 2A), demonstrating that the LDL uptake by macrophages directly correlates with LDLr expression. In hyperlipidemic mice, as well as humans, lipoprotein particles accumulate not only in LDL fractions but also in VLDL fractions. For example, in the plasma of LDLr−/− mice with human apoE, VLDL particles are enriched with cholesterol and triglyceride, as well as apoE protein (7). We therefore incubated the MPM with VLDL fractions enriched in apoE3 or apoE4 from 3ko and 4ko mice. Uptake of the mouse apoE3-enriched VLDL particles by 3h MPM was modestly but significantly increased compared with that by 3ko MPM (9%, p = 0.02; Fig. 2B). Uptake of apoE3-enriched VLDL by the 4h MPM was not different from the uptake by the 4m MPM. In marked contrast, both 3h and 4h MPM incorporated significantly more apoE4-enriched VLDL than 3ko and 4ko MPM (33% (p ≤ 0.0001) and 26% (p ≤ 0.0001), respectively) (Fig. 2C). This suggests that the apoE isoform present on the VLDL particle and the LDLr in the macrophages, but not macrophage apoE, influences uptake and that apoE4 in the particles promotes VLDL uptake more than apoE3. Taken together, the data suggest that increased LDLr in macrophages by mRNA stabilization causes increased uptake of LDL particles as well as VLDL particles, and the VLDL uptake would be more pronounced when the particles contain apoE4 rather than apoE3.
Figure 2. Lipoprotein uptake.
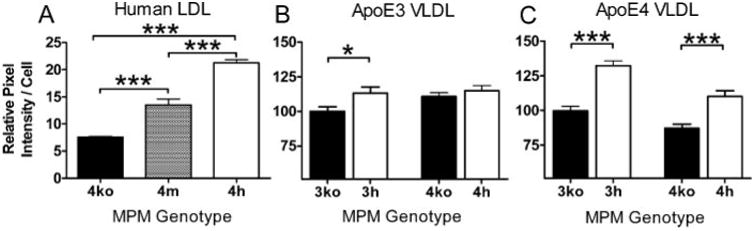
A, MPM from 4ko, 4m, and 4h mice were cultured and incubated for 2 h with 1 μg/ml DiI-labeled human LDL Cells were washed three times with PBS, and fluorescent intensity was measured using NIH Image J. MPM from 3ko, 3h,4ko, and 4h mice were incubated for 2 h with 1 μg/ml each of DiI-labeled 3ko VLDL(B)or4ko VLDL(C). Pixel intensities reflecting VLDLuptake were measured from four wells per experiment with cells pooled from at least three mice of each group, and averages of three experiments are shown as mean uptake of 3ko cells as 100%. Effect of VLDL is as follows: *, p≤ 0.05; ***, p ≤ 0.0005 with Tukey-Kramer HSD.
Effects of ApoE4 and LDLr Expression on Cholesterol Efflux from Macrophages
To examine whether the different apoE isoforms facilitate cholesterol efflux from macrophages differently and whether the effect is dependent on the levels of LDLr expression in macrophages, we measured cholesterol efflux from MPM isolated from mice expressing human apoE isoforms with high levels of LDLr (3h and 4h) or without LDLr (3ko and 4ko). In unloaded cells expressing apoE3, cholesterol efflux was not influenced by the LDLr expression when cells were incubated without apoA-I. In contrast, the LDLr expression had a significant effect when cells expressed apoE4, and the efflux from the 4h cells was 30% less than from 4ko cells (p ≤ 0.0005; Fig. 3A). The addition of apoA-I to the culture medium increased cholesterol efflux from cells regardless of apoE isoforms (Fig. 3B). ApoA-I-mediated efflux from the 4h cells with elevated expression of LDLr was significantly higher than 4ko cells not expressing LDLr. The difference was larger and significant between 4ko and 4h cells (p < 0.003) than between 3ko and 3h cells (not significant). In contrast, efflux of cholesterol from cholesterol-loaded foam cells after incubation with AcLDL depended on LDLr but not on apoE genotype (Fig. 3C). These data demonstrate that the LDLr expression in macrophages influences the cholesterol efflux in an apoE isoform-dependent fashion when macrophages are cultured in lipid-free medium and suggest that both apoE isoform and LDLr expression may be important determinants for foam cell formation. However, when cells were loaded with AcLDL, apoE isoform effects were no longer detectable.
Figure 3. Cholesterol efflux from MPM.
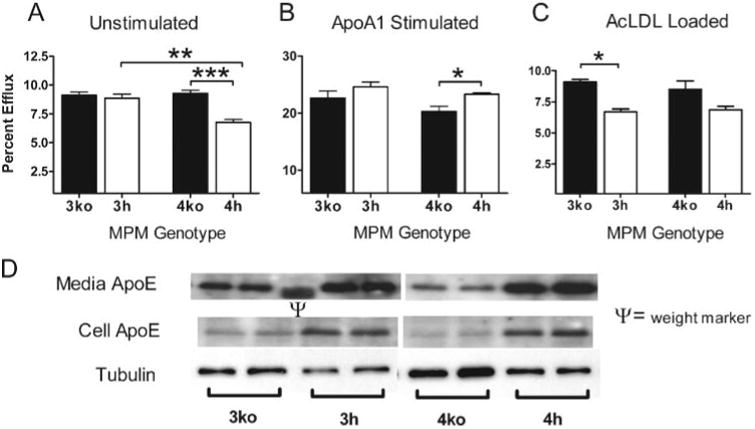
Thioglycolate-elicited MPM were isolated from mice and incubated with [3H]cholesterol for 24 h. Cells were then washed and incubated 24 h in medium without fetal bovine serum (A) or with the same medium containing 10/ng/ml apoA-I for 24 h (B). Radiolabel in medium and the cellular isopropyl alcohol extract were quantified, and the percentage of efflux was calculated as the ratio of radioactivity in the medium divided by total radioactivity (cells + medium) × 100%. Data are mean ± S.E. for three mice of the indicated genotypes assayed in quadruplicate. C, cholesterol efflux from MPM loaded with 50 μg/ml AcLDL and [3H]cholesterol for 24 h. D, ApoE that is cell-associated or secreted into culture medium from MPM incubated for 24 h in lipid-free medium.*, p≤ 0.05;**, p ≤ 0.005;***, p ≤ 0.0005.
ApoE Secretion from Cultured MPM
To address whether cholesterol efflux is related to the intracellular levels of apoE isoforms, we examined the apoE contents associated with cells and in medium 24 h after culturing in lipid-free F-10 medium by Western blot analysis. The intracellular apoE contents in both 3h and 4h cells were consistently higher than in 3ko and 4ko cells (Fig. 3D). Similarly, apoE protein contents in culture medium of 3ko and 4ko cells were lower than those of 3h and 4h cells. Relative amounts between apoE3 cells and apoE4 cells somewhat varied culture by culture and were not consistently different. We conclude that LDLr expression in MPM significantly affects apoE uptake and secretion and probably influences the cholesterol efflux.
VLDL Oxidation
We next tested whether lipoprotein oxidation in our mice is apoE isoform-dependent, which may be due to the free cysteinyl groups on apoE3 and apoE2 (9, 16). ApoE2 has two free cysteinyl groups, and apoE3 has one, and both have been shown to function as a better antioxidant than apoE4 protein, which has no free cysteinyl group. We isolated human apoE-enriched VLDL particles from 2ko, 3ko, and 4ko mice and tracked conjugated diene formation during Cu2+-mediated oxidation of these lipoprotein particles. The lag time for conjugated diene formation was 24 min in VLDL isolated from 4ko mice compared with 49 min in VLDL from 3ko mice and the longest, 70 min, in 2ko VLDL (Fig. 4A). Measurement of TBARS after overnight oxidation showed that apoE4-VLDL contained 3-fold more TBARS than did apoE3-VLDL (Fig. 4B).
Figure 4. VLDL oxidation.
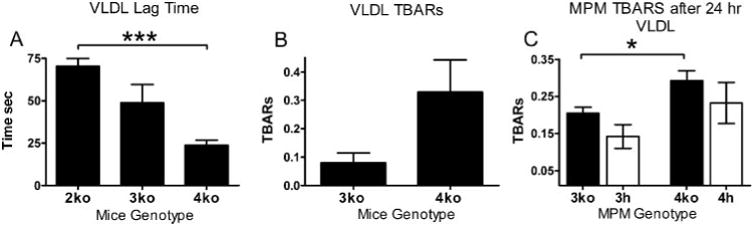
A, VLDL from 3ko and 4ko mice (1 μg) was incubated with 10 μm Cu2+. The ordinate shows lag time (average of three experiments) until the inflection point on 234 nm absorption curves. B, TBARS after Cu2+ oxidation of VLDL isolated from 3ko and 4ko mice. TBARS were measured after 16 h of incubation with 10 μm Cu2+ at 0.5 mg protein/ml. C, TBARS in the medium of MPM incubated with VLDL in culture. Thioglycolate-elicited MPM isolated from either 4ko or 3ko mice was incubated with 50 μg/ml apoE-deficient VLDL. TBARS in medium were measured after 24 h. *, p ≤ 0.05; **, p ≤ 0.005; ***, p ≤ 0.0005 with Tukey-Kramer HSD.
To test whether expression of LDLr in macrophages influences VLDL oxidation in an apoE-dependent manner, we next cultured MPM in medium with apoE-deficient VLDL for 24 h and measured TBARS in medium as an indicator for oxidized lipids. The amounts of TBARS were borderline detectable and were slightly higher in medium cultured with apoE4 macrophages than with apoE3 and lower with elevated LDLr than with no LDLr, but neither apoE genotype nor LDLr had statistically significant effects by two-way analysis of variance. These data suggest that the apoE4 isoform does not protect lipoprotein particles from oxidation as well as apoE3 when a strong oxidative stress is applied in vitro. However, effects of apoE isoforms in macrophages on oxidation in culture are not significant, even under conditions of elevated triglycerides and cholesterol. In addition, we did not detect any statistical differences in the amount of TBARS or GSH in the plasma of 3ko and 4ko recipient mice (data not shown).
BMT and Plasma Lipids
The experiments above indicate that apoE4 proves to be impaired relative to apoE3 in several pathways important to lipid homeostasis in macrophages and perhaps atherosclerosis lesion formation, and the effects are influenced by the levels of LDLr expression in macrophages. To test whether the observed differences could alter overall atherogenesis in vivo, we carried out BMT in mice lacking LDLr but expressing human apoE3 (3ko) or apoE4 (4ko). To alter macrophage expression of LDLr, 3ko or 4ko females were lethally irradiated and transplanted with BM isolated from male mice with the same ApoE genotype, having no LDLr (3ko, 4ko), wild type LDLr (3m, 4m), or elevated expression of LDLr (3h, 4h). Three months after BMT, PCR of the Ldlr gene in the blood cells of the recipient animals showed only the products corresponding to the genotype of the donor cells, and no visible products corresponding to the recipient genotype (ko) were detected in the mice that received m or h BM cells (Fig. 5A). This confirmed that the replacement of the BM-derived cells in the recipients by the donor cells was probably complete. The total Ldlr mRNA levels in the peritoneal cells isolated from representative recipient mice showed that the macrophages in the 3ko mice receiving 3h BM cells had 4-fold higher Ldlr gene expression than the macrophages in the 3ko mice receiving 3m BM cells. Similarly, total expression of the Ldlr gene in macrophages from the 4ko mice receiving 4h BM cells was 4-fold higher than in those receiving 4m cells (Fig. 5B). Thus the levels of Ldlr mRNA in mice that received 3h or 4h bone marrow are elevated compared with those in mice that received wild type LDLr to similar degrees as the MPM cells in culture as shown in Fig. 1A
Figure 5. Bone marrow transfer.
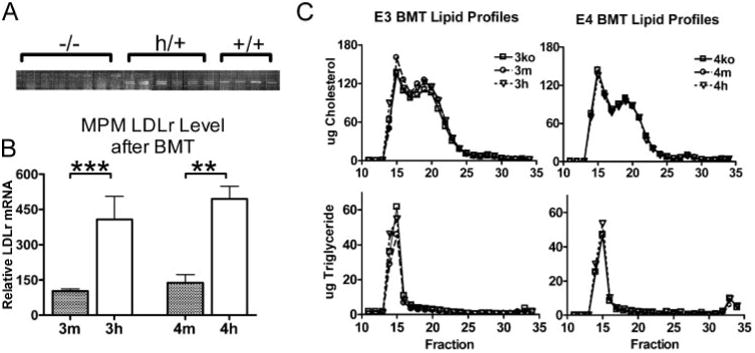
A, verification of bone marrow engraftment. DNA was isolated from buffy coat cells of recipient mice, and LDLr genotype was determined by multiplex PCR. The first seven lanes were recipients of ko cells showing a single PCR fragment. The next six lanes were recipients of hLdlr cells showing two bands; the upper band corresponds to the wild type mouse Ldlr allele, and the lower band corresponds to the hLdlr allele. The last four lanes are recipients of m-Ldlr cells showing only the wild type allele. No bands corresponding to the ko allele were detected in the recipients of h or m cells. B, macrophage LDLr mRNA level. Thioglycolate-elicited peritoneal cells were collected from bone marrow transfer recipients. Real time quantitative PCR was used for LDLr mRNA levels using exon 1 sequence that is common to hLdlr and wild type alleles. Averages from mice are shown relative to the expression of 3m donor. **, p ≤ 0.005; ***, p ≤ 0.0005. C, lipoprotein distribution in plasma isolated from the 3ko or 4ko recipient mice that received BM cells with different LDLr expression levels. Plasma (100 μl) pooled from at least five mice of each group was size-fractionated on a fast protein liquid chromatography Superose 6 column, and the cholesterol and triglyceride contents of each fraction were measured with an enzymatic assay.
Feeding HFW diet increased plasma cholesterol levels ∼3-fold in both 3ko and 4ko mice regardless of donor Ldlr genotype (Table 2). Both 3ko and 4ko mice have elevated cholesterol in VLDL fractions as well as LDL fractions. HDL-cholesterol were very low in both 3ko and 4ko mice on HFW diet, and the plasma apoA-I levels were not different between them (not shown). The transfer of bone marrows with differing expression levels of LDLr did not cause any significant changes in plasma lipoprotein distribution in either the 3ko or 4ko mice by fast protein liquid chromatography analyses (Fig. 5C). We conclude that the increased macrophage LDLr expression is not sufficient to alter plasma VLDL clearance in the LDLr−/− mice with apoE3 or apoE4. Consistent with previous work, the LDLr expression in BM-derived cells had no influence over plasma lipid profiles (18).
Table 2. Plasma lipids in LDLr−/− mice expressing human apoE3 or apoE4 after BMT and fed HFW diet.
| 3ko BM recipient/3ko BM donor | 3ko BM recipient/3m BM donor | 3ko BM recipient/3h BM donor | 4ko BM recipient/4ko BM donor | 4ko BM recipient/4m BM donor | 4ko BM recipient/4h BM donor | |
|---|---|---|---|---|---|---|
| Number | 15 | 15 | 15 | 14 | 15 | 12 |
| TC (mg/dl) | 1,324 ± 57 | 1,313 ± 46 | 1,394 ± 23 | 1,245 ± 49 | 1,217 ± 43 | 1,184 ± 97 |
| TG (mg/dl) | 186 ± 61 | 177 ± 43 | 221 ± 16 | 111 ± 17 | 95 ± 22 | 133 ± 17 |
Macrophage LDLr Expression and Atherosclerosis in Mice with ApoE4
Both 3ko and 4ko mice develop lesions in the aortic sinus as summarized in Table 3. Feeding the recipient mice with the HFW diet for 12 weeks increased atherosclerotic plaque size in both 3ko and 4ko mice that received either control 3ko or 4ko BM. There were no significant apoE genotype effects on the lesion area (346 ± 19 × 103 μm2 (n = 12) in 3ko versus 386 ± 28 × 103 μm2 (n = 14) in 4ko, p = 0.3) (Fig. 6).
Table 3. Plasma lipids and atherosclerosis in LDLr−/− mice expressing human apoE3 or apoE4 fed a NC diet.
| Male 3ko | Female 3ko | Male 4ko | Female 4ko | |
|---|---|---|---|---|
| Number | 11 | 12 | 12 | 12 |
| TC (mg/dl) | 241 ± 18 | 386 ± 12 | 313 ± 25 | 427 ± 32 |
| TG (mg/dl) | 109 ± 12 | 102 ± 6 | 225 ± 22 | 114 ± 2 |
| Lesion size (103 μm2) | 13.4 ± 3.1 | 55.5 ± 7.2 | 12.0 ± 2.7 | 71.0 ± 14.4 |
| Occlusion (%) | 1.3 ± 0.3 | 5.1 ± 0.5 | 1.0 ± 0.2 | 5.4 ± 0.8 |
Figure 6. Atherosclerotic lesions.
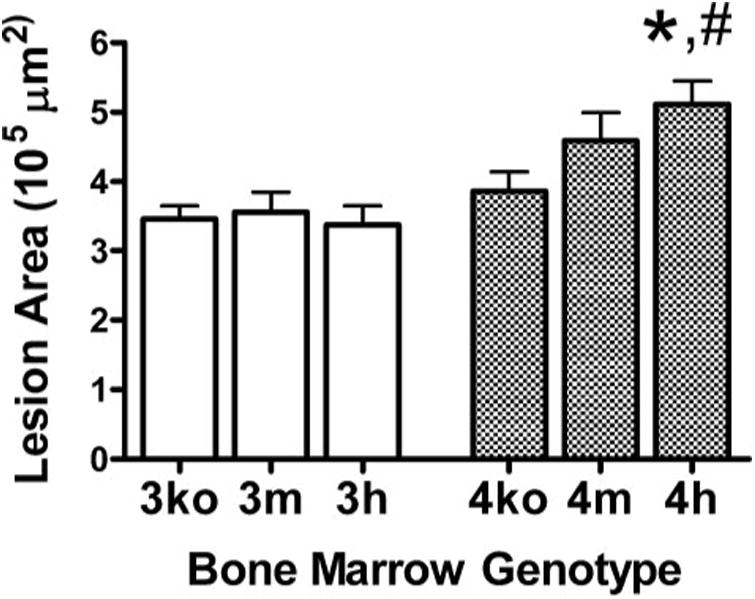
Mean aortic sinus atherosclerotic lesion areas of 3ko mice that received 3m, 3h, and 3ko BM cells and of 4ko mice that received 4m, 4h, and 4ko BM cells are shown. *, p≤ 0.05 (4ko versus 4h); #, p ≤ 0.01 (3h versus 4h) with Tukey-Kramer HSD.
We also found no change in the lesion area of 3ko mice as macrophage LDLr level increased (Fig. 6). Thus, 3ko mice that received 3m and 3h BM had a mean area of 356 ± 28 × 103 μm2 (n = 13) and 338 ± 28 × 103 μm2 (n = 13), respectively (p = 0.9). In contrast, 4ko mice developed larger lesions as their macrophages expressed increasing amounts of LDLr. Thus, 4ko mice that received 4m BM and 4h BM had mean lesion areas of 459 ± 41 × 103 μm2 (n = 15) and 511 ± 34 × 103 μm2 (n = 13), respectively. The difference between the lesion sizes in mice that received 4h donor cells and 4ko donor cells was significant (p ≤ 0.05). When all six groups were analyzed together, the difference in atherosclerosis between 3ko mice with 3h BM and 4ko mice with 4h BM was also significant (p ≤ 0.01).
Taken together, our data demonstrate that there is an interaction between the apoE genotype and LDLr expression of macrophages in determining atherosclerosis development. In the LDLr−/− mice, increased expression of LDLr in BM-de-rived cells has no effect when animals are expressing apoE3, whereas it is detrimental in mice expressing apoE4.
Gene Expression in the Peritoneal Macrophages of Mice after BMT
To gain insight into the mechanism of the interaction between macrophage LDLr expression and apoE expression in atherosclerosis development, we isolated MPM from mice receiving BM transfer 4 days after stimulation with thioglycollate. Quantitative RT-PCR of peritoneal cells showed that the expression of genes coding for MCP1 (monocyte chemotactic protein-1), CD36, LDLr, apoE, and SR-A (scavenger receptor A) were not significantly different among recipients who received the donor cells with different LDLr levels (Table 4). Thus, the effect of apoE4 isoform and LDLr level in atherogen-esis does not appear to involve pathways affecting expression of MCP1, CD36, or SR-A in macrophages. In contrast, ABCA1 expression was increased in MPM that expressed elevated LDLr compared with ko MPM regardless of apoE genotype, although the increase was statistically significant only in 3h cells compared with 3ko cells. The up-regulation of ABCA1 in 3h and 4h macrophages may indicate higher intracellular cholesterol content in these cells relative to 3ko and 4ko cells (19, 20). This is also consistent with an increased cholesterol efflux from 3h and 4h macrophages compared with 3ko and 4ko macrophages when exogenous apoA-I acceptor protein is given to the culture (Fig. 3B).
Table 4. Gene expression of peritoneal macrophage isolated from BM recipient mice.
Relative mRNA levels of the genes were calculated by the ratio of expression to the levels of β-actin mRNA. Values are mean ± S.E. and relative to 3ko, which was adjusted to 100. ABCA1 levels of 3h cells were significantly different from 3ko.
| Recipient 3ko/macrophage 3ko | Recipient 3ko/macrophage 3m | Recipient 3ko/macrophage 3h | Recipient 4ko/macrophage 4ko | Recipient 4ko/macrophage 4m | Recipient 4ko/macrophage 4h | |
|---|---|---|---|---|---|---|
| Number | 6 | 7 | 8 | 6 | 2 | 8 |
| ABCA1 | 100 ±9 | 126 ± 22 | 210 ± 32a | 111 ± 14 | 76 ±4 | 156 ± 20 |
| MCP1 | 102 ±8 | 74 ± 17 | 97 ±11 | 79 ±22 | 107 ± 22 | 62 ±33 |
| CD36 | 100 ± 23 | 102 ± 27 | 118 ± 25 | 102 ± 12 | 70 ±6 | 111 ± 12 |
| SR-A | 104 ± 13 | 64 ±10 | 72 ±23 | 67 ±14 | 78 ±7 | 52 ± 10 |
| ApoE | 101 ±7 | 125 ± 27 | 116 ± 21 | 119 ± 30 | 108 ± 44 | 156 ± 28 |
p ≤ 0.05.
Discussion
ApoE, as a ligand for the removal of lipoproteins, is generally considered atheroprotective; however, the extent to which this depends on its isoform, concentration, and location (1, 21-23) is not known. In this study, we tested the interaction of apoE isoforms with LDLr on several atherosclerosis pathways in macrophages. We then used BMT in mice to investigate whether their interaction in macrophages affect atherosclerosis development. Our results showed that expression of macrophage LDLr in the presence of apoE4 enhanced atherosclerosis. In contrast, these same LDLr increases in mice with apoE3 did not affect atherosclerosis. Thus, our results demonstrate, for the first time in animal models, that some of the risk associated with human apoE4 isoform may be due to its interaction with the LDLr in macrophages as well as with hepatic LDLr.
We found that apoE isoforms expressed by the macrophages had only a modest, nonsignificant effect on apoA-I-stimulated cholesterol efflux from MPM but that the levels of LDLr expressed by the macrophages significantly increased apoA-I-assisted cholesterol efflux from apoE4 macrophages and less so from apoE3 macrophages. Cholesterol efflux, when apoA-I was not present, was generally low but affected by LDLr expression only in apoE4 macrophages and in an inverse direction. Thus, high LDLr expression did not alter efflux from macrophages with apoE3 but significantly inhibited efflux from macrophages with apoE4. A likely explanation for this is that apoE4 is more efficient than apoE3 for capturing and reuptake of cholesterol containing particles by macrophages. Alternatively, the possible increased amount of intracellular apoE4 compared with apoE3 may retard cholesterol secretion with the endogenous apoE, thus yielding the apparent difference in efflux. In contrast, in cholesterol-loaded foam cells, high LDLr expression inhibited efflux in both apoE3 and apoE4 macrophages. The interaction between LDLr and apoE4 resulted in significant effects only in unloaded cells in which LDLr expression is high. The difference in efflux between apoE3 and apoE4 is small in loaded foam cells, in which expression of LDLr is down-regulated. In the present work, we observed that cultured MPM expressing LDLr have more cell-associated apoE than MPM without LDLr regardless of apoE genotype. Nevertheless, the difference between apoE3 and apoE4 in intracellular trafficking and accumulation has been established (24) and may be contributing to the difference in cholesterol efflux. Supporting this possibility, we reported earlier, in collaboration with Dr. Mazzone and colleagues at the University of Illinois, that MPM with increased LDLr expression showed reduced apoE secretion compared with MPM with basal LDLr expression and that the relative reduction was greater with apoE4 than with apoE3. Further studies are necessary to elucidate the possible relationships between cholesterol efflux and intracellular localization of apoE3 and apoE4 in MPM (25).
Increased cholesterol uptake increases ABCA1 expression and efflux to acceptor proteins (19,20). Thus, we observed that macrophages isolated from Ldlr−/− mice receiving BM cells with high Ldlr gene expression showed elevated ABCA1 expression. This increase in ABCA1 expression probably accounts for the increased apoA-I-mediated cholesterol efflux in the presence of LDLr in macrophages. In addition, the presence of a large amount of apoA-I could interfere with the apoE-mediated reuptake of lipid particles. Our observation of iso-form-specific effects of apoE on cholesterol efflux is consistent with previous reports by other investigators. For example, Cullen et al. (10) observed using macrophages isolated from human subjects with different apoE isoforms, that apoE4 macrophages effluxed less cholesterol than apoE2 or apoE3 and suggested that a combination of enhanced uptake and degradation of apoE4 by macrophages leads to lower cholesterol efflux. Similarly, in a murine macrophage cell line, Hara et al. demonstrated that adenovirus-mediated expression of apoE isoforms reduced cellular cholesterol in a manner inversely proportional to the LDLr affinity of apoE (26). Effects of LDLr were not investigated in either study by Cullen et al. (10) or Hara et al. (26).
The apoE3 and apoE2 proteins carry either one or two free cysteinyl groups, respectively, and may function as a better antioxidant than apoE4 protein, which does not have a free cysteinyl group. It has been shown in vitro that lipoprotein oxidation is apoE isoform-dependent (9,16). Similarly, we show that lipid peroxidation in vitro by Cu2+ of mouse VLDL particles enriched with apoE4 is more susceptible to lipid peroxidation than particles with apoE3 or apoE2. However, we were not able to demonstrate the antioxidant effects of apoE in vivo. Antioxidant effects of apoE in vivo are likely to be small. Nevertheless, differences in the antioxidant capacity of apoE isoforms could be significant when excess oxidative stress is applied, such as in lipid-loaded macrophages in the atherosclerotic plaques.
Many of the functions of apoE in vascular tissues are isoform-specific and have been implicated as contributing factors to the well established increased atherosclerosis risk associated with apoE4 in humans. For example, apoE has been shown to inhibit smooth muscle cell proliferation, with apoE4 being less effective than apoE2 or apoE3 (27). This antiproliferative function may also be related to isoform-dependent nitric oxide release (28). ApoE has been shown to reduce atherosclerosis when expressed in the arterial wall without affecting lipid levels (29). Human apoE3 expressed in macrophages binds LDLr and is internalized, and a proportion of the internalized apoE is recycled in both hepatocytes and macrophages (30, 31). Consistent with its increased LDLr affinity, apoE4 recycling is impaired relative to apoE3 in hepatocytes (24). Decreased recycling of apoE4 in the subendothelial space of blood vessels could result in prolonged retention and modification of lipoproteins. In contrast, apoE3 and apoE2 may exert their antiatherosclerotic effects multiple times if recycled back to the intimal space.
Macrophage LDLr metabolizes LDL and VLDL/chylomicron remnants (1, 15, 32, 33), and uptake of LDL by macrophages through the LDLr can induce foam cell formation in culture (32, 34). Thus, previous studies have shown that WT mice that received BM cells with WT LDLr develop larger diet-induced plaques compared with the WT mice that received cells lacking LDLr (18, 34). However, macrophage expression of the LDLr did not significantly increase atherosclerosis in the hyperlipidemic setting of LDLr−/− mice recipients when their macrophages expressed WT LDLr (11, 18, 35). All of these experiments used mice with wild type mouse apoE and LDLr. We now show, in the present paper, that the proatherogenic effects of human apoE4 isoform are LDLr expression-dependent even under the severe hyperlipidemic setting. Mice with apoE3 showed no difference in atherosclerosis when macrophage LDLr expression level was changed under the same hyperlipidemic condition.
Together, our results show that there is an important interaction between the apoE genotypes and LDLr expression levels in macrophages in determining cholesterol regulation in the cell (Fig. 7). Thus, the higher LDLr binding affinity of apoE4 increases cholesterol delivery to macrophages compared with apoE3. This effect is enhanced when lipoprotein particles are enriched with apoE4. The proatherogenic effects of apoE4 are further enhanced by a reduction in sterol efflux that is also apoE4-specific and LDLr-dependent. This is particularly important when cholesterol acceptors, such as HDL and apoA-I, are reduced in hyperlipidemia. Additionally, the reduced capacity of apoE4 to protect lipoproteins from oxidation compared with that of apoE3 could increase the atherogenicity of apoE4-containing lipoproteins. Although differences in the individual effects of apoE4 compared with apoE3 are small, the combined effects are of importance and probably contribute to the adverse effects of apoE4 on plaque development in an LDLr-dependent fashion in macrophages. A similar mechanism is likely to operate in humans during the development of atherosclerosis.
Figure 7. Hypothetical interactions between macrophage LDLr and apoE isoforms in mice that underwent BMT.
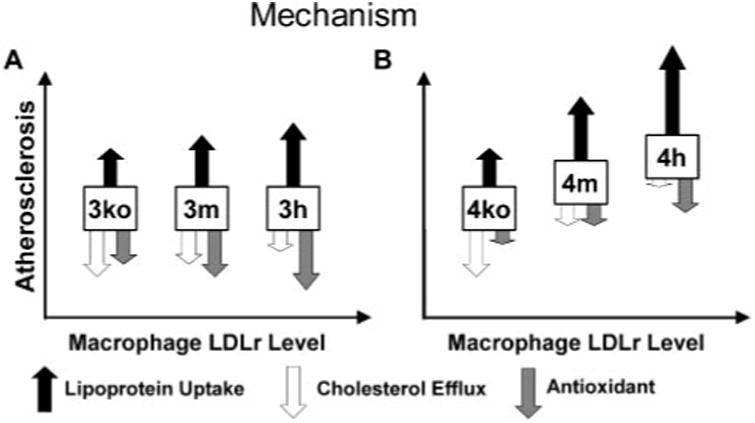
Increasing LDLr results in increased macrophage uptake of apoE containing VLDL and LDL (solid arrow). The relative uptake of apoE3-containing lipoproteins (A) is less than the uptake of apoE4 containing lipoproteins (B). Elevated LDLr reduced efflux in macro-phages that were loaded with cholesterol as well as unloaded apoE4 macro-phages (open arrow). ApoE3 has greater antioxidant capacity than apoE4. The antioxidant capacity increases as more apoE is retained by LDLr (shaded arrow). Overall, the combined effects are balanced in apoE3 macrophages, and their atherogenicity is not influenced by LDLr expression. In contrast, the combined effects are not balanced in apoE4 macrophage, and the atherogenicity is increased as LDLr expression increases.
Acknowledgments
We thank Shin-Ja Kim and Svetlana Zhilicheva for technical help; Drs. Theodore Mazzone, John Parks, and Patrick Sullivan for valuable discussions; and Drs. Kumar Pandya, Jose Arbones-Mainar, and Raymond Givens for reviewing the manuscript.
*This work was supported by National Institutes of Health (NIH) Grant HL42630.
Footnotes
The abbreviations used are: VLDL, very low density lipoprotein(s); LDL, low density lipoprotein(s); LDLr, LDL receptor(s); apo, apolipoprotein; AcLDL, acetylated LDL; BM, bone marrow; BMT,bone marrow transfer;DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; MPM, mouse peritoneal macrophage(s); TBARS, thiobarbiturate-reactive substances; HFW, high fat Western-type; RT, reverse transcription; WT, wild type; HSD, honestly significantly difference.
References
- 1.Mahley RW, Rall SC., Jr Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 2.Bohnet K, Pillot T, Visvikis S, Sabolovic N, Siest G. J Lipid Res. 1996;37:1316–1324. [PubMed] [Google Scholar]
- 3.Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. J Clin Invest. 1999;103:1579–1586. doi: 10.1172/JCI6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 5.Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. J Clin Invest. 1998;102:130–135. doi: 10.1172/JCI2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malloy SI, Altenburg MK, Knouff C, Lanningham-Foster L, Parks JS, Maeda N. Arterioscler Thromb Vasc Biol. 2004;24:91–97. doi: 10.1161/01.ATV.0000094963.07902.FB. [DOI] [PubMed] [Google Scholar]
- 7.Knouff C, Briand O, Lestavel S, Clavey V, Altenburg M, Maeda N. Biochim Biophys Acta. 2004;1684:8–17. doi: 10.1016/j.bbalip.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Ishigami M, Swertfeger DK, Hui MS, Granholm NA, Hui DY. Arterioscler Thromb Vasc Biol. 2000;20:1020–1026. doi: 10.1161/01.atv.20.4.1020. [DOI] [PubMed] [Google Scholar]
- 9.Miyata M, Smith JD. Nat Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 10.Cullen P, Cignarella A, Brennhausen B, Mohr S, Assmann G, von Eckardstein A. J Clin Invest. 1998;101:1670–1677. doi: 10.1172/JCI119887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtiss LK, Boisvert WA. Curr Opin Lipidol. 2000;11:243–251. doi: 10.1097/00041433-200006000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 13.Knouff C, Malloy S, Wilder J, Altenburg MK, Maeda N. J Biol Chem. 2001;276:3856–3862. doi: 10.1074/jbc.M009423200. [DOI] [PubMed] [Google Scholar]
- 14.Stephan ZF, Yurachek EC. J Lipid Res. 1993;34:325–330. [PubMed] [Google Scholar]
- 15.Basu SK, Goldstein JL, Anderson GW, Brown MS, editors. Proc Natl Acad Sci U S A. 1976;73:3178–3182. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mabile L, Lefebvre C, Lavigne J, Boulet L, Davignon J, Lussier-Cacan S, Bernier L. J Cell Biochem. 2003;90:766–776. doi: 10.1002/jcb.10697. [DOI] [PubMed] [Google Scholar]
- 17.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 18.Linton MF, Babaev VR, Gleaves LA, Fazio S. J Biol Chem. 1999;274:19204–19210. doi: 10.1074/jbc.274.27.19204. [DOI] [PubMed] [Google Scholar]
- 19.Costet P, Luo Y, Wang N, Tall AR. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 20.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Proc Natl Acad Sci U S A. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahley RW. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 22.Rall SC, Jr, Mahley RW. J Intern Med. 1992;231:653–659. doi: 10.1111/j.1365-2796.1992.tb01254.x. [DOI] [PubMed] [Google Scholar]
- 23.Davignon J, Gregg RE, Sing CF. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, Beisiegel U. J Biol Chem. 2004;279:55483–55492. doi: 10.1074/jbc.M409324200. [DOI] [PubMed] [Google Scholar]
- 25.Lucic D, Huang ZH, Gu DS, Altenburg MK, Maeda N, Maz-zone T. J Lipid Res. 2006;48:366–372. doi: 10.1194/jlr.M600259-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Hara M, Matsushima T, Satoh H, Iso-o N, Noto H, Togo M, Kimura S, Hashimoto Y, Tsukamoto K. Arterioscler Thromb Vasc Biol. 2003;23:269–274. doi: 10.1161/01.atv.0000054199.78458.4b. [DOI] [PubMed] [Google Scholar]
- 27.Zeleny M, Swertfeger DK, Weisgraber KH, Hui DY. Biochemistry. 2002;41:11820–11823. doi: 10.1021/bi026202k. [DOI] [PubMed] [Google Scholar]
- 28.Sacre SM, Stannard AK, Owen JS. FEBS Lett. 2003:540, 181–187. doi: 10.1016/s0014-5793(03)00261-8. [DOI] [PubMed] [Google Scholar]
- 29.Shimano H, Ohsuga J, Shimada M, Namba Y, Gotoda T, Harada K, Katsuki M, Yazaki Y, Yamada N. J Clin Invest. 1995;95:469–476. doi: 10.1172/JCI117687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasty AH, Plummer MR, Weisgraber KH, Linton MF, Fazio S, Swift LL. J Lipid Res. 2005;46:1433–1439. doi: 10.1194/jlr.M400418-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Farkas MH, Swift LL, Hasty AH, Linton MF, Fazio S. J Biol Chem. 2003;278:9412–9417. doi: 10.1074/jbc.M208026200. [DOI] [PubMed] [Google Scholar]
- 32.Tabas I, Boykow GC, Tall AR. J Clin Invest. 1987;79:418–426. doi: 10.1172/JCI112828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koo C, Wernette-Hammond ME, Garcia Z, Malloy MJ, Uauy R, East C, Bilheimer DW, Mahley RW, Innerarity TL. J Clin Invest. 1988;81:1332–1340. doi: 10.1172/JCI113460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herijgers N, Van Eck M, Groot PH, Hoogerbrugge PM, Van Berkel TJ. Arterioscler Thromb Vasc Biol. 2000;20:1961–1967. doi: 10.1161/01.atv.20.8.1961. [DOI] [PubMed] [Google Scholar]
- 35.Herijgers N, Van Eck M, Groot PH, Hoogerbrugge PM, Van Berkel TJ. Arterioscler Thromb Vasc Biol. 1997;17:1995–2003. doi: 10.1161/01.atv.17.10.1995. [DOI] [PubMed] [Google Scholar]