

Abstract
The majority of transmembrane proteins are integrated into the endoplasmic reticulum (ER) by virtue of a signal sequence‐mediated co‐translational process. However, a substantial portion of transmembrane proteins fails to reach the ER, leading to mislocalized cytosolic polypeptides. Their appropriate recognition and removal are of the utmost importance to avoid proteotoxic stress. Here, we identified UBQLN4 as a BAG6‐binding factor that eliminates newly synthesized defective polypeptides. Using a truncated transmembrane domain protein whose degradation occurs during a pre‐ER incorporation process as a model, we show that UBQLN4 recognizes misassembled proteins in the cytoplasm and targets these to the proteasome. We suggest that the exposed transmembrane segment of the defective polypeptides is essential for the UBQLN4‐mediated substrate discrimination. Importantly, UBQLN4 recognizes not only the defective model substrate but also a pool of endogenous defective proteins that were induced by the depletion of the SRP54 subunit of the signal recognition particle. This study identifies a novel quality control mechanism for newly synthesized and defective transmembrane domain polypeptides that fail to reach their correct destination at the ER membrane.
Keywords: BAG6, proteasome, signal sequence, SRP, ubiquitin
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Recent progress in genome analysis revealed that more than one‐fifth of proteins encoded in the mammalian genome are putative transmembrane (TM) proteins 1, 2, 3, 4. Since TM proteins contain an almost 20‐residue long bulky hydrophobic stretch called the transmembrane domain (TMD), their biogenesis in the aqueous cytosol requires specialized machinery dedicated to TM protein assembly 5. Indeed, many TM proteins possess a characteristic signal sequence (SS) at the N‐terminus of the nascent polypeptide for endoplasmic reticulum (ER) targeting 6, 7, 8, 9, 10. The timely and accurate capture of the SS in the ribosome–nascent chain complex is essential for the proper initiation of membrane protein assembly in the ER 10. At the initial stage of translation on cytosolic ribosomes, the SS of nascent chain polypeptides is recognized by a signal recognition particle (SRP), a novel class of GTPase whose activity is regulated by nucleotide‐dependent dimerization cycles 11, 12, 13, 14, 15. Subsequently, the SRP‐ribosome complex is recruited to the surface of the rough ER, where newly synthesized polypeptides are assembled in the ER membrane through SRP receptor and translocon complexes 6, 9, 10, 16.
The fidelity of SS recognition by the SRP, however, is challenged by the kinetic difficulties of targeting nascent chain polypeptides elongating from the ribosome 10, 17, 18. Indeed, the SRP must identify the targeted substrates within several seconds in the cytosol; a nascent polypeptide reportedly loses its competence to be targeted by the SRP when it exceeds a critical length of 140 residues 10, 19. Furthermore, the concentration of the SRP is estimated to be more than 100‐fold lower than that of the translating ribosomes 10. Thus, difficulties of SRP recognition within a limited time window in the crowded cytosol might result in the probable production of defective SRP substrates with an orphan TMD 20, 21. Similar consequences might occur when SRP subunits are compromised or when the ratio of the SRP to its client substrates is overwhelmed 18, 22.
In accordance with these studies, it was reported that some classes of TM proteins fail frequently to access the ER membrane 21, 23. Indeed, a surprisingly low efficiency of ER targeting (as low as 60% success for some newly synthesized ER proteins) was reported, resulting in the production of defective (and mislocalized) proteins 21, 24, 25. Their appropriate recognition and removal before they reach the lumen of the ER are of real significance for the maintenance of cellular homeostasis 26, 27. Defects in SS machinery can lead to various human diseases such as prion disease, amyloid precursor protein‐derived neurodegeneration, and autosomal dominant late‐onset diabetes 28, 29, 30, 31. Nevertheless, the molecular mechanism of the elimination pathway that targets newly synthesized and mistargeted TMD protein is not well understood.
A hydrophobic TMD that is normally buried in the lipid bilayer tends to be exposed in the aqueous cytoplasm by mistargeting of TM proteins during the course of their biogenesis. BAG6, a major histocompatibility complex (MHC)‐encoded gene product, was identified as an essential factor in the metabolism of newly synthesized defective and/or mistargeted TM proteins 25, 32, 33, 34, 35, 36. The BAG6 complex recognizes the exposed hydrophobicity of TM proteins just after their ribosomal release and leads to the ER insertion of these newly synthesized polypeptides in the case of tail‐anchored (TA) proteins 37, 38, 39, 40, 41, 42, while it enhances the targeted degradation of the remainder (including mislocalized proteins) by the ubiquitin/proteasome‐mediated degradation pathway 25, 32, 33. Thus, the BAG6 complex provides an essential quality control system for TMD substrates and newly synthesized defective polypeptides with exposed hydrophobic stretches 43, 44, 45, 46, 47. However, how BAG6 and its associated proteins determine the fate of client membrane proteins has not been elucidated adequately.
In this study, we explored the potential function of BAG6‐associated proteins and thus identified UBQLN4 as an essential factor for the elimination of newly synthesized polyubiquitinated proteins. Using a model TMD protein whose degradation occurs at a pre‐ER incorporation process, we show that UBQLN4 specifically associates with misassembled TM protein in the cytoplasm for targeted degradation via the ubiquitin pathway. Importantly, UBQLN4 recognized not only this model defective substrate but also a pool of endogenous defective proteins that were induced by compromising the SRP. We suggest that UBQLN4 acts as a substrate‐discriminating ubiquitin receptor for newly synthesized and defective TM polypeptides before their ER entry.
Results
Newly synthesized polypeptides are major clients of UBQLN4
The BAG6 complex was identified recently as a receptor for TA and defective TM proteins 25, 32, 34, 37, 38, 42, 43, 44, 48. During the course of our BAG6 study, we identified UBQLN4/UBIN, the main subject of this paper, as a novel binding protein of BAG6 (Appendix Fig S1). As shown in Fig EV1A, UBQLN4 co‐precipitates with endogenous BAG6 protein from the extract of cells that were treated with the proteasome inhibitor MG‐132, while the UBQLN4‐related protein UBQLN1/PLIC1 did not. UBQLN family proteins belong to a typical class of ubiquitin‐like (UBL) and ubiquitin‐associated (UBA) domain proteins that can interact with proteasomes, chaperones, and polyubiquitinated species 49, 50, 51, 52, 53, 54, 55, 56, 57. In accordance with the nature of UBA domain proteins 58, 59, 60, 61, 62, 63, 64, we confirmed that a significant amount of polyubiquitinated proteins co‐immunoprecipitated with UBQLN4 (Fig 1A). Since co‐precipitated polyubiquitinated species are induced by treatment with MG‐132, the cellular function of UBQLN4 could be linked to protein degradation.
Figure EV1. UBQLN4 possesses a distinct preference over UBQLN1 for endogenous BAG6.
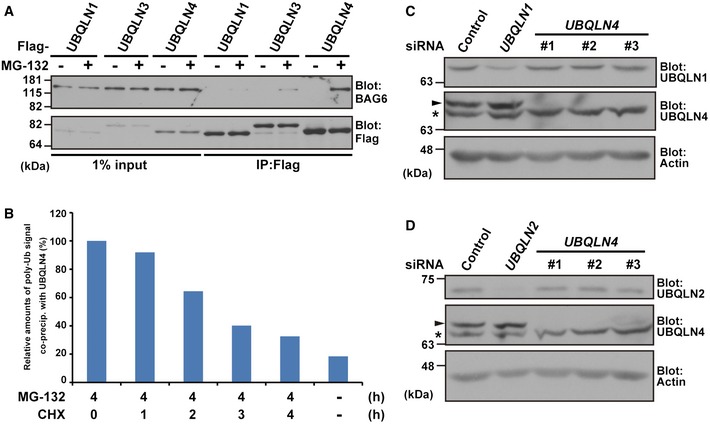
-
AUBQLN4, but not UBQLN1, interacts with endogenous BAG6 in an MG‐132 treatment‐dependent manner. Flag‐tagged UBQLN1, UBQLN3, and UBQLN4 were expressed in HeLa cells, and the cells were treated with (+) or without (−) 20 μM MG‐132 for 4 h. UBQLNs were immunoprecipitated, and the precipitates were blotted with anti‐BAG6 and anti‐Flag antibodies.
-
BPolyubiquitinated proteins associated with UBQLN4 are CHX‐sensitive. The anti‐polyubiquitin signals that co‐precipitated with UBQLN4 in Fig 1D were quantified.
-
C, DTransfection of three independent UBQLN4 siRNA duplexes (#1, #2, and #3) as in Fig 4A did not affect the expression levels of UBQLN1 (C) or UBQLN2 (D) protein. Arrowheads indicate the UBQLN4 signal, and asterisks indicate a non‐specific band. Actin was used as a loading control.
Source data are available online for this figure.
Figure 1. UBQLN4 associates with newly synthesized polyubiquitinated proteins.
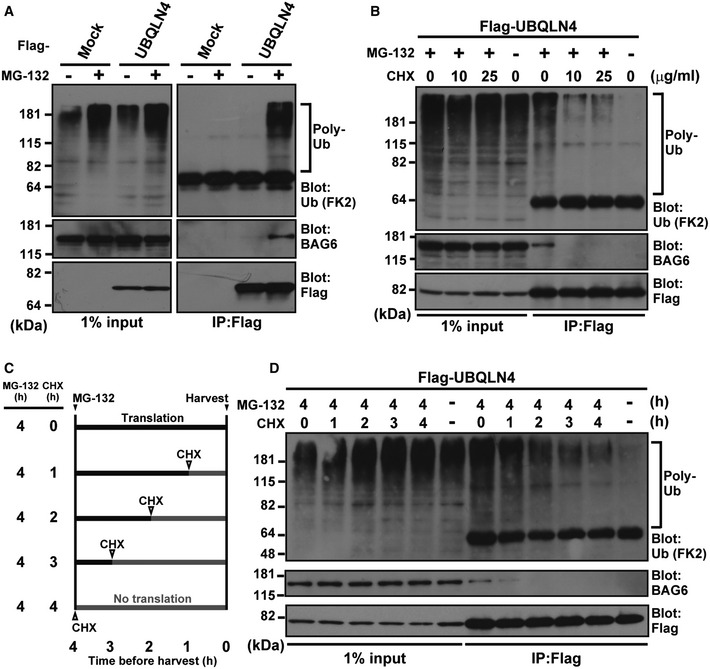
- UBQLN4 associates with polyubiquitinated proteasomal substrates. Flag‐tagged UBQLN4 protein was affinity‐purified from extracts of HeLa cells that were treated with (+) or without (−) 20 μM MG‐132 for 4 h before harvesting, and the Flag precipitates (IP:Flag) were blotted with anti‐polyubiquitin (FK2), anti‐BAG6, and anti‐Flag antibodies. Mock indicates empty vector transfection. Note that UBQLN4‐BAG6 co‐precipitation can be observed exclusively in the presence of polyubiquitinated substrates.
- Polyubiquitinated proteins associated with UBQLN4 are sensitive to the translation inhibitor cycloheximide (CHX). HeLa cells expressing Flag‐tagged UBQLN4 were treated with 5 μM MG‐132 as well as CHX (either 10 or 25 μg/ml) for 4 h as indicated and then subjected to immunoprecipitation with an anti‐Flag antibody as in (A).
- Schematic representation of MG‐132 and CHX chase experiments in (D). During the course of MG‐132 (5 μM) treatment for 4 h, CHX (10 μg/ml) was added at the indicated time points before harvesting.
- HeLa cells were treated as in (C), and Flag precipitates were blotted as in (B).
Source data are available online for this figure.
Since BAG6 reportedly associates with newly synthesized polyubiquitinated proteins that are sensitive to the protein synthesis inhibitor cycloheximide (CHX) 32, we examined whether polyubiquitinated species associated with UBQLN4 are also sensitive to CHX 65, 66. We found that treatment with CHX largely abolished the polyubiquitin conjugates associated with UBQLN4 in MG‐132 treated cells (Fig 1B). To verify further whether the UBQLN4‐associated polyubiquitinated species are newly synthesized polypeptides, we performed CHX chase experiments with the time course shown in Fig 1C. The results showed that the amounts of polyubiquitinated proteins on UBQLN4 decreased successively as CHX treatment was extended (Figs 1D and EV1B). These observations suggest that a significant part of the polyubiquitinated proteins associated with UBQLN4 consists of neo‐synthesized polypeptides that are targeted for proteasomal degradation just after their ribosomal synthesis.
UBQLN4 associates with cytoplasmic aggregates composed of defective polypeptides
Puromycin‐induced defective nascent polypeptides reportedly tend to form ubiquitin‐positive cytoplasmic aggregates called aggresome‐like induced structures (ALIS) 32, 67. To verify whether UBQLN4 indeed associates with newly synthesized defective polypeptides, we performed co‐immunostaining with UBQLN4 and polyubiquitin, a marker for ALIS. We found that UBQLN4 co‐localized with puromycin‐induced ALIS (Fig 2A, arrowhead), supporting the hypothesis that UBQLN4 possesses an intrinsic affinity for newly synthesized defective polypeptides.
Figure 2. UBQLN4 suppresses puromycin‐induced ubiquitin‐positive aggregates.

-
AUBQLN4 co‐localizes with cytoplasmic aggregates after puromycin treatment (indicated by arrowheads). HeLa cells expressing Flag‐tagged UBQLN4 were treated with 5 μg/ml puromycin for 2 h and then followed by 1% Triton X‐100 extraction and 4% paraformaldehyde fixation. Fixed cells were stained with anti‐polyubiquitin FK2 (shown in red to detect ubiquitin‐positive cytoplasmic aggregates, ALIS) and anti‐Flag antibodies (shown in green).
-
B, CPuromycin‐treated cells with UBQLN4 siRNA contain increased number of ALIS. At 72 h after transfection of the two distinct siRNA duplexes for UBQLN4 (UBQLN4 siRNA#1 and #2) or control siRNA (5 nM each), the cells were treated with 5 μg/ml puromycin for 2 h and then subjected to immunostaining with an anti‐polyubiquitin FK2 antibody (shown in green, B) as in (A). The number of ALIS was determined. The quantified data in (C) represent mean ± SEM. n = 76 cells for control siRNA, n = 89 cells for UBQLN4 siRNA#1, and n = 81 cells for UBQLN4 siRNA#2. P‐values were calculated by Welch's t‐test between the siRNA‐transfected and the respective control condition (*P < 0.01).
-
DDepletion of UBQLN4 makes HeLa cells more sensitive to puromycin‐induced cell death. After 48 h of UBQLN4 siRNA treatment, the cells were treated with 5 μg/ml puromycin for 15.5 h, and then, viability was measured. The data represent mean ± SD calculated from three independent experiments (n = 3). P‐values were calculated by Student's t‐test compared with puromycin‐treated control siRNA cells (*P < 0.01).
To estimate the impact of UBQLN4 on the formation of ALIS, we compared the number of ALIS in the presence or absence of UBQLN4 small interfering RNA (siRNA). We found that UBQLN4 knockdown (using two independent double‐stranded siRNAs) stimulated the formation of polyubiquitin‐positive ALIS with puromycin treatment (Fig 2B and C). Additionally, UBQLN4 knockdown accelerated puromycin‐induced cell death (Fig 2D), indicating that UBQLN4 has a protective role against defective polypeptide‐induced cell toxicity. To examine the possible off‐target effects of UBQLN4 knockdown on other UBQLN family proteins, we examined the effects of our double‐stranded siRNAs for UBQLN4 on the levels of UBQLN1 and UBQLN2 proteins (UBQLN3 is not expressed in HeLa cells). We provided experimental evidence that UBQLN4 siRNAs did not interfere with the expression of endogenous UBQLN1 nor UBQLN2 proteins (Fig EV1C and D), while endogenous UBQLN4 was clearly knocked down (Fig EV1C and D, arrowheads). Thus, it is obvious that UBQLN4 knockdown scarcely affects the expression of other UBQLN family proteins. Collectively, these results indicate that UBQLN4 is an essential element for the metabolism of aggregation‐prone neo‐synthesized polypeptides.
Establishing a model defective TMD polypeptide that is degraded immediately after its synthesis
Recently, the interesting concept of mislocalized proteins (MLPs) was suggested 21, 25, 68. An MLP is a kind of newly synthesized defective protein that fails to be assembled correctly in the ER membrane, thus being targeted to the BAG6‐mediated protein degradation pathway in the cytosol 25, 68. To explore whether such MLPs can be utilized as potential clients of UBQLN4, we examined a model for a misassembled TM protein.
The IL‐2 receptor α‐subunit (IL‐2Rα) is a single‐pass TM protein that contains a typical SS for ER insertion at its N‐terminus and a TMD at its C‐terminus (Fig 3A). Eisenlohr and colleagues reported recently that truncating the SS from IL‐2Rα destabilized it for proteasomal destruction and its degradation products are the source of the peptide ligands presented on the cell surface as MHC class I molecules 69. To examine whether the SS‐deleted form of IL‐2Rα (IL‐2Rα ΔSS) could be a model for MLPs, we prepared truncated IL‐2Rα proteins (Fig 3A).
Figure 3. ΔSS mutant form of IL‐2Rα as a model for mislocalized proteins.
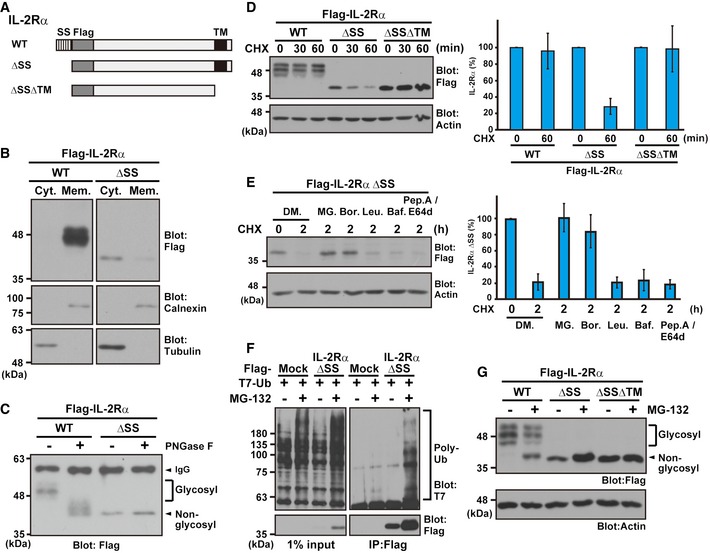
- Schematic representation of the IL‐2Rα proteins used in this study. WT: wild type, ΔSS: signal sequence (SS)‐deleted mutant, and ΔSSΔTM: SS and transmembrane domain (TM)‐deleted mutant.
- IL‐2Rα ΔSS mislocalized to the cytosolic fraction. HeLa cells, expressing Flag‐tagged IL‐2Rα WT or ΔSS, were fractionated to the cytosolic (Cyt.) and membrane (Mem.) fractions. Tubulin was used as a cytoplasmic marker, while calnexin was used for the ER membrane fraction.
- IL‐2Rα ΔSS was not glycosylated. Flag‐tagged IL‐2Rα WT or ΔSS proteins were expressed in HeLa cells and immunoprecipitated with an anti‐Flag antibody. The precipitates were incubated with (+) or without (−) 10 units of the deglycosylation enzyme PNGase F for 2 h and subjected to Western blot analysis with an anti‐Flag antibody. Glycosylated and non‐glycosylated signals are indicated.
- The glycosylated form of IL‐2Rα WT is a stable protein, while mislocalized IL‐2Rα ΔSS is degraded rapidly via its TMD. A series of IL‐2Rα deletion proteins were expressed in HeLa cells and then chased with 20 μg/ml CHX for the indicated periods. Actin was used as a loading control. The right panel is a quantification graph of the left blot signals, and data represent mean ± SD calculated from four independent experiments (n = 4).
- IL‐2Rα ΔSS is degraded in a proteasome‐dependent manner. HeLa cells expressing Flag‐tagged IL‐2Rα ΔSS were treated with 20 μg/ml CHX as well as 0.1% DMSO (DM.), 10 μM MG‐132 (MG.), 2 μM bortezomib (Bor.), 10 μM leupeptin (Leu.), 0.1 μM bafilomycin A1 (Baf.), or 10 μg/ml pepstatin A (pep. A)/E64d for the indicated periods. The right panel indicates the quantified data of the left blot signals presented as mean ± SD calculated from three independent experiments (n = 3).
- The Flag‐tagged IL‐2Rα ΔSS mutant and T7‐tagged ubiquitin were co‐expressed in HeLa cells, and the cells were treated with (+) or without (−) 10 μM MG‐132. After 4 h, Flag precipitates were blotted with anti‐T7 and anti‐Flag antibodies.
- HeLa cells expressing Flag‐tagged IL‐2Rα WT, ΔSS, and ΔSSΔTM proteins were treated with (+) or without (−) 10 μM MG‐132. At 4 h after MG‐132 treatment, the cells were lysed and analyzed by immunoblotting using the indicated antibodies. Note that a higher migrating form of non‐glycosylated (and thus defective) IL‐2Rα WT (indicated by the arrowhead) is detected strongly in the presence of MG‐132.
Source data are available online for this figure.
To demonstrate directly that IL‐2Rα ΔSS fails to assemble in the membrane, we examined its mislocalization to the cytosolic fraction. As shown in Fig 3B, wild‐type IL‐2Rα (IL‐2Rα WT) was detected in the membrane fraction (Mem), while the majority of IL‐2Rα ΔSS was present in the cytosolic soluble fraction (Cyt), even though it possesses a highly hydrophobic TMD. To investigate further whether IL‐2Rα ΔSS fails to translocate to the ER membrane, we analyzed the glycosylation state of IL‐2Rα protein. In the case of IL‐2Rα WT expression, smear bands were detected (Fig 3C, indicated as glycosyl), and these signals were shifted down by treatment with PNGase F, a deglycosylation enzyme (Fig 3C, indicated as non‐glycosyl). This result suggests that the majority of IL‐2Rα WT is indeed glycosylated and thus assembled in the ER membrane fraction. On the contrary, glycosyl modification was detected scarcely for the ΔSS form of mutant IL‐2Rα (Fig 3C), indicating that IL‐2Rα ΔSS is not inserted properly into the ER membrane.
A previous study suggested that IL‐2Rα ΔSS is a highly unstable protein 69, and we provide additional evidence for its ubiquitin–proteasome‐mediated degradation (Figs 3D–F and EV2A and B, ΔSS). Indeed, the proteasome inhibitor bortezomib blocked its degradation as efficiently as MG‐132‐treatment, while both leupeptin and bafilomycin A1, lysosomal inhibitors, failed to stabilize IL‐2Rα ΔSS (Fig 3E). We also confirmed that the C‐terminal TMD is responsible for the rapid cytosolic degradation of IL‐2Rα ΔSS (Fig 3D), and removal of the TMD from IL‐2Rα ΔSS largely abolished the MG‐132‐induced increase in its levels (Fig 3G). Thus, the exposed hydrophobic domain of misassembled IL‐2Rα in the cytosol functions as a major degradation signal for the ubiquitin‐dependent degradation pathway. However, we do not exclude the possibility of the existence of another potential “degron sequence” in IL‐2Rα ΔSSΔTM, since residual effects of MG‐132 were observed occasionally for IL‐2Rα ΔSSΔTM protein (Fig EV2C, ΔSSΔTM).
Figure EV2. Defective forms of IL‐2Rα protein are stabilized by MG‐132.
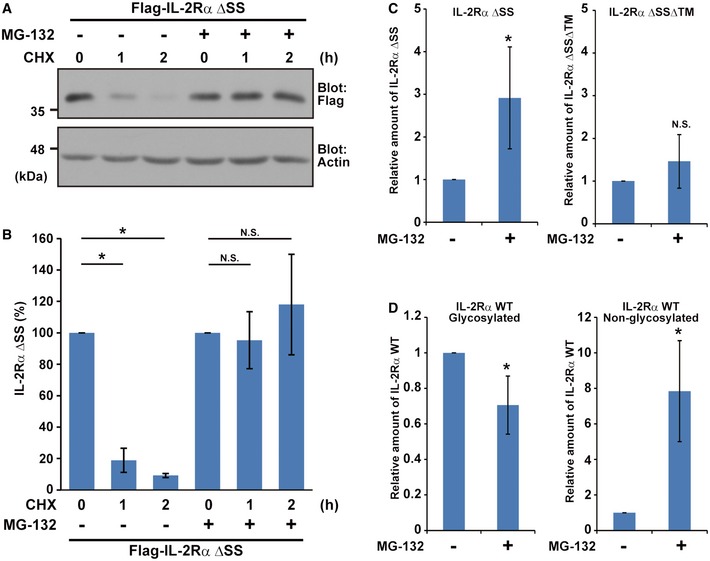
-
A, BMislocalized IL‐2Rα protein was stabilized by MG‐132. Anti‐Flag immunosignals with (+) or without (−) MG‐132 were quantified at the indicated time points. The data represent mean ± SD calculated from three independent experiments (n = 3). Statistical analysis by Welch's t‐test (*P < 0.01; N.S., non‐significant).
-
C, DAnti‐Flag immunosignals of IL‐2Rα ΔSS and IL‐2Rα ΔSSΔTM (C) as well as those of low‐mobility (indicated as glycosylated) and high‐mobility (indicated as non‐glycosylated) signals of IL‐2Rα WT (D) (shown in Fig 3G as a representative example) were quantified. The data represent mean ± SD calculated from four independent experiments (n = 4). Statistical analysis by Welch's t‐test (*P < 0.05; N.S., non‐significant).
Source data are available online for this figure.
It should be noted that the levels of the non‐glycosylated form of IL‐2Rα WT, a high‐mobility band close to that of IL‐2Rα ΔSS, were sensitive to MG‐132 (Fig 3G, arrowheads and Fig EV2D, non‐glycosylated), as is the case for defective IL‐2Rα, IL‐2Rα ΔSS, although those of the low‐mobility glycosylated forms of IL‐2Rα WT, representing mature folded ER species, were not (Fig 3G, glycosyl and Fig EV2D, glycosylated). These observations suggest that some portion of IL‐2Rα WT fails to translocate to the ER and thus remains in the cytosol as a defective protein. These results indicate that deleting the SS from IL‐2Rα, as well as naturally occurring defective assembly of TM proteins, leads to the production of client proteins for the ubiquitin‐dependent degradation system. We suggest that IL‐2Rα ΔSS can be considered as an appropriate model for such newly synthesized (and rapidly degraded) cytosolically mislocalized TM proteins.
UBQLN4 is essential for misassembled membrane protein degradation
Identification of the mechanism for the targeted degradation of misassembled TM proteins is crucial for the quality control of newly synthesized TMD polypeptides. Hegde and colleagues reported that the mislocalized prion protein (PrP) is a client for the BAG6‐mediated cytosolic degradation pathway 25, 68. In agreement with their findings for PrP, we found that BAG6 co‐precipitated with the defective TM protein IL‐2Rα ΔSS (Fig EV3A). Furthermore, this interaction depended on the presence of a C‐terminal TMD (Fig EV3A). In addition, knockdown of BAG6 blocked the proteasome‐mediated degradation of IL‐2Rα ΔSS (Fig EV3B). Since BAG6 is now found to be critical for the degradation of IL‐2Rα ΔSS, we examined the possible participation of UBQLN4, a BAG6‐binding ubiquitin receptor protein associated with newly synthesized polyubiquitinated polypeptides (Figs 1 and EV1A), in the metabolism of the misassembled IL‐2Rα ΔSS model substrate. We designed three independent siRNA constructs for UBQLN4 to avoid possible adventitious off‐target effects of each double‐stranded RNA and provided evidence that all three blocked the degradation of IL‐2Rα ΔSS effectively (Fig 4A and B, siRNA#1, #2, and #3). Furthermore, we showed that three scrambled double‐stranded RNAs for each knockdown construct (Fig EV4A, siRNA#1scr, #2scr, and #3scr), in addition to the universal negative control siRNA construct (Fig 4A and B), did not affect IL‐2Rα ΔSS stability. These results suggest that endogenous UBQLN4 is indeed essential for IL‐2Rα ΔSS metabolism. It is interesting to note that UBQLN4 knockdown did not abolish, but rather strengthened, the polyubiquitin modification of IL‐2Rα ΔSS (Fig 4C, compare lanes 1 and 3, Appendix Fig S3A), while BAG6 knockdown abolished ubiquitination (Fig 4C, compare lanes 2 and 6, Appendix Fig S3A). These observations support the idea that UBQLN4 could be essential for the degradation of misassembled TM proteins, possibly at the post‐ubiquitination step of IL‐2Rα ΔSS degradation.
Figure EV3. BAG6 is involved in the degradation of mislocalized IL‐2Rα ΔSS .
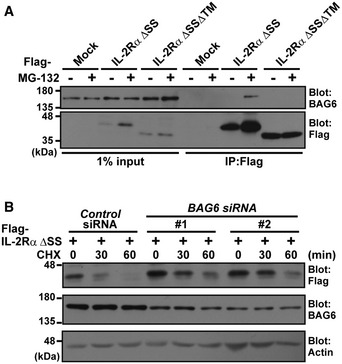
- BAG6 interacts with misassembled IL‐2Rα through its TMD. HeLa cells expressing Flag‐tagged IL‐2Rα ΔSS or IL‐2Rα ΔSSΔTM were treated with (+) or without (−) 10 μM MG‐132 for 4 h, and then, Flag‐IL‐2Rα substrates were immunoprecipitated and their co‐precipitation with endogenous BAG6 was analyzed with an anti‐BAG6 antibody.
- BAG6 associates with the mislocalized IL‐2Rα ΔSS degradation pathway. HeLa cells were transfected with two distinct siRNA duplexes for BAG6 (BAG6 siRNA#1 and #2) or control siRNA. At 48 h after siRNA transfection, Flag‐tagged IL‐2Rα ΔSS was detected as in Fig 4A. Anti‐Flag immunosignals in control or BAG6 siRNA‐treated cells were detected at the indicated time points. Actin was used as a loading control.
Source data are available online for this figure.
Figure 4. UBQLN4 is essential for the degradation of mislocalized IL‐2Rα ΔSS .
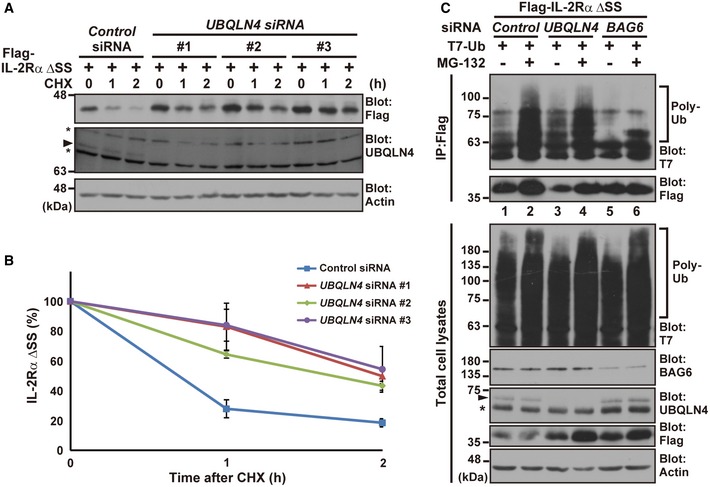
-
A, BIL‐2Rα ΔSS is stabilized in UBQLN4 knockdown cells. HeLa cells were transfected with three distinct siRNA duplexes for UBQLN4 (UBQLN4 siRNA#1–#3) or control siRNA. At 48 h after siRNA transfection, Flag‐tagged IL‐2Rα ΔSS was expressed in the cells. At 24 h after IL‐2Rα ΔSS transfection, the cells were chased with 20 μg/ml CHX and harvested at the indicated time after CHX addition. Anti‐Flag signals in the control or UBQLN4 siRNA‐treated cells (siRNA#1–#3) were quantified at the indicated time points. The data represent mean ± SEM calculated from four independent experiments (n = 4). The UBQLN4 signal is marked by an arrowhead, while the band just below it (indicated by an asterisk) is a non‐specific band, since this signal was never affected by any of the UBQLN4 siRNAs. Actin was used as a loading control.
-
CPolyubiquitin modification of IL‐2Rα ΔSS was not diminished, but rather strengthened, in UBQLN4 knockdown cells. At 48 h after transfection of HeLa cells with siRNA for UBQLN4 or BAG6, Flag‐tagged IL‐2Rα ΔSS and T7‐tagged ubiquitin were expressed with (+) or without (−) 10 μM MG‐132. From these cells, Flag‐tagged IL‐2Rα ΔSS was affinity‐purified using hot lysis analysis, and Flag precipitates were blotted with an anti‐T7 antibody to detect polyubiquitination of IL‐2Rα ΔSS. The UBQLN4 signal is marked by an arrowhead, and the asterisk indicates a non‐specific band. Actin was used as a loading control.
Source data are available online for this figure.
Figure EV4. UBQLN4‐mediated metabolism for mislocalized proteins.
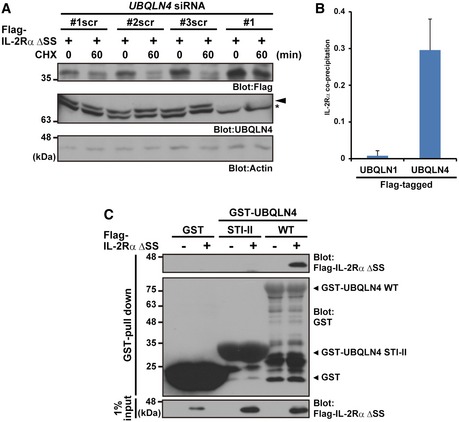
- HeLa cells were transfected with three independent scrambled siRNA duplexes (UBQLN4 siRNA#1scr, #2scr, and #3scr) as negative controls for the UBQLN4 siRNA#1‐#3 experiments shown in Fig 4A. The arrowhead indicates the UBQLN4 signal, and the asterisk indicates a non‐specific band. Actin was used as a loading control.
- T7‐tagged IL‐2Rα ΔSS signals (shown in Fig 6B as a representative example) were quantified. The data represent mean ± SD calculated from three independent experiments (n = 3).
- Purified UBQLN4 WT protein co‐precipitated with Flag‐IL‐2Rα ΔSS, while the STI‐II fragment of UBQLN4 did not. Bacterially expressed GST‐UBQLN4 WT and GST‐STI‐II were incubated with extracts of HeLa cells expressing Flag‐tagged IL‐2Rα ΔSS and treated with MG‐132 for 4 h. After in vitro GST pull‐down, the precipitates were probed with anti‐Flag and anti‐GST antibodies.
Source data are available online for this figure.
An unembedded TMD is essential for UBQLN4 recognition
To reveal how UBQLN4 regulates the turnover of misassembled TM proteins, we examined its physical interaction with IL‐2Rα ΔSS. We immunoprecipitated a series of IL‐2Rα deletion mutants with full‐length UBQLN4 and examined their interaction (Fig 5A). We found that UBQLN4 co‐precipitated with IL‐2Rα ΔSS far more efficiently than with IL‐2Rα WT (Fig 5A, lanes 8 and 10). Furthermore, UBQLN4 associated with IL‐2Rα ΔSS even more under proteasome inhibition (Fig 5A, lanes 9 and 10). In addition, the TMD of IL‐2Rα ΔSS was found to be indispensable for UBQLN4 recognition, since IL‐2Rα ΔSSΔTM did not interact with UBQLN4 very much, even in the presence of MG‐132 (Fig 5A, lane 12). Conversely, similar results were observed when we immunoprecipitated UBQLN4 (Fig 5B, Appendix Fig S3B); it co‐precipitated IL‐2Rα ΔSS effectively, while it scarcely precipitated the TMD‐deleted mutant (Fig 5B, ΔSSΔTM). Note that the glycosylated 48 kDa form of IL‐2Rα WT co‐precipitated scarcely with UBQLN4 (Fig 5B). These observations suggest that UBQLN4 preferentially targets misassembled TM proteins for proteasome‐mediated degradation through an exposed TMD‐dependent mechanism.
Figure 5. UBQLN4 recognizes misassembled IL‐2Rα through its unembedded TMD .
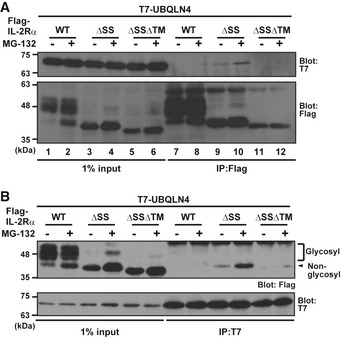
- A series of Flag‐tagged truncated mutants of IL‐2Rα substrates were expressed in HeLa cells with T7‐tagged UBQLN4 and treated with (+) or without (−) 10 μM MG‐132 for 4 h. Flag‐IL‐2Rα substrates were immunoprecipitated, and their co‐precipitation with UBQLN4 was analyzed.
- T7‐UBQLN4 was immunoprecipitated, and its interactions with a series of IL‐2Rα proteins were detected.
Source data are available online for this figure.
The STI‐II region of UBQLN4 is necessary for its recognition of unembedded IL‐2Rα
All UBQLN family proteins possess N‐terminal UBL and C‐terminal UBA domains (Fig 6A) 50, 51, 52, 70, 71, 72. The overall amino acid sequence homology between UBQLN1 and UBQLN4 is high (61%), suggesting they might share common functions. To determine whether UBQLN1 also recognizes misassembled TM proteins, we examined the affinity of UBQLN1 for IL‐2Rα ΔSS. Although UBQLN1 reportedly interacts with polyubiquitinated substrates and proteasomes 50, UBQLN1 poorly precipitated IL‐2Rα ΔSS substrates (and BAG6) compared to UBQLN4 (Figs 6B, E and F, and EV1A). Indeed, our quantified data show the clear preference of IL‐2Rα ΔSS binding for UBQLN4 compared to that for UBQLN1 (Fig EV4B), providing convincing evidence for their specificity.
Figure 6. The STI‐II region of UBQLN4 determines its specificity to misassembled IL‐2Rα.
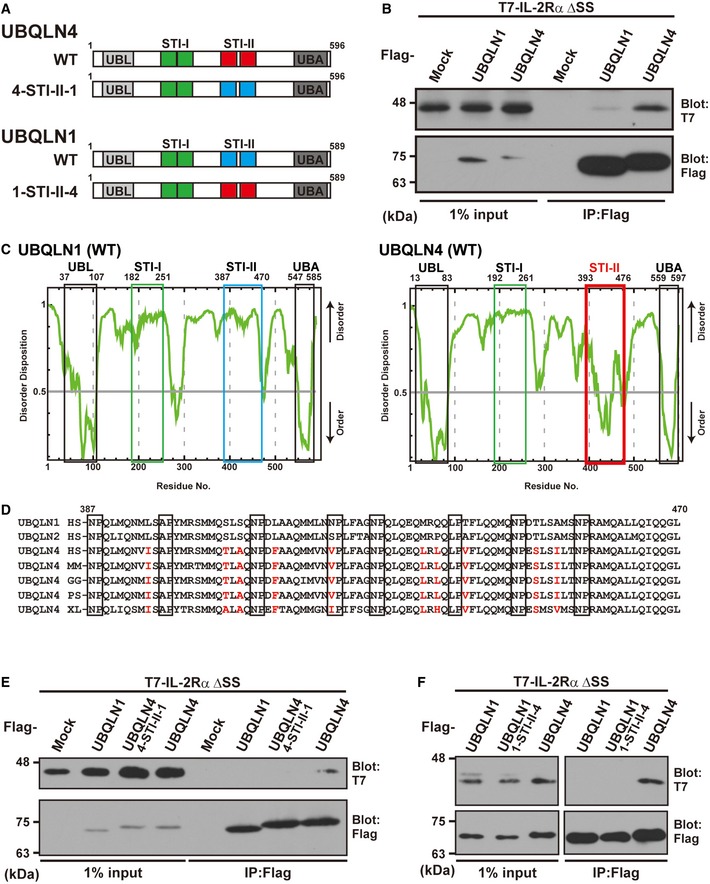
- Schematic representation of the UBQLN1, UBQLN4, and STI‐II mutant proteins used in this study.
- UBQLN4 dominantly co‐precipitates IL‐2Rα ΔSS over UBQLN1. Precipitates of Flag‐tagged UBQLN1 or UBQLN4 from HeLa cell lysates were probed with IL‐2Rα ΔSS.
- The complete amino acid sequences of human UBQLN1 and UBQLN4 were analyzed using the disorder/order structure prediction program PONDR‐FIT. The STI‐II region of UBQLN4 is indicated by the red box, and that of UBQLN1 is boxed blue. Note that the disorder tendency of the UBL, STI‐I, and UBA regions is nearly identical between UBQLN1 and UBQLN4 (indicated by black and green boxes), while that of the STI‐II region is quite distinct, and the STI‐II of UBQLN4 shows a high probability of an ordered structure. The numbers denote the corresponding amino acid positions in these proteins.
- Amino acid sequence alignments of the STI‐II regions of UBQLN1 and UBQLN4. UBQLN4‐specific residues are shown as red characters. HS: Homo sapiens (Mammalia), MM: Mus musculus (Mammalia), GG: Gallus gallus (Aves), PS: Pelodiscus sinensis (Reptilia), and XL: Xenopus laevis (Amphibia).
- Substitution of the STI‐II sequence of UBQLN4 with that of UBQLN1 (designated as the 4‐STI‐II‐1 mutant protein) abolished its high affinity to the IL‐2Rα ΔSS client protein.
- Substitution of the STI‐II sequence of UBQLN1 with that of UBQLN4 (designated as the 1‐STI‐II‐4) did not enable UBQLN1 to recognize IL‐2Rα ΔSS.
Source data are available online for this figure.
In order to obtain insights into the differences between UBQLN family proteins, we performed comprehensive disorder/order structural prediction analysis. When the amino acid sequences of UBQLN1 and UBQLN4 were analyzed using the secondary structure prediction program PONDR‐FIT, their overall disorder/order tendency was nearly identical. For example, the UBL and UBA domains of UBQLN1 and UBQLN4 had a very low disorder score (Fig 6C, black boxes), exactly matching the fixed structures of these domains as determined by X‐ray crystal diffraction analysis (PDB ID codes 2KNZ for UBQLN4 UBA, 2JY5 for UBQLN1 UBA and 2KLC for UBQLN1 UBL). Both UBQLN family proteins contain putative STI1‐like motifs between the UBL and UBA domains, and we designated two tandem STI1 repeats in the N‐terminal half as STI‐I, and two other tandem repeats in the C‐terminal half as STI‐II (Fig 6A). While STI‐I (amino acids 182–251 and 192–261 for human UBQLN1 and UBQLN4, respectively) had a high disorder score in all UBQLN family members (Fig 6C, green boxes), we found that the STI‐II sequence of UBQLN4 (amino acids 393–476 for human UBQLN4) had the characteristic lowest disorder score (Fig 6C, red box), making a clear contrast with the high disorder score of STI‐II in UBQLN1 (Fig 6C, blue box) and UBQLN2. Similar results were obtained using the structural prediction algorithms RONN and Displot, with the STI‐II region demonstrating the lowest disorder tendency in UBQLN4. The structured feature of UBQLN4 STI‐II is evolutionarily conserved from Amphibia, Reptilia, and Aves to their mammalian orthologs (Appendix Fig S2). These analyses suggest that the STI‐II region of UBQLN4 forms a unique, distinct, and evolutionarily conserved structural domain.
When we compared the sequences of the STI‐II regions of UBQLN1 and UBQLN4, they are highly conserved with only 10 residue characteristic substitutions with increased hydrophobicity (Fig 6D). To investigate whether the structural and sequential features of the UBQLN4 STI‐II domain might be linked with its functional properties, we prepared a mutant protein in which UBQLN4‐type STI‐II was replaced with UBQLN1‐type STI‐II (Fig 6A and E). We found that such a substituted mutant, designated as 4‐STI‐II‐1, showed a greatly reduced affinity for IL‐2Rα ΔSS when compared to WT UBQLN4 (Fig 6E). When STI‐II was transferred from UBQLN4 into UBQLN1, this reverse chimera (designated 1‐STI‐II‐4) failed to stimulate binding with IL‐2Rα ΔSS (Fig 6F), suggesting that STI‐II is not the sole determinant of binding to specific substrates. In agreement with this view, a recombinant STI‐II fragment fused with GST failed to associate with IL‐2Rα ΔSS, while recombinant GST‐UBQLN4 WT co‐precipitated with this client protein in vitro (Fig EV4C).
To determine whether the STI‐II domain is indeed required for IL‐2Rα ΔSS recognition by UBQLN4, we prepared a series of UBQLN4 deletion mutants (Fig 7A). While deletion of either the UBL or STI‐I region of UBQLN4 did not affect its affinity for IL‐2Rα ΔSS (Fig 7B, ΔUBL and ΔSTI‐I, respectively), we found that truncation of the STI‐II domain, as well as the C‐terminal UBA domain, greatly compromised its affinity for IL‐2Rα ΔSS (Fig 7B, ΔSTI‐II and ΔUBA, respectively). Furthermore, we provide evidence that the C‐terminal half of the UBQLN4 fragment (from STI‐II to the C‐terminal UBA domain, residues 378–596) sufficiently co‐precipitated IL‐2Rα ΔSS substrates, while its STI‐II‐truncated mutant (residues 471–596) showed reduced affinity (Fig 7C). These results suggest that the STI‐II region might be necessary, but not sufficient, for substrate recognition in UBQLN4 (Fig 7B and C). It is also apparent that UBL domain is dispensable for substrate binding.
Figure 7. STI‐II region of UBQLN4 is essential for its binding to client proteins.
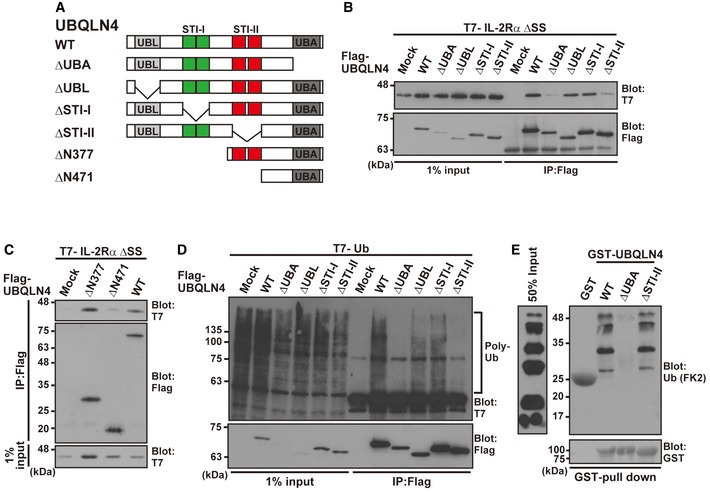
-
ASchematic representation of the UBQLN4 deletion mutant proteins used in this study.
-
B–DThe wild‐type (WT) form of Flag‐tagged UBQLN4 and its truncated derivatives were expressed in HeLa cells with T7‐tagged IL‐2Rα ΔSS (B and C) and T7‐tagged ubiquitin (D). At 4 h after the addition of 10 μM MG‐132, UBQLN4 was immunoprecipitated with an anti‐Flag antibody and the precipitates were blotted with an anti‐T7 antibody.
-
EGST‐tagged and purified UBQLN4 and its truncated derivatives (ΔUBA and ΔSTI‐II) were incubated with K48‐linked polyubiquitin chains. After in vitro GST pull‐down, the precipitates were probed with an anti‐polyubiquitin (FK2) antibody.
Source data are available online for this figure.
The STI‐II domain of UBQLN4 is essential for endogenous client recognition
Since both the UBA and STI‐II domains of UBQLN4 are essential for its binding with the misassembled model substrate protein IL‐2Rα ΔSS (Fig 7B), which can be polyubiquitinated for proteasomal degradation (Fig 3F), we examined whether deletion of these regions affects its co‐precipitation with not only the defective model protein but also with endogenous newly synthesized polyubiquitinated substrates (Fig 1D). We immunoprecipitated a series of UBQLN4 deletion mutants from MG‐132‐treated cell extracts, and the precipitates were probed with an anti‐T7‐ubiquitin antibody (Fig 7D, Appendix Fig S3C). It is quite natural that deletion of the UBA domain results in a reduction of polyubiquitinated proteasomal substrate co‐precipitation (Fig 7D, ΔUBA), since the UBA domain is known to recognize the polyubiquitin moiety 58, 60, 61, 62. Our notable finding is that deletion of the STI‐II region also compromised co‐precipitation of polyubiquitinated client proteins from the cell extracts as effectively as UBA domain truncation (Fig 7D, ΔSTI‐II), suggesting that the STI‐II domain of UBQLN4 possesses an essential function for substrate recognition. It is also apparent that the UBA domain itself is not sufficient to recruit endogenous polyubiquitinated substrates in the case of UBQLN4. It is of note that deletion of STI‐II did not affect the direct polyubiquitin chain recognition ability of the C‐terminal UBA domain, since our in vitro experiments showed that the STI‐II‐deleted form of purified UBQLN4 protein bound to a K48‐linked polyubiquitin chain as efficiently as to the WT protein (Fig 7E). These observations suggest that the ubiquitin‐binding ability of the UBQLN4 UBA domain itself is not sufficient to recognize newly synthesized client proteins. Collaboration between the UBA and STI‐II domains of UBQLN4 might be necessary for the efficient recognition of misassembled TM protein substrates.
Endogenous substrates of SRP are clients of UBQLN4
Successful biogenesis of TM proteins is mediated through the SRP, which recognizes the SS of client TM proteins to bring them to the SRP receptor on the rough ER 6, 7, 8, 13, 14, 15, 22. Since we identified the ΔSS truncated form of IL‐2Rα as a client of UBQLN4, we examined whether depletion of the SRP results in the production of defective cytoplasmic species derived from WT IL‐2Rα. As shown in Fig 8A, knockdown of SRP54, the SS‐binding subunit of the SRP, indeed led to a reduction of the glycosylated form of mature IL‐2Rα (Fig 8A, lanes 3 and 4, indicated as assembled). Furthermore, SRP54 depletion induced the accumulation of the non‐glycosylated form of IL‐2Rα WT in the presence of MG‐132 (Fig 8A, lane 4, indicated as defective), similar to the case for the truncation of its SS (Fig 3C), suggesting that cytosolically mislocalized TM proteins can be induced by defects in the SRP. Importantly, the non‐glycosylated form of IL‐2Rα WT in SRP‐suppressed cells was recognized efficiently by UBQLN4 (Fig 8A, lane 8, indicated by an arrowhead). These observations suggest that UBQLN4 contributes to the quality control of misassembled cytoplasmic TM proteins under SRP defective conditions, as for IL‐2RαΔSS.
Figure 8. SRP deficiency enhances the interaction between UBQLN4 and its client proteins.
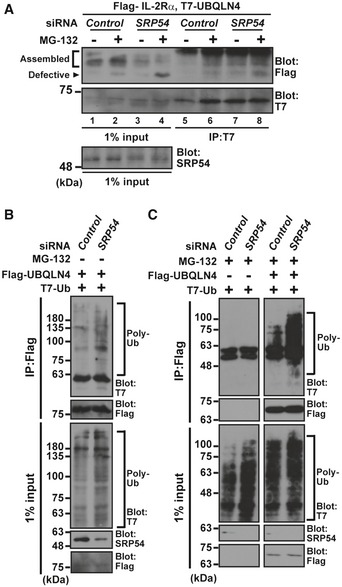
-
AUBQLN4 interacts with non‐glycosylated (and thus defective) IL‐2Rα WT in SRP54 knockdown cells. After treatment of HeLa cells with SRP54 siRNA (10 nM) for 48 h, Flag‐tagged IL‐2Rα WT and T7‐tagged UBQLN4 proteins were expressed, and the cells were treated with (+) or without (−) 10 μM MG‐132 at 4 h before harvesting. UBQLN4 was affinity‐purified from cell extracts and probed with an anti‐Flag antibody. Note that the levels of non‐glycosylated IL‐2Rα WT (indicated as defective) increased, while the glycosylated form of this protein (indicated as assembled) decreased in SRP54 knockdown cells.
-
B, CSRP54 knockdown stimulates polyubiquitinated proteins co‐precipitation of UBQLN4. Flag‐tagged UBQLN4 and T7‐tagged ubiquitin were expressed in SRP54 siRNA‐treated cells, and Flag precipitates were probed with an anti‐T7 antibody to detect polyubiquitinated client co‐precipitation with UBQLN4 under no addition of MG‐132 (B). Note that the interaction of UBQLN4 with polyubiquitinated substrates was further increased in SRP54 knockdown cells treated with MG‐132 for 4 h before harvesting (C).
Source data are available online for this figure.
In the condition of SRP54 knockdown, it is probable that a number of endogenous TMD proteins with an N‐terminal SS also fail to assemble in the ER and thus accumulate as polyubiquitinated defective TM proteins. Since UBQLN4 associates with a model misassembled TM protein (Fig 5) as well as endogenous polyubiquitinated newly synthesized polypeptides (Fig 1B and D) through a common STI‐II region (Fig 7B–D), we reasoned that not only IL‐2Rα but also a variety of endogenous defective TM proteins might be general clients of UBQLN4. To investigate whether UBQLN4 might be involved with a variety of SRP substrates, we inactivated SRP54 using siRNA. As shown in Fig 8B, SRP54 knockdown resulted in a slight accumulation of polyubiquitinated proteins, even in the absence of MG‐132, and we found an increased level of UBQLN4‐associated polyubiquitinated species that were induced by SRP54 knockdown (Fig 8B, Appendix Fig S3D). In the presence of MG‐132, SRP54 siRNA further stimulated the association of polyubiquitinated substrates with UBQLN4 (Fig 8C, IP lane, with MG‐132). These results suggest that at least a part of the endogenous pool of SRP substrates became a client of UBQLN4 for targeted degradation when the function of the SRP was compromised.
Discussion
The majority of nascent TMD proteins are integrated into the ER membrane by virtue of an SS‐ and SRP‐mediated co‐translational process 6, 8, 9, 12, 14. In accordance with this notion, we showed that SS truncation or SRP suppression resulted in the production of a defective form of IL‐2Rα. With such a defective protein model, we identified the UBL‐UBA ubiquitin receptor UBQLN4 as a binding protein whose function is closely linked with the metabolism of newly synthesized and misassembled TMD polypeptides in the cytoplasm.
Accumulating evidence suggests that the substrates of both the co‐ and post‐translational ER targeting pathways are polyubiquitinated in the aqueous cytosol in a BAG6‐dependent manner when targeting fails 25, 43, 44, 47, 68. Mammalian PrP also showed preferential stabilization as a non‐glycosylated precursor species when BAG6 was knocked down 25. In the present study, we found that UBQLN4 is included in the BAG6 complex, while other UBQLN paralog family proteins were found to have a much weaker cellular interaction with BAG6 (Fig EV1A). This observation suggests that BAG6 collaborates preferentially with UBQLN4 and their function might be closely related. Indeed, both BAG6 25 and UBQLN4 (this study) were shown to be essential for targeting improperly assembled TM proteins for proteasomal degradation. However, the function of UBQLN4 is not identical to that of BAG6, because the knockdown phenotype of UBQLN4 with polyubiquitinated protein accumulation was the opposite of that of BAG6. Indeed, UBQLN4 knockdown stimulated the cellular accumulation of polyubiquitinated IL‐2Rα ΔSS, while BAG6 knockdown reduced it (Fig 4C). Thus, they might play partly distinct roles in their complex.
How UBQLN4 recognizes client substrates is an important issue to understand the quality control mechanism of newly synthesized TM proteins. UBQLN4 possesses two tandem repeats of the STI1 motif in its central region 49, 73, 74. STI‐I motifs and their upstream region in the N‐terminal half of UBQLN4 are involved in SS recognition of various ER luminal proteins 51 as well as in the interaction with LC3, an autophagic regulator 74, while the functions of the STI‐II region in UBQLN4 remain completely elusive to date. In this study, we found that the STI‐II region of UBQLN4 is necessary, but not sufficient, for the recruitment of mislocalized TMD proteins (Fig 7B). We also showed that the STI‐II region of UBQLN4 is critical for co‐precipitation with newly synthesized polyubiquitinated proteins (Fig 7D). While all UBQLN family proteins possess an STI‐II motif, UBQLN1 associates with neither IL‐2Rα ΔSS (Fig 6B, E and F) nor BAG6 (Fig EV1A), in contrast to UBQLN4. These observations suggest that the STI‐II region of UBQLN4 is a distinct domain, as suggested by disorder/order prediction analysis (Fig 6C), which plays a key role in the discrimination of unassembled TM proteins.
Groettrup et al 23 reported that an MHC class I‐restricted peptide, mainly derived from the degradation products of newly synthesized defective polypeptides 66, 75, 76, 77, 78, contains the cleavage site of the ER signal peptidase 23. As signal peptides for ER targeting are cleaved off immediately after their co‐translational insertion into the lumen of the ER 8, this uncleaved antigenic peptide did not originate from its retro‐translocation from the ER 23 and thus could not be derived from ER‐associated protein degradation (ERAD), a major TM protein quality control machinery in eukaryotic cells 77, 79, 80, 81, 82, 83, 84, 85. Instead, this endogenous antigenic protein could be degraded by cytoplasmic proteasomes before it reaches the lumen of the ER 23. In accordance with this idea, ablating the SS of a type I membrane protein reportedly results in its default delivery to the cytosol and accelerated turnover for MHC‐mediated antigen presentation, in contrast to the substantially delayed presentation from the ERAD‐derived counterpart pathway 69, 86. Although we do not exclude the possibility that a portion of IL‐2Rα ΔSS is an ERAD substrate, this seems largely unlikely since the typical ERAD pathway is effective only after proteins are incorporated into the ER membrane. It should be noted that UBQLN4 also recognizes the non‐glycosylated (and thus mislocalized) form of wild‐type IL‐2Rα that possesses a non‐processed SS (with a T7‐tag) at its N‐terminus in the cytoplasm (Fig EV5). These observations also support our hypothesis that UBQLN4 eliminates MLP via a non‐ERAD pathway. It was reported that the cytosolic face of the ER membrane serves as a “platform” for the rapid degradation of cytosolic substrates that exposed hydrophobic 87, and ERAD‐C components are responsible for such “non‐ERAD” protein degradation 87, 88. We speculate that the ERAD‐C machinery might also be utilized for defective IL‐2Rα degradation on the cytosolic surface of the ER. An interesting challenge for the future will be to determine how the UBQLN4 ubiquitin receptor collaborates with chaperones/E3 ligases/BAG6/ERAD‐C machineries. Clarification of the linkage between defects in the UBQLN4‐BAG6 complex and various human diseases such as immune disorders, neurodegeneration, and diabetes should also be a relevant prospect for future extensive investigations, since the quality control system for MLP is intimately linked with these diseases. Our findings in this study will provide important clues to reveal these critical points.
Figure EV5. UBQLN4 recognizes ER assembly defective forms of IL‐2Rα.

- Schematic representation of the IL‐2Rα proteins used in this experiment. WT: wild type, ΔSS: SS‐deleted mutant, and ΔSSΔTM: SS‐ and TM‐deleted mutant. Note that the position of the T7‐tag in WT IL‐2Rα is upstream (N‐terminal) of the SS: thus, SS cleavage by signal peptidase that occurs immediately after ER luminal incorporation would result in the loss of the T7 signal.
- A series of T7‐tagged IL‐2Rα substrates were expressed in HeLa cells with Flag‐tagged UBQLN4 and treated with (+) or without (−) 10 μM MG‐132 for 4 h. Flag‐UBQLN4 was immunoprecipitated, and its co‐precipitation with IL‐2Rα was analyzed. In contrast to the case in Fig 5, glycosylated bands of WT IL‐2Rα were never detected, since T7‐tagged SS for ER targeting is cleaved off immediately after its successful co‐translational insertion into the lumen of the ER. Thus, only cytoplasmic defective forms of IL‐2Rα can be detected as T7‐positive polypeptides in this experiment. Asterisks indicate non‐specific signals.
Source data are available online for this figure.
Materials and Methods
Plasmid construction
Full‐length cDNAs of UBQLN4 (Mus musculus and Homo sapiens), ubiquitin (Mus musculus) and those of UBQLN1 and UBQLN3 (Homo sapiens) were amplified by PCR from cDNA libraries derived from HeLa and NIH3T3 cells, respectively. The IL‐2Rα cDNA was amplified from mouse thymic cDNA library. The PCR fragments were cloned into pCI‐neo‐based mammalian expression vectors (Promega) with N‐terminal 3xFlag‐ or 3xT7‐tags with their products. Truncated and mutated versions of UBQLN4 and IL‐2Rα were prepared by PCR. Vectors were used for experiments after verification of the sequence of inserted DNA.
Mammalian cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Wako) supplemented with 10% heat‐inactivated calf serum at 37°C under 5% CO2 atmosphere. Transfections of the expression vectors were performed with HilyMax (Dojindo, Japan) or polyethylenimine “MAX” transfection reagent (Polysciences, Inc.) according to the protocols supplied by the manufacturers. At 18–24 h after transfection, the cells were harvested and subjected to immunological analysis unless otherwise noted.
RNA interference
For knockdown analysis of UBQLN4, three independent duplex siRNAs covering the targeted sequences
5′‐CAAACAGCAGGGUGACUUUtt‐3′ (UBQLN4 siRNA#1),
5′‐ CUCAAUAACCCUGAACUCAtt‐3′ (UBQLN4 siRNA#2),
5′‐CUCUUCAGAUGCUGGCAGUtt‐3′ (UBQLN4 siRNA#3).
were synthesized (SIGMA Genosys). UBQLN4 siRNA#1 was designed to target the 3′‐untranslated region of UBQLN4 where the sequence is quite diverged in all UBQLN family genes. The other two siRNA sequences were also designed to avoid matching higher sequence conserved regions within UBQLN family genes. We also synthesized three scrambled duplex siRNA sequences as negative controls to the above UBQLN4 knockdown sequences, respectively,
5′‐GAUGAAACAUAGCGUCCGUtt‐3′ (UBQLN4 siRNA#1scr),
5′‐AACACAAAUCCGCACUCUUtt‐3′ (UBQLN4 siRNA#2scr),
5′‐UUCCUAUAUGUACCGGGGCtt‐3′ (UBQLN4 siRNA#3scr).
In addition, MISSION siRNA Universal Negative Control 1 (Sigma‐Aldrich) was used as a general negative control in every experiment. The duplex siRNA sequences of UBQLN1 and UBQLN2 are 5′‐GGTGCTGGCGCCCCCGCGGCCG‐3′ and 5′‐CAUGUACACUGACAUUCAAtt‐3′, respectively. For SRP54 depletion, the duplex siRNA sequence 5′‐CACUUAUAGAGAAGUUGAAtt‐3′ was synthesized. BAG6 depletion was performed as described previously 32 with two independent duplex siRNA sequences of
5′‐UUUCUCCAAGAGCAGUUUAtt‐3′ (BAG6 siRNA#1).
5′‐CAGAAUGGGUCCCUAUUAUtt‐3′ (BAG6 siRNA#2).
Transfections of HeLa cells with duplex siRNA were performed using Lipofectamine 2000 (Invitrogen) according to the protocol provided by the manufacturer. The efficacy of each siRNA was verified by immunoblot with their specific antibodies listed in the next section.
Immunological analysis
The anti‐UBQLN4 antibody was prepared for this study as follows. Bacterially produced full‐length Mus musculus UBQLN4 was mixed and emulsified with TiterMax Gold (TiterMax USA, Inc.) and then inoculated into a rabbit with 2‐week intervals. After 2 weeks from the third inoculation, anti‐UBQLN4 IgG in the serum was affinity‐purified with Protein A‐Sepharose (GE Healthcare). We confirmed that the endogenous immunosignal of this antibody in HeLa cells was suppressed by three independent UBQLN4 siRNA duplex (UBQLN4 siRNA#1, #2, and #3).
For immunoprecipitation analysis, HeLa cells were washed with ice‐cold phosphate‐buffered saline (PBS) and lysed with immunoprecipitation (IP) buffer containing 20 mM Tris–HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Nonidet P‐40, 10 mM N‐ethylmaleimide, 25 μM MG‐132, and protease inhibitor cocktail (Nacalai tesque). In the case of Figs 3C and 5A, HeLa cells were lysed with chaperone buffer containing 10 mM Tris–HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Tween‐20, 10% glycerol, 10 mM N‐ethylmaleimide, 25 μM MG‐132, and protease inhibitor cocktail. The lysates were sonicated (except for Fig 8B), centrifuged at 20,630× g for 20–30 min at 4°C, and mixed with 4–10 μl of anti‐Flag M2 affinity gel (Sigma) or anti‐T7 tag antibody agarose (Novagen) for 10 min to 2 h at 4°C. After the beads had been washed five times with the IP buffer or chaperone buffer, the immunocomplexes were eluted by SDS sample buffer.
For hot lysis analysis, HeLa cells were washed by PBS and lysed with hot lysis buffer containing 1% SDS, 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 25 μM MG‐132, 10 mM N‐ethylmaleimide, and inhibitor cocktail. The lysates were heated in 90°C for 15 min and sonicated. After the lysates were centrifuged at 20,630× g for 20 min at room temperature, the supernatant was diluted for fourfold with buffer A (containing 1% Triton X‐100, 50 mM Tris–HCl pH 7.5, and 150 mM NaCl) and mixed with 5 μl anti‐Flag M2 affinity gel for 10 min at 4°C. After the beads had been washed five times with buffer A, the immunocomplexes were eluted by SDS sample buffer.
For Western blot analyses, whole‐cell lysates and the immunoprecipitates were subjected to SDS–PAGE and transferred onto polyvinylidene fluoride transfer membrane (GE Healthcare, Pall Corporation). The membranes were then immunoblotted with specific antibodies as indicated and then incubated with horseradish peroxidase‐conjugated antibody against mouse or rabbit immunoglobulin (GE Healthcare), followed by detection with ECL Western blotting detection reagents (GE Healthcare), Clarity™ Western ECL Substrate (Bio‐RAD), or Immobilon Western chemiluminescent HRP Substrate (Merck Millipore).
The following antibodies were used in this study: anti‐UBQLN4 (prepared in this study), anti‐BAG6 32, anti‐polyubiquitin FK2 (MBL or Nippon Bio‐Test Laboratories Inc.), anti‐SRP54 (BD Biosciences), anti‐UBQLN1 (Lifespan Biosciences, Inc.), anti‐UBQLN2 (Abnova), anti‐Rpt5 (Enzo Life Sciences), anti‐Flag M2 monoclonal (Sigma), anti‐Flag polyclonal (Sigma), anti‐T7‐tag monoclonal (Novagen), anti‐β‐actin (Sigma), anti‐calnexin (Sigma), anti‐tubulin (Santa Cruz Biotech.), and anti‐GST (Santa Cruz Biotech.).
Subcellular fractionation
The expression vectors of IL‐2Rα WT with N‐terminal Flag‐tags and its ΔSS derivative were transfected into HeLa cells. After 22 h of transfection, subcellular fractionation was performed using a EzSubcell Extract (ATTO) according to the protocol provided by the manufacturer.
Microscopic observations
For immunocytochemical observations, HeLa cells that grown on micro‐cover glass (Matsunami) were washed twice with PBS, fixed by incubating in 4% paraformaldehyde for 20 min at 4°C, and were permeabilized with 0.1% Triton X‐100 for 3 min in room temperature. Fixed cells were blocked with 3% heat‐inactivated calf serum in PBS and then reacted with a series of primary antibodies at room temperature for 1 h (Fig 2A and B). Alexa FluorR 488‐conjugated anti‐mouse IgG, Alexa Fluor 594‐conjugated anti‐mouse IgG, and Alexa Fluor 488‐conjugated anti‐rabbit IgG antibodies (Invitrogen) were used as secondary antibodies at dilutions of 1:500. To observe the nucleus, cells were stained with Hoechst 33342. Immunofluorescent images were obtained with BIOREVO BZ9000 fluorescence microscope (Keyence, Japan). BZ‐II Analyzer application (Keyence, Japan) was used to quantify the number of ALIS. For the quantification of ALIS formation, polyubiquitin‐positive dots with a luminance value > 150 were regarded as ALIS.
GST pull‐down and ubiquitin‐binding assay
For the expression of GST‐fused recombinant proteins, cDNAs encoding Mus musculus UBQLN4, as well as their truncated derivatives, were subcloned into pGEX6P1 expression vector (GE Healthcare). GST‐fused proteins were expressed in E. coli BL21 (DE3) by adding 0.1 mM IPTG. At 4 h from IPTG addition, E. coli were harvested and lysed with PBS‐T (0.1% Tween‐20 in PBS) and sonicated. The lysates were centrifuged at 20,630× g for 20 min at 4°C. The supernatant was mixed with 10 μl glutathione‐immobilized beads (GE Healthcare) for 10–30 min at 4°C. After the beads had been washed three times with PBS‐T, the beads were mixed with 3 μg K48‐linked polyubiquitin chains (R&D Systems, Inc.), incubated at 4°C for 30 min, and washed three times with PBS‐T. Polyubiquitin chain co‐precipitations were analyzed by Western blot analysis with anti‐ubiquitin (FK2) and anti‐GST antibodies. In the case of Fig EV4C, the beads with purified GST‐UBQLN4 or those with GST‐STI‐II were mixed with HeLa cells extracts that expressing Flag‐tagged IL‐2Rα ΔSS for 30 min at 4°C. After the beads had been washed three times with the IP buffer, the immunocomplexes were eluted by SDS sample buffer.
Cell viability assay
After 48 h of UBQLN4‐specific siRNA treatment, HeLa cells were treated with 5 μg/ml puromycin for the indicated time. Cell viability was determined using a Cell Counting kit 8 (Dojindo) according to the protocol provided by the manufacturer.
Statistical analysis
Evaluation of data was performed either by Welch's t‐test or Student's t‐test as indicated. All data in the figures are presented as the mean ± SEM or SD. P‐value of < 0.05 is considered statistically significant.
Author contributions
RS and HK conceived and designed the experiments. RS performed all the experiments. RS and HK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Expanded View
Acknowledgements
We thank Dr. R. Minami, Ms A. Hayakawa, and Ms Y. Yanagi for their contribution in the initial stage of this work. We also thank Prof. Jack Taunton (UCSF), Prof. K. Nagata (Kyoto Sangyo Univ.), Prof. K. Tanaka (Tokyo Metropol. Inst. Med. Sci.), and Dr. N. Yokota (Tokyo Metropol. Univ.) for constructive suggestions. We thank Ms A. Tojima, Mr K. Yamamoto, Ms Y. Fukuda, and Mr K. Nakajima for technical assistance. This work was supported in part by grants from the Ministry of Education, Culture, Science, and Technology of Japan (Ubiquitin neo‐biology, No. 24112007), the Uehara Memorial Foundation, and Naito Foundation to HK. RS is a recipient of JSPS Research Fellowship for Young Scientists and supported by grant‐in‐aid (No. 265957).
EMBO Reports (2016) 17: 842–857
References
- 1. Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305: 567–580 [DOI] [PubMed] [Google Scholar]
- 2. Istrail S, Florea L, Halldórsson BV, Kohlbacher O, Schwartz RS, Yap VB, Yewdell JW, Hoffman SL (2004) Comparative immunopeptidomics of humans and their pathogens. Proc Natl Acad Sci USA 101: 13268–13272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kahsay RY, Gao G, Liao L (2005) An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 21: 1853–1858 [DOI] [PubMed] [Google Scholar]
- 4. Cross BC, Sinning I, Luirink J, High S (2009) Delivering proteins for export from the cytosol. Nat Rev Mol Cell Biol 10: 255–264 [DOI] [PubMed] [Google Scholar]
- 5. Shao S, Hegde RS (2011) Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol 27: 25–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walter P, Johnson AE (1994) Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu Rev Cell Biol 10: 87–119 [DOI] [PubMed] [Google Scholar]
- 7. Ng DT, Brown JD, Walter P (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol 134: 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martoglio B, Dobberstein B (1998) Signal sequences: more than just greasy peptides. Trends Cell Biol 8: 410–415 [DOI] [PubMed] [Google Scholar]
- 9. Rapoport TA (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450: 663–669 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X, Shan SO (2014) Fidelity of cotranslational protein targeting by the signal recognition particle. Annu Rev Biophys 43: 381–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walter P, Blobel G (1980) Purification of a membrane‐associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci USA 77: 7112–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walter P, Blobel G (1981) Translocation of proteins across the endoplasmic reticulum II. Signal recognition protein (SRP) mediates the selective binding to microsomal membrane of in vitro assembled polysomes synthesizing secretory protein. J Cell Biol 91: 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keenan RJ, Freymann DM, Stroud RM, Walter P (2001) The signal recognition particle. Annu Rev Biochem 70: 755–775 [DOI] [PubMed] [Google Scholar]
- 14. Halic M, Beckmann R (2005) The signal recognition particle and its interactions during protein targeting. Curr Opin Struct Biol 15: 116–125 [DOI] [PubMed] [Google Scholar]
- 15. Akopian D, Shen K, Zhang X, Shan SO (2013) Signal recognition particle: an essential protein‐targeting machine. Annu Rev Biochem 82: 693–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson AE, van Waes MA (1999) The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol 15: 799–842 [DOI] [PubMed] [Google Scholar]
- 17. Zhang X, Rashid R, Wang K, Shan SO (2010) Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science 328: 757–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D, Shan S (2012) Translation elongation regulates substrate selection by the signal recognition particle. J Biol Chem 287: 7652–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siegel V, Walter P (1988) The affinity of signal recognition particle for presecretory proteins is dependent on nascent chain length. EMBO J 7: 1769–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim SJ, Mitra D, Salerno JR, Hegde RS (2002) Signal sequences control gating of the protein translocation channel in a substrate‐specific manner. Dev Cell 2: 207–217 [DOI] [PubMed] [Google Scholar]
- 21. Levine CG, Mitra D, Sharma A, Smith CL, Hegde RS (2005) The efficiency of protein compartmentalization into the secretory pathway. Mol Biol Cell 16: 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogg SC, Walter P (1995) SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell 81: 1075–1084 [DOI] [PubMed] [Google Scholar]
- 23. Schlosser E, Otero C, Wuensch C, Kessler B, Edelmann M, Brunisholz R, Drexler I, Legler DF, Groettrup M (2007) A novel cytosolic class I antigen‐processing pathway for endoplasmic‐reticulum‐targeted proteins. EMBO Rep 8: 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM (2006) Sequential quality‐control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126: 571–582 [DOI] [PubMed] [Google Scholar]
- 25. Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, Hegde RS (2011) Protein targeting and degradation pathway are coupled for elimination of misfolded proteins. Nature 475: 394–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10: S10–S17 [DOI] [PubMed] [Google Scholar]
- 27. Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS (2006) Substrate‐specific translocational attenuation during ER stress defines a pre‐emptive quality control pathway. Cell 127: 999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmermann R, Müller L, Wullich B (2006) Protein transport into the endoplasmic reticulum: mechanisms and pathologies. Trends Mol Med 12: 567–573 [DOI] [PubMed] [Google Scholar]
- 29. Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS (2008) Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell 15: 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS (2010) Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol 188: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braunstein I, Zach L, Allan S, Kalies KU, Stanhill A (2015) Proteasomal degradation of preemptive quality control (pQC) substrates is mediated by an AIRAPL‐p97 complex. Mol Biol Cell 26: 3719–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minami R, Hayakawa A, Kagawa H, Yanagi Y, Yokosawa H, Kawahara H (2010) BAG6 is essential for selective elimination of defective proteasomal substrates. J Cell Biol 190: 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, Ye Y (2011) A ubiquitin ligase‐associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol Cell 42: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Claessen JHL, Ploegh HL (2011) BAT3 guides misfolded glycoproteins out of the endoplasmic reticulum. PLoS ONE 6: e28542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leznicki P, High S (2012) SGTA antagonizes BAG6‐mediated protein triage. Proc Natl Acad Sci USA 109: 19214–19219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kadowaki H, Nagai A, Maruyama T, Takami Y, Satrimafitrah P, Kato H, Honda A, Hatta T, Natsume T, Sato T et al (2015) Pre‐emptive quality control protects the ER from protein overload via the proximity of ERAD components and SRP. Cell Rep 13: 944–956 [DOI] [PubMed] [Google Scholar]
- 37. Leznicki P, Clancy A, Schwappach B, High S (2010) Bat3 promotes the membrane integration of tail‐anchored proteins. J Cell Sci 123: 2170–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, Keenan RJ, Hegde RS (2010) A ribosome‐associating factor chaperones tail‐anchored membrane proteins. Nature 466: 1120–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hegde R, Keenan RJ (2011) Tail‐anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol 12: 787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson N, Powis K, High S (2013) Post‐translational translocation into the endoplasmic reticulum. Biochem Biophys Acta 1833: 2403–2409 [DOI] [PubMed] [Google Scholar]
- 41. Kuwabara N, Minami R, Yokota N, Matsumoto H, Senda T, Kawahara H, Kato R (2015) Structure of a BAG6 (bcl‐2‐associated athanogene 6)–Ubl4a (ubiquitin‐like protein 4a) complex reveals a novel binding interface that functions in tail‐anchored protein biogenesis. J Biol Chem 290: 9387–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mock J, Chartron JW, Zaslaver M, Xu Y, Ye Y, Clemons WM Jr (2015) Bag6 complex contains a minimal tail‐anchor‐targeting module and a mock BAG domain. Proc Natl Acad Sci USA 112: 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawahara H, Minami R, Yokota N (2013) BAG6/BAT3; emerging roles in quality control for nascent polypeptides. J Biochem 153: 147–160 [DOI] [PubMed] [Google Scholar]
- 44. Lee JG, Ye Y (2013) Bag6/Bat3/Scythe: a novel chaperone activity with diverse regulatory functions in protein biogenesis and degradation. BioEssays 35: 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Payapilly A, High S (2014) BAG6 regulates the quality control of a polytopic ERAD substrate. J Cell Sci 127: 2898–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Binici J, Koch J (2014) BAG‐6, a jack of all trades in health and disease. Cell Mol Life Sci 71: 1829–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wunderley L, Leznicki P, Payapilly A, High S (2014) SGTA regulates the cytosolic quality control of hydrophobic substrates. J Cell Sci 127: 4728–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y, Liu Y, Lee JG, Ye Y (2013) A ubiquitin‐like domain recruits an oligomeric chaperone to a retrotranslocation complex in endoplasmic reticulum‐associated degradation. J Biol Chem 288: 18068–18076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaye FJ, Modi S, Ivanovska I, Koonin EV, Thress K, Kubo A, Kornbluth S, Rose MD (2000) A family of ubiquitin‐like proteins binds the ATPase domain of Hsp70‐like Stch. FEBS Lett 467: 348–355 [DOI] [PubMed] [Google Scholar]
- 50. Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 6: 409–419 [DOI] [PubMed] [Google Scholar]
- 51. Matsuda M, Koide T, Yorihuzi T, Hosokawa N, Nagata K (2001) Molecular cloning of a novel ubiquitin‐like protein, UBIN, that binds to ER targeting signal sequences. Biochem Biophys Res Commun 280: 535–540 [DOI] [PubMed] [Google Scholar]
- 52. Ko HS, Uehara T, Tsuruma K, Nomura Y (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin‐associated and ubiquitn‐like domains. FEBS Lett 566: 110–114 [DOI] [PubMed] [Google Scholar]
- 53. Hartmann‐Petersen R, Gordon C (2004) Integral UBL domain proteins: a family of proteasome interacting proteins. Semin Cell Dev Biol 15: 247–259 [DOI] [PubMed] [Google Scholar]
- 54. Saeki Y, Saitoh A, Toh‐e A, Yokosawa H (2002) Ubiquitin‐like proteins and Rpn10 play cooperative roles in ubiquitin‐dependent proteolysis. Biochem Biophys Res Commun 293: 986–992 [DOI] [PubMed] [Google Scholar]
- 55. Elsasser S, Chandler‐Militell D, Muller B, Hanna J, Finley D (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J Biol Chem 279: 26817–26822 [DOI] [PubMed] [Google Scholar]
- 56. Grabbe C, Dikic I (2009) Functional roles of ubiquitin‐like domain (ULD) and ubiquitin‐binding domain (UBD) containing proteins. Chem Rev 109: 1481–1494 [DOI] [PubMed] [Google Scholar]
- 57. Rothenberg C, Monteiro MJ (2010) Ubiquilin at a crossroads in protein degradation pathways. Autophagy 6: 979–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilkinson CR, Seeger M, Hartmann‐Petersen R, Stone M, Wallace M, Semple C, Gordon C (2001) Proteins containing the UBA domain are able to bind to multi‐ubiquitin chains. Nat Cell Biol 3: 939–943 [DOI] [PubMed] [Google Scholar]
- 59. Hartmann‐Petersen R, Gordon C (2004) Protein degradation: recognition of ubiquitinylated substrates. Curr Biol 14: R754–R756 [DOI] [PubMed] [Google Scholar]
- 60. Raasi S, Varadan R, Fushman D, Pickart CM (2005) Diverse polyubiquitin interaction properties of ubiquitin‐associated domains. Nat Struct Mol Biol 12: 708–714 [DOI] [PubMed] [Google Scholar]
- 61. Kang Y, Vossler RA, Diaz‐Martinez LA, Winter NS, Clarke DJ, Walters KJ (2006) UBL/UBA ubiquitin receptor proteins bind a common tetra ubiquitin chain. J Mol Biol 356: 1027–1035 [DOI] [PubMed] [Google Scholar]
- 62. Zhang D, Raasi S, Fushman D (2008) Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin‐1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol 377: 162–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Finley D (2009) Recognition and processing of ubiquitn‐protein conjugates by the proteasome. Annu Rev Biochem 78: 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Husnjak K, Dikic I (2012) Ubiquitin‐binding proteins: decoders of ubiquitin‐mediated cellular functions. Annu Rev Biochem 81: 291–322 [DOI] [PubMed] [Google Scholar]
- 65. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404: 770–774 [DOI] [PubMed] [Google Scholar]
- 66. Qian S‐B, Princiotta MF, Bennink JR, Yewdell JW (2006) Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J Biol Chem 281: 392–400 [DOI] [PubMed] [Google Scholar]
- 67. Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, Gatti E, Pierre P (2004) Dendritic cell aggresome‐like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 164: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rodrigo‐Brenni MC, Gutierrez E, Hegde RS (2014) Cytosolic quality control of mislocalized proteins requires RNF126 recruitment to Bag6. Mol Cell 55: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang L, Kuhls MC, Eisenlohr LC (2011) Hydrophobicity as a driver of MHC class I antigen processing. EMBO J 30: 1634–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mah AL, Perry G, Smith MA, Monteiro MJ (2000) Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol 151: 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kleijnen MF, Alarcon RM, Howley PM (2003) The ubiquitin‐associated domain of hPLIC‐2 interacts with the proteasome. Mol Biol Cell 14: 3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li X, Su V, Kurata WE, Jin C, Lau AF (2008) A novel connexin43‐interacting protein, CIP75, which belongs to the UbL‐UBA protein family, regulates the turnover of connexin43. J Biol Chem 283: 5748–5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lässle M, Blatch GL, Kundra V, Takatori T, Zetter BR (1997) Stress‐inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J Biol Chem 272: 1876–1884 [DOI] [PubMed] [Google Scholar]
- 74. Lee DY, Arnott D, Brown EJ (2013) Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep 14: 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reits EA, Vos JC, Gromme M, Neefjes J (2000) The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature 404: 774–778 [DOI] [PubMed] [Google Scholar]
- 76. Khan S, Giuli R, Schmidtke G, Bruns M, Buchmeier M, van den Broek M, Groettrup M (2001) Cutting edge: neosynthesis is required for the presentation of a T cell epitope from a long‐lived viral protein. J Immunol 167: 4801–4804 [DOI] [PubMed] [Google Scholar]
- 77. Goldberg AL (2007) Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 35: 12–17 [DOI] [PubMed] [Google Scholar]
- 78. Yewdell JW (2011) DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol 32: 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899 [DOI] [PubMed] [Google Scholar]
- 80. Goldberg AL (2012) Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol 199: 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Meusser B, Hirsch C, Jarosch E, Sommer T (2005) ERAD: the long road to destruction. Nat Cell Biol 7: 766–772 [DOI] [PubMed] [Google Scholar]
- 82. Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T (2009) The ubiquitylation machinery of the endoplasmic reticulum. Nature 458: 453–460 [DOI] [PubMed] [Google Scholar]
- 83. Buchberger A, Bukau B, Sommer T (2011) Protein quality control in the cytosol and the endoplasmic reticulum: brother in arms. Mol Cell 40: 238–252 [DOI] [PubMed] [Google Scholar]
- 84. Claessen JH, Kundrat L, Ploegh HL (2012) Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol 22: 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum‐associated degradation in human physiology. Physiol Rev 92: 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Townsend AR, Bastin J, Gould K, Brownlee GG (1986) Cytotoxic T lymphocytes recognize influenza haemagglutinin that lacks a signal sequence. Nature 324: 575–577 [DOI] [PubMed] [Google Scholar]
- 87. Metzger MB, Maurer MJ, Dancy BM, Michaelis S (2008) Degradation of a cytosolic protein requires endoplasmic reticulum‐associated degradation machinery. J Biol Chem 283: 32302–32316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Swanson R, Locher M, Hochstrasser M (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER‐associated and Matα2 repressor degradation. Genes Dev 15: 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Expanded View