

Abstract
The oxysterol‐binding protein (OSBP)‐related proteins ORP5 and ORP8 have been shown recently to transport phosphatidylserine (PS) from the endoplasmic reticulum (ER) to the plasma membrane (PM) at ER–PM contact sites. PS is also transferred from the ER to mitochondria where it acts as precursor for mitochondrial PE synthesis. Here, we show that, in addition to ER–PM contact sites, ORP5 and ORP8 are also localized to ER–mitochondria contacts and interact with the outer mitochondrial membrane protein PTPIP51. A functional lipid transfer (ORD) domain was required for this localization. Interestingly, ORP5 and ORP8 depletion leads to defects in mitochondria morphology and respiratory function.
Keywords: endoplasmic reticulum, lipid transfer proteins, membrane contact sites, mitochondria, ORP
Subject Categories: Membrane & Intracellular Transport
Introduction
Mitochondria play a major role in several cellular processes including energy and lipid metabolism, calcium (Ca2+) homeostasis and apoptosis. Like other organelles, mitochondria can be closely associated with the endoplasmic reticulum (ER). ER membrane subdomains closely apposed to the mitochondria, within less than 30 nm, are called mitochondria‐associated ER membranes (MAM) and facilitate exchange between the two organelles including Ca2+ and lipids. Increasing lines of evidence suggest that lipid transfer proteins (LTPs) play a major role in regulating lipid composition of membranous organelles and could facilitate non‐vesicular lipid transport at membrane contact sites (MCS) between the ER and other intracellular organelles 1, 2, 3.
The oxysterol‐binding protein (OSBP)‐related proteins (ORPs) constitute a large family of lipid‐binding/transfer proteins conserved from yeast (Osh) to humans (ORP) and localized to different subcellular sites, shown in several cases to be MCS 4, 5, 6. ORP proteins usually contain dual targeting determinants for the ER and the partner membrane, such as an FFAT motif that binds ER‐localized VAP proteins and a pleckstrin homology (PH) domain that allow their interaction with distinct non‐ER organelle membranes 4.
ORP proteins also contain an OSBP‐related lipid‐binding (ORD) domain that can bind sterols 7 and have been thought for long time to act as sterol sensor or transport proteins 4. There is increasing evidence that they can also harbor other lipids such as PI4P and mediate exchange of sterols with PI4P between the ER and sterol‐enriched membranes 5, 6, 8, 9, 10. ORP5 and ORP8 are two similar ORPs with the unique feature of being anchored to the ER membranes via a C‐terminal hydrophobic tail sequence 10 (Fig 1A). ORP5 has been originally reported to transfer cholesterol in vitro with specific and direct competition by PI4P 10. Recently, ORP5 and ORP8 have also been shown to specifically bind PS and to transfer PS from the cortical ER (ER juxtaposed to the PM) to the PM in a counter‐exchange mechanism with PM PI4P 11, 12. Interestingly, transport of PS occurs also at ER–mitochondria MCS 13, 14. At these sites, the newly synthesized PS is shuttled from the ER to the mitochondria membranes where it is rapidly converted to phosphatidylethanolamine (PE) and it represents the major source for the mitochondrial PE 14. PE, as well as cardiolipin, plays crucial roles in maintaining mitochondrial tubular morphology and therefore in mitochondrial respiratory functions 15, 16, 17.
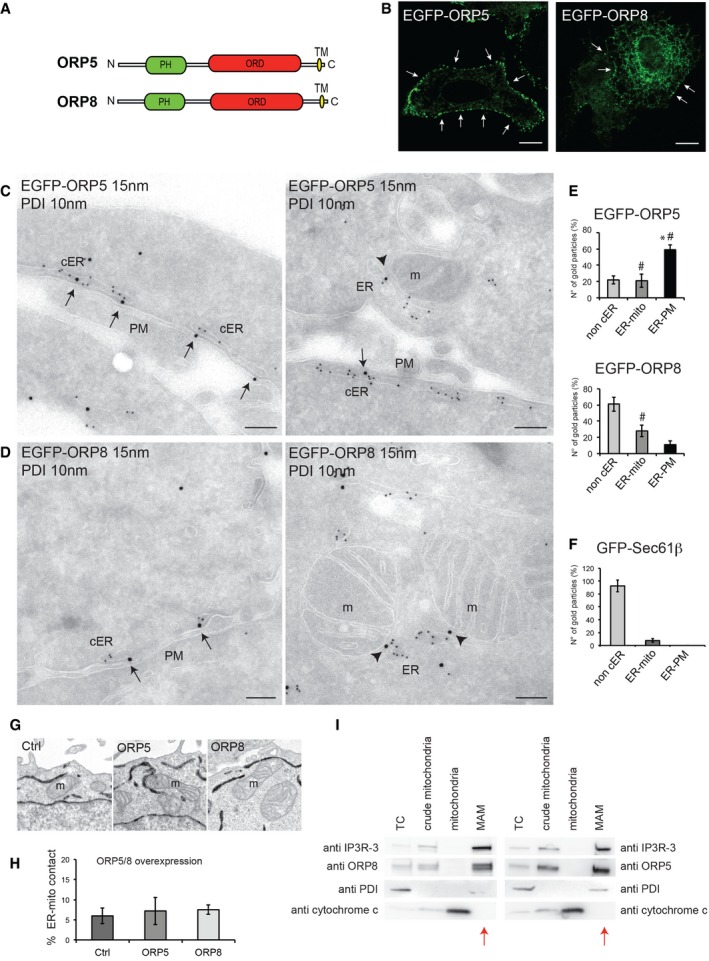
Figure 1. ORP5 and ORP8 localize at ER–PM and ER–mitochondria contact sites.
-
ADomain structures of human ORP5 and ORP8. PH, Pleckstrin homology domain; ORD, OSBP‐related lipid‐binding domain; TM, transmembrane domain.
-
BConfocal images of HeLa cells transfected with EGFP‐ORP5 or EGFP‐ORP8 showing variable expression levels. Arrows indicate ORP5 and ORP8 localization to cortical ER (cER). Scale bar, 10 μm.
-
C, DElectron micrographs of ultrathin cryosections of HeLa cells transfected with EGFP‐ORP5 or EGFP‐ORP8 and immunogold stained with anti‐GFP (15 nm gold) to detect ORP5 or ORP8 and anti‐PDI (10 nm gold) to label the ER lumen. ORP5 and ORP8 localize at ER–PM (arrows) and ER–mitochondria contact sites (arrowheads). m, mitochondria; cER, cortical ER; PM, plasma membrane. Scale bar, 200 nm.
-
E, FQuantification of the IEM labeling for EGFP‐ORP5, EGFP‐ORP8 (E), and GFP‐Sec61β (F) in transfected HeLa cells. Results are presented as the percentage of the total number of gold particles (800 per condition, n = 35–45 cells) for ORP5, ORP8, and Sec61β in the indicated compartments (non‐cER, non‐cortical ER; ER‐PM, ER–PM contact sites; ER‐mito, ER–mitochondria contact sites). % gold particles ± SEM. Data are the mean of three independent replicates. *P < 0.001 compared to non‐cER and ER‐mito (EGFP‐ORP5) and to ER‐PM and ER‐mito (EGFP‐ORP8 and GFP‐Sec61β), # P < 0.001 compared to ER‐PM and ER‐mito (GFP‐Sec61β).
-
G, HRepresentative electron micrographs of mitochondria (G) and quantification of the percentage of ER–mitochondria contact sites (ER‐mito contact) per mitochondria (H) of HeLa cells expressing EGFP‐ORP5, EGFP‐ORP8 together with HRP‐KDEL, to visualize the ER. Control (Ctrl) consisted of HeLa cells overexpressing only HRP‐KDEL. % ER–mitochondria contact sites ± SEM. Data are representative of three independent replicates.
-
ICrude mitochondria, mitochondria, and MAM fractions were purified from HeLa cells, and equal amounts of protein (25 μg) from each fraction were loaded on a 4–20% gradient SDS–PAGE gel and immunoblotted using anti‐ORP5, anti‐ORP8 and anti‐IP3R‐3 (MAM protein), anti‐PDI (ER protein), and anti‐cytochrome c (mitochondrial protein). Red arrows indicate the MAM fraction lane. TC, total cell; MAM, mitochondria‐associated ER membrane
Source data are available online for this figure.
Here, we show that ORP5 and ORP8 localize to ER–mitochondria MCS, in addition to ER–PM contacts, where they interact with the outer mitochondrial membrane protein PTPIP51. This localization is dependent on a functional lipid transfer (ORD) domain. Our data also show that ORP5 and ORP8 depletion leads to altered mitochondria morphology and function.
Results and Discussion
ORP5 and ORP8 localize to ER–PM and ER–mitochondria contact sites
As ORP5 and ORP8 are ER‐anchored proteins, we first analyzed the distribution of EGFP‐tagged ORP5 and ORP8 within the ER by confocal microscopy. EGFP‐ORP5 showed a predominant localization to cortical ER structures (punctae), and a minor localization to non‐cortical ER membranes in all cells analyzed (Fig 1B). The majority of EGFP‐ORP8 localized to the non‐cortical ER, with a minor pool of the protein detected in cortical ER punctae (Fig 1B). Using TIRF microscopy, both ORP5‐ and ORP8‐positive cortical ER structures were visible in the evanescent field (Fig EV4A), confirming their localization within a ~100 nm zone extending from the PM. Consistent with confocal imaging, the abundance of ORP5 cortical ER structures was higher as compared to ORP8. These results were consistent with the data shown in a report published very recently 12 and suggest that ORP5 and, to a lesser extent, ORP8, are localized at sites of close apposition between the ER and the PM.
Figure EV4. Recruitment of ORP5 and ORP8 to ER–mitochondria contact sites.
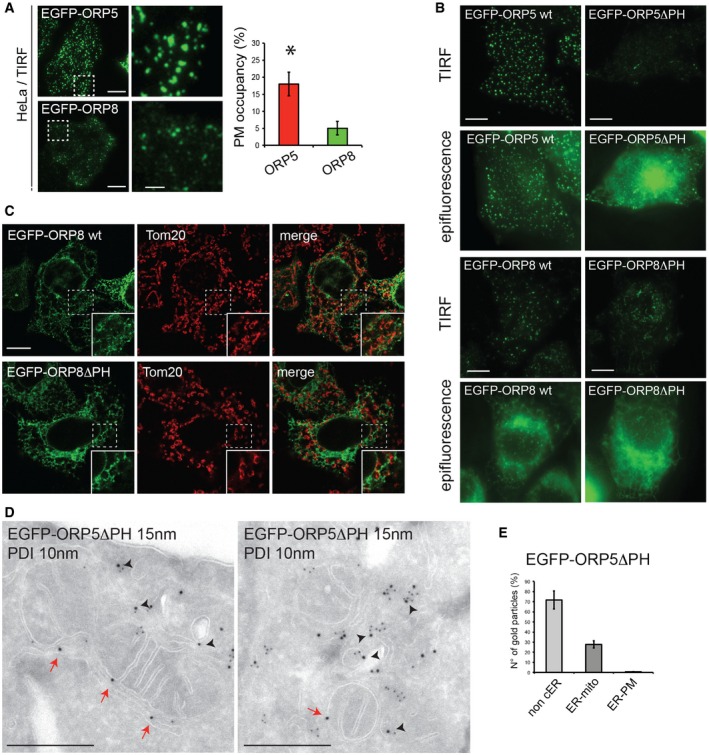
- TIRF microscopy of HeLa cells overexpressing EGFP‐ORP5 or EGFP‐ORP8 showing the localization of these proteins within the evanescent field (100 nm from the PM). Scale bar, 10 μm. Quantifications of the TIRF microscopy showing the percentage of PM occupied by cER (% ± SEM, n = 8 for each of 3 biological replicates, *P < 0.001).
- Representative TIRF microscopy images (upper panel) and corresponding epifluorescence images (lower panel) of HeLa cells expressing EGFP‐ORP5, EGFP‐ORP5ΔPH, EGFP‐ORP8, or EGFP‐ORP8ΔPH. Scale bar, 10 μm.
- Confocal images of HeLa cells transfected with EGFP‐ORP8 or EGFP‐ORP8ΔPH and immunostained using anti‐TOM20 antibody to visualize mitochondria. Scale bar, 10 μm.
- Electron micrographs of ultrathin cryosections of HeLa cells transfected with EGFP‐ORP5ΔPH and immunogold stained with anti‐GFP (15 nm gold) to detect ORP5ΔPH and anti‐PDI (10 nm gold) to detect the ER. Red arrows indicate EGFP‐ORP5ΔPH at ER–mitochondria contact sites. Black arrowheads indicate EGFP‐ORP5ΔPH in the reticular ER. Scale bar, 200 nm.
- Quantification of the IEM labeling for EGFP‐ORP5ΔPH in transfected HeLa cells. Results are presented as the percentage of the total number of gold particles (800 per conditions) for ORP5ΔPH in the indicated compartments (non‐cER, non‐cortical ER; ER‐mito, ER–mitochondria contact sites; ER‐PM, ER–PM contact sites). % gold particles ± SEM, n = 32 cells, *P < 0.001 compared to non‐cER and ER‐mito and to ER‐PM.
To test this hypothesis, immunogold labeling was performed on ultrathin frozen sections of EGFP‐ORP5‐ and EGFP‐ORP8‐expressing HeLa cells. The bulk of EGFP‐ORP5 (59%) was localized to areas of apposition between the PM and the ER, the latter identified by co‐immunolabeling of endogenous protein disulfide isomerase (PDI) (Figs 1C (arrows) and E, and EV1A); 20% of ORP5 was found on internal ER membranes (Figs 1E and EV1A (arrowhead)). In contrast, the majority of EGFP‐ORP8 (60%) was detected on internal ER membranes distributed throughout the cell (Fig EV1B (arrowheads)), with only 8% found at ER in close proximity to the PM (Figs 1D (black arrows) and E, and EV1B). Surprisingly, we found both EGFP‐ORP5 (21%) (Figs 1C (arrowheads) and E, and EV1A (red arrow)) and EGFP‐ORP8 (28%) (Figs 1D (arrowheads) and E, and EV1B (red arrows)) localized to sites of apposition between ER and mitochondria. To exclude that their localization to these sites is not a consequence of their distribution throughout non‐cortical ER membranes, we sought to determine if other ER proteins have a similar frequency of localization to regions where ER and mitochondria are juxtaposed. EGFP‐Sec61β is a subunit of the Sec61 complex involved in protein translocation in the ER. EGFP‐Sec61β was present in internal ER elements distributed throughout the cell (92%) (Fig 1F). In line with this, very little EGFP‐Sec61β immunogold labeling was found at ER–mitochondria contact sites (7%), as compared to ORP5 and ORP8 (Fig 1F). Hence, our results support the conclusion that ORP5 and ORP8 are localized to both ER–PM and ER–mitochondria contact sites.
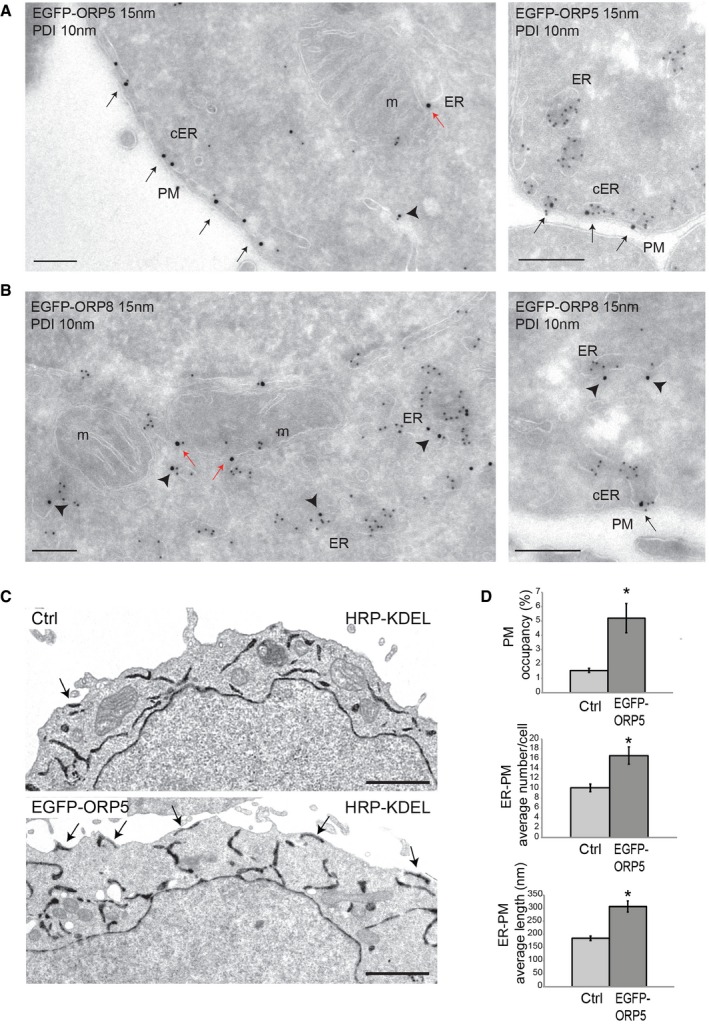
Figure EV1. ORP5 and ORP8 localization at ER–PM and ER–mitochondria contact sites.
-
A, BIEM micrographs of HeLa cells transfected with EGFP‐ORP5 (A) or EGFP‐ORP8 (B) and immunogold labeled with anti‐GFP (15 nm gold) and anti‐PDI (10 nm gold), showing ORP5 and ORP8 localization at both ER–PM (black arrows) and ER–mitochondria contact sites (red arrows). Note that the bulk of ORP5 localizes to ER–PM contacts while the bulk of ORP8 localizes to non‐cER membranes (arrowheads). Scale bar, 200 nm.
-
CRepresentative electron micrographs of HeLa cells transfected with HRP‐KDEL (Ctrl) or co‐transfected with EGFP‐ORP5. Arrows indicate ER–PM contact sites. Scale bar, 2 μm.
-
DQuantification of the percentage of ER–PM contact sites per cell (upper panel), the average number (middle panel) or length (lower panel) of ER–PM contact sites per cell of HeLa cells transfected with HRP‐KDEL (Ctrl) or co‐transfected with EGFP‐ORP5 (n = 20 cells). % ER–PM contact sites ± SEM. *P < 0.001 compared to Ctrl.
To test whether ORP5 and ORP8 targeting to mitochondria is specific or simply due to rearrangements of the ER and/or to an increase of ER–mitochondria contacts induced by their overexpression, we carried out a morphological analysis of the ER by EM in HeLa cells co‐transfected with EGFP‐ORP5 or EGFP‐ORP8 and horseradish peroxidase (HRP) tagged with an ER retention motif (HRP‐KDEL) (Figs 1G and H, and EV1C and D). In accord with a previous study 18, control cells displayed some contacts between the ER and the PM in individual cell profile sections, with approximately 2% of the PM perimeter within 30 nm of ER elements (Fig EV1C and D). Cells overexpressing EGFP‐ORP5 showed a twofold to threefold increase of ER underlying the PM in a range of less than 30 nm, due to an increase in the number but also in the length of ER–PM contacts (Fig EV1C and D), as recently reported 12. Nevertheless, in cells overexpressing either ORP5 or ORP8 we did not observe rearrangements of the overall ER ultrastructure and the abundance of ER–mitochondria contact sites did not change (Figs 1G and H, and EV1C). These results indicate that ORP5 and ORP8 targeting to the mitochondria is specific and not due to an increase in ER–mitochondria appositions induced by their overexpression.
To confirm the localization of ORP5 and ORP8 at ER–mitochondria contact sites at endogenous level, we analyzed the presence of these proteins in the mitochondria‐associated ER membranes (MAMs) (Fig 1I). Percoll gradient‐based purification of mitochondria and MAMs revealed that both ORP5 and ORP8 were present and enriched in the MAM fraction, as compared to the total cell lysate, and were excluded from the mitochondria fraction. As controls for the purity of subcellular fractions, the samples were also probed for cytochrome c as mitochondrial marker, IP3R‐3 as a MAM‐enriched marker and PDI as a luminal ER marker that should not be enriched in the MAM. All markers were highly enriched in their respective fractions while PDI was not, showing that in addition to their localization at ER–PM contact sites, a significant proportion of ORP5 and ORP8 specifically localize at ER–mitochondria contact sites at endogenous levels.
ORP5 and ORP8 targeting to ER–mitochondria contact sites and interaction with the mitochondrial protein PTPIP51
Since both ORP5 and ORP8 endogenously and specifically localize at ER–mitochondria contact sites, we first addressed whether these proteins interact with each other. Mass spectrometry (MS) analysis of endogenous ORP8 immunoprecipitated from HeLa cell lysates revealed ORP5 as a major interaction partner (Fig EV2A, left table). Endogenous ORP5 or ORP8 were also identified as a major interaction partners upon IP‐MS analysis of HeLa cells overexpressing EGFP‐ORP8 or EGFP‐ORP5, respectively (Fig EV2A, right and lower table). To validate the MS data, co‐immunoprecipitations from HeLa cells followed by immunoblot analysis were carried out. Endogenous ORP5 was found to co‐immunoprecipitate with endogenous ORP8 from untransfected HeLa cells (Fig EV2B (arrow)), and 3XFLAG‐ORP8 was specifically immunoprecipitated with EGFP‐ORP5 when both were co‐overexpressed (Fig EV2C). These results indicate that endogenous ORP5 and ORP8 are physically associated in cells.
Figure EV2. ORP5 and ORP8 interaction.
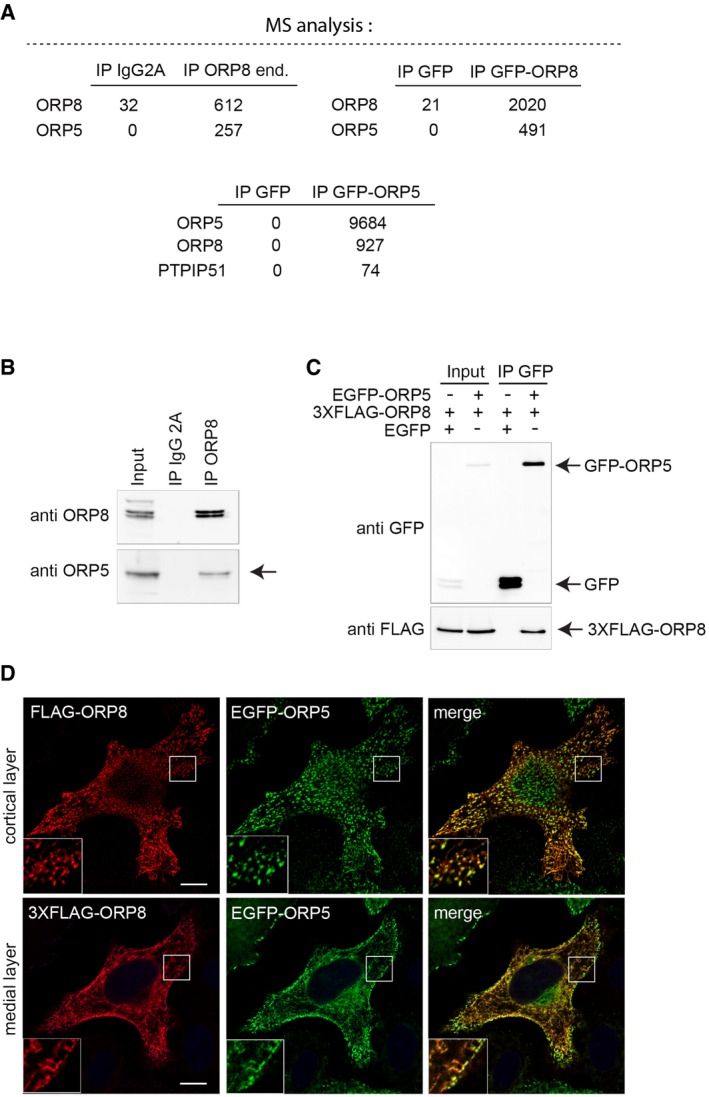
- Mass spectrometry (MS) analysis on immunoprecipitated endogenous ORP8 or overexpressed EGFP‐ORP8 or EGFP‐ORP5 indicates ORP5 or ORP8 as the major interacting partners. Note the presence of PTPIP51 in the MS of EGFP‐ORP5 immunoprecipitates.
- Immunoprecipitation of endogenous ORP8 reveals interaction with endogenous ORP5 (arrow).
- EGFP‐ORP5 or EGFP alone were immunoprecipitated from lysates of HeLa cells co‐expressing EGFP or EGFP‐ORP5 with 3XFLAG‐ORP8 and analyzed by Western blot using antibodies against GFP (for ORP5) or FLAG (for ORP8).
- Fluorescent images of HeLa cells co‐transfected with EGFP‐ORP5 and 3XFLAG‐ORP8 and stained with anti‐FLAG. Cortical layer (upper panel) and medial layer (lower panel) of the same cell are shown. Scale bar, 10 μm.
Since endogenous proteins are not detectable by microscopy, to assess their localizations when both proteins are expressed at similar levels, we co‐expressed 3XFLAG‐ORP8 and EGFP‐ORP5 (Fig EV2D). Indeed, when expressed together, EGFP‐ORP5 and 3XFLAG‐ORP8 displayed a similar localization pattern with an increase in the fraction of ORP8 located in ORP5‐positive cortical ER (Fig EV2D, upper panel), as compared to ORP8 expressed alone, and an increase of ORP5 in the non‐cortical ER as compared to ORP5 alone (Fig EV2D, lower panel) in accordance with previous studies 10, 12. These data show that ORP5 and ORP8 interact at both ER–PM and ER–mitochondria contact sites.
To further characterize the ER–mitochondria contact site localization of ORP5 and ORP8 described above, we sought to investigate whether ORP5 and ORP8 targeting to ER–mitochondria contact sites could be mediated by the interaction with specific mitochondrial proteins. Interestingly, among the major specific hits detected by our MS analysis of immunoprecipitated EGFP‐ORP5 (Fig EV2A, lower table) was the outer mitochondrial membrane protein PTPIP51, recently shown to promote ER–mitochondria junctions through interaction with the ER‐localized VAPB 19. Using confocal microscopy, we found that co‐expression of PTPIP51‐HA and EGFP‐tagged ORP5/ORP8 causes a robust increase of ORP5‐ and ORP8‐containing ER regions in proximity to mitochondria (Fig 2A). Such increase was also accompanied by a dramatic decrease in both ORP5 and ORP8 cortical ER localization. To exclude that this effect was due only to the overexpression of PTPIP51 and consequent aspecific recruitment of the ER, we analyzed the localization of EGFP‐Sec61β (Fig 2A) in co‐expression with PTPIP51‐HA. When co‐expressed with PTPIP51, EGFP‐Sec61β was only slightly redistributed in proximity to mitochondria and still remained predominantly localized to the reticular ER, in contrast to ORP5/ORP8.
Figure 2. ORP5 and ORP8 targeting to ER–mitochondria contact sites and interaction with the mitochondrial protein PTPIP51.
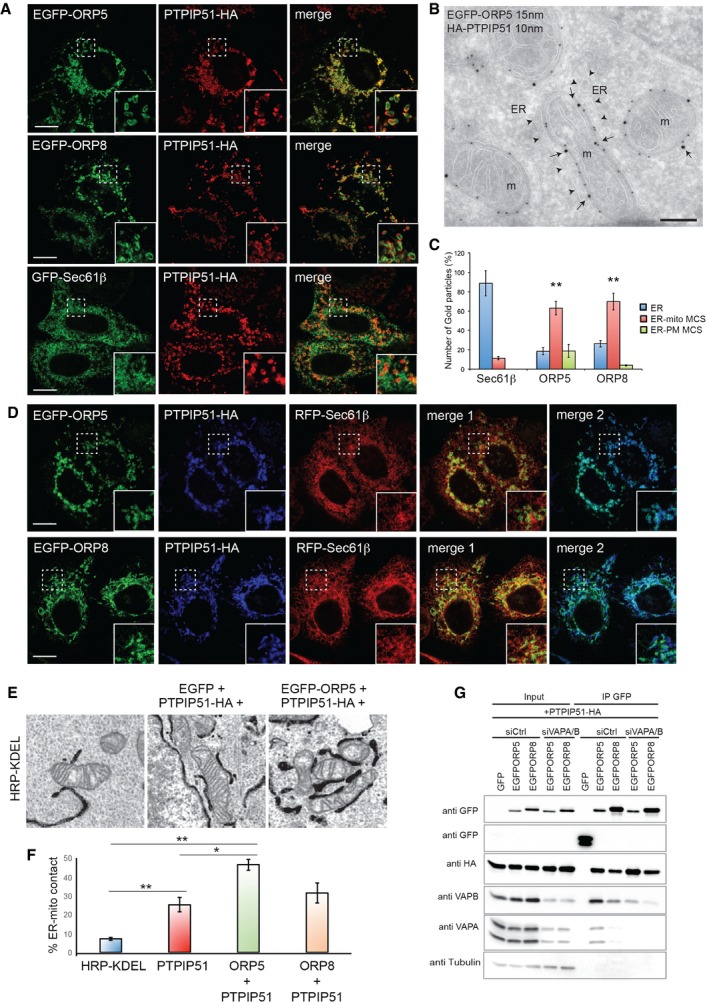
- Confocal images of HeLa cells transfected with EGFP‐ORP5, EGFP‐ORP8, or GFP‐Sec61β together with PTPIP51‐HA and immunostained using HA antibody to detect PTPIP51. Scale bar, 10 μm.
- Electron micrograph of ultrathin cryosections of HeLa cells co‐transfected with EGFP‐ORP5 and PTPIP51‐HA and immunogold stained with anti‐GFP (15 nm gold) and anti‐HA (10 nm gold) showing EGFP‐ORP5 localization at ER–mitochondria contacts (arrows). Arrowheads indicate the highly expanded ER membrane juxtaposed to mitochondria. Scale bar, 200 nm.
- Quantification of the IEM labeling for EGFP‐ORP5, EGFP‐ORP8, and GFP‐Sec61β in HeLa cells co‐transfected with PTPIP51‐HA. Results are presented as the percentage of the total number of gold particles (800 per condition, n = 30–35 cells) for ORP5, ORP8, and Sec61β in the indicated compartments (ER, reticular ER; ER‐mito MCS, ER–mitochondria membrane contact sites; ER‐PM MCS, ER–PM membrane contact sites). % gold particles ± SEM. Data are representative of three independent replicates. **P < 0.001.
- Confocal images of HeLa cells transfected with EGFP‐ORP5 or EGFP‐ORP8 together with PTPIP51‐HA and RFP‐Sec61β and immunostained using HA antibody to detect PTPIP51. Scale bar, 10 μm.
- Representative EM images of ER–mitochondria contacts in the indicated overexpression conditions.
- Quantification of the percentage of ER–mitochondria contact sites (ER‐mito contact) per mitochondria of HeLa cells transfected with HRP‐KDEL (Ctrl) or co‐transfected with PTPIP51‐HA and EGFP or PTPIP51‐HA and EGFP‐ORP5 or PTPIP51‐HA and EGFP‐ORP8 (n = 30 cells and 1,082–1,223 mitochondria). % ER–mitochondria contact sites ± SEM. Data are representative of three independent replicates. **P < 0.001, *P < 0.05.
- EGFP, EGFP‐ORP5, or EGFP‐ORP8 were immunoprecipitated from lysates of HeLa cells co‐expressing PTPIP51‐HA and treated with control (siCtrl) or VAPA and VAPB (SiVAPA/B) siRNAs and analyzed by Western blot using antibodies against GFP (ORPs), HA (PTPIP51), VAPA, VAPB, and tubulin.
Source data are available online for this figure.
Overexpression of PTPIP51 alone has been shown to increase the amount of ER–mitochondria contact 19, leaving open the possibility that ORP5 and ORP8 localization to ER regions close to mitochondria observed upon co‐expression with PTPIP51 could be simply due to their localization to the ER. Thus, we further analyzed the localization of EGFP‐tagged ORP5, ORP8, and Sec61β in cells co‐expressing PTPIP51‐HA by immuno‐electron microscopy (IEM). The bulk of EGFP‐ORP5 (63%) and EGFP‐ORP8 (69%) localized at the highly expanded ER–mitochondria contact sites, also labeled by PTPIP51 (Figs 2B (arrows) and C, and EV3A (arrows)), while only 18% of EGFP‐ORP5 and 3% of EGFP‐ORP8 were found at ER–PM contacts (Figs 2C and EV3A). In contrast, the majority of EGFP‐Sec61β (88%) was detected on internal ER membranes distributed throughout the cell (Fig 2C), with only 11% found at ER–mitochondria contact sites, showing that ORP5 and ORP8 but not Sec61β are specifically increased at ER–mitochondria contact sites when co‐expressed with PTPIP51. In parallel experiments, immunogold labeling of endogenous PDI on ultrathin cryo‐sections of PTPIP51‐HA overexpressing cells showed that also PDI was enriched in ER elements dispersed throughout the cells that were sometimes found in proximity but not in close contact with mitochondria (Fig EV3A).
Figure EV3. ORP5 and ORP8 localization at ER–mitochondria contacts is promoted by the mitochondrial protein PTPIP51.
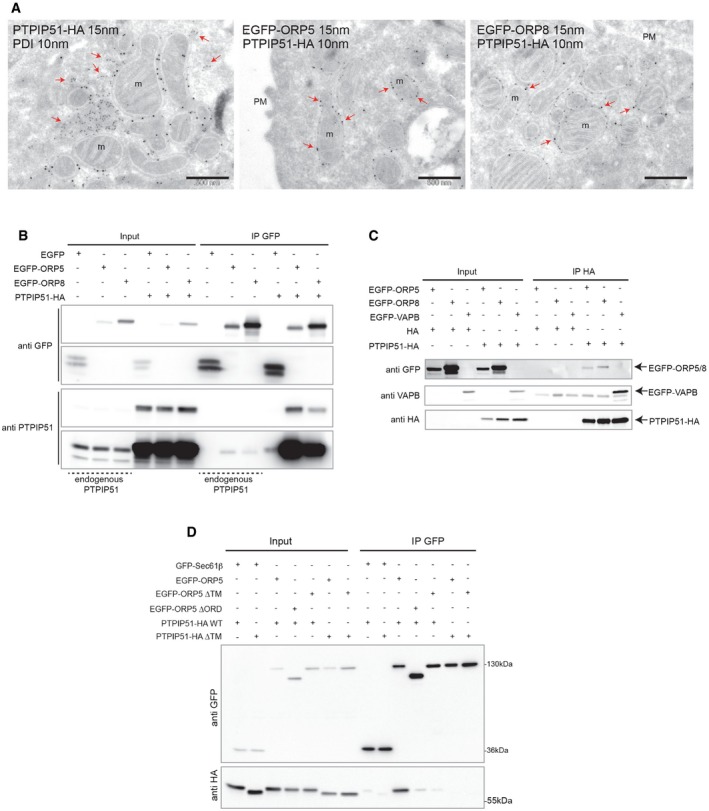
- Electron micrographs of ultrathin cryosections of HeLa cells transfected with PTPIP51‐HA alone, or with EGFP‐ORP5 and EGFP‐ORP8 together with PTPIP51‐HA and double‐immunogold stained with anti‐PDI to detect the ER (10 nm gold) and anti‐HA (15 nm gold) to detect PTPIP51 or with anti‐GFP (15 nm gold) to detect ORP5 or ORP8 and anti‐HA (10 nm gold) to detect PTPIP51. Red arrows indicate PDI in the lumen of ER structures not in contact with mitochondria, and ORP5 or ORP8 at ER–mitochondria contact sites. Scale bar, 500 nm.
- EGFP, EGFP‐ORP5, or EGFP‐ORP8 were immunoprecipitated from lysates of HeLa cells co‐expressing or not PTPIP51‐HA and analyzed by Western blot using antibodies against GFP and PTPIP51 to detect endogenous and overexpressed proteins.
- PTPIP51‐HA was immunoprecipitated from lysates of HeLa cells co‐expressing EGFP, EGFP‐ORP5, or EGFP‐ORP8 and analyzed by Western blot using antibodies against GFP and HA.
- GFP‐Sec61β, EGFP‐ORP5, EGFP‐ORP5ΔTM, or EGFP‐ORP5ΔORD were immunoprecipitated from lysates of HeLa cells co‐expressing PTPIP51‐HA or PTPIP51‐HA ΔTM and analyzed by Western blot using antibodies against GFP and HA.
The specific enrichment of ORP5/ORP8 at ER–mitochondria contacts in co‐expression with PTPIP51 was further confirmed in cells triple transfected with either EGFP‐ORP5 or EGFP‐ORP8, mRFP‐Sec61β, and PTPIP51‐HA by confocal imaging. EGFP‐ORP5 and EGFP‐ORP8 strongly co‐localized with PTPIP51‐HA at ER–mitochondria contacts, while the majority of mRFP‐Sec61β remained reticular in HeLa triple‐transfected cells (Fig 2D). We also carried out a morphological analysis of the ER–mitochondria contact sites in cells triple transfected with EGFP‐ORP5 or EGFP‐ORP8, PTPIP51‐HA, and HRP‐KDEL and compared to cells transfected with PTPIP51‐HA and HRP‐KDEL or with HRP‐KDEL alone (Fig 2E and F). In agreement with 19 PTPIP51 overexpression increased of about threefold the surface of mitochondria engaged in contact with the ER (23%), as compared to cells expressing only HRP‐KDEL (6%) (Fig 2E and F). Interestingly, co‐transfection of EGFP‐ORP5 and PTPIP51‐HA resulted in a significant increase of ER–mitochondria contacts (45%) similarly to what was previously observed in cells co‐expressing PTPIP51 and VAPB 19. Co‐expression of PTPIP51‐HA and EGFP‐ORP8 also induced an increase, but smaller (31%), of ER–mitochondria contacts. Altogether, these data show that ORP5 and ORP8 are specifically recruited to ER–mitochondria contact sites when co‐transfected with PTPIP51 and suggest a possible interaction between these proteins.
To confirm such interaction, co‐immunoprecipitation experiments from HeLa cells transfected with EGFP, EGFP‐ORP5 or EGFP‐ORP8, and with or without PTPIP51‐HA were carried out. PTPIP51‐HA was specifically immunoprecipitated with both EGFP‐ORP5 and EGFP‐ORP8 when co‐overexpressed with either one of these proteins (Fig EV3B). Endogenous PTPIP51 was also immunoprecipitated from HeLa cells overexpressing either EGFP‐ORP5 or EGFP‐ORP8. Note that the interaction of PTPIP51 with EGFP‐ORP8 is lower compared to EGFP‐ORP5. Likewise, EGFP‐ORP5 and EGFP‐ORP8 were co‐immunoprecipitated with PTPIP51‐HA (Fig EV3C). These results support the conclusion that ORP5 and ORP8 are localized to ER–mitochondria contact sites and interact with PTPIP51.
Since PTPIP51 promotes ER–mitochondria junctions through its interaction with VAPB, we investigated whether ORP5/ORP8–PTPIP51 interactions were dependent on VAP proteins. Co‐immunoprecipitation experiments in control cells or in cells knocked down for VAPA and VAPB and co‐transfected with EGFP, EGFP‐ORP5, or EGFP‐ORP8 together with PTPIP51‐HA revealed that ORP5/ORP8 interaction with PTPIP51 was conserved in the absence of both VAPA and VAPB (Fig 2G). They also revealed that ORP5 (and to a lesser extent ORP8) interacts with endogenous VAPs in addition to PTPIP51, suggesting that ORP5/ORP8 may be part of the VAP–PTPIP51 complex. Interestingly, more VAPB than VAPA appeared to be co‐immunoprecipitated in complexes with EGFP‐ORP5/ORP8. Overall, these results show that ORP5 and ORP8 are enriched at ER–mitochondria contact sites and interact physically with PTPIP51.
The lipid‐binding/transfer domain of ORP5 and ORP8 is required for their targeting to ER–mitochondria contact sites and for their interaction with PTPIP51
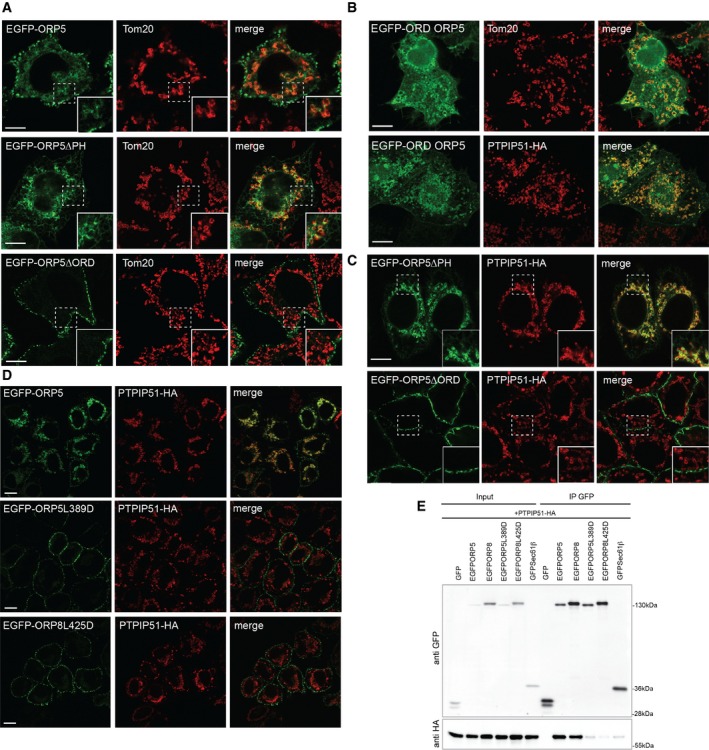
Both ORP5 and ORP8 contain a PH domain, which was recently shown to mediate their targeting to ER–PM contact sites, and an ORD domain involved in lipid transfer 12 (Fig 1A). To further characterize the ER–mitochondria contact site localization of ORP5 and ORP8 described above, we studied the localization of EGFP‐tagged ORP5 and ORP8 constructs lacking the PH domain (EGFP‐ORP5ΔPH and EGFP‐ORP8ΔPH) at non‐cortical ER membranes. In agreement with a role of the PH domain in PM targeting 12, EGFP‐ORP5ΔPH and EGFP‐ORP8ΔPH were no longer found at the cell periphery, as assessed by TIRF microscopy (Fig EV4B). The localization of EGFP‐ORP5, EGFP‐ORP8, EGFP‐ORP5ΔPH, and EGFP‐ORP8ΔPH at non‐cortical ER membranes in HeLa cells was analyzed by confocal microscopy (Figs 3A and EV4C). As expected and shown in Fig 1, EGFP‐ORP5 was found predominantly at the cortical ER elements near the PM, with a minor localization to ER adjacent to mitochondria, visualized by the outer mitochondrial membrane protein TOM20 labeling (Fig 3A). Loss of the PH domain redistributed ORP5 to the reticular ER, with loss of localization adjacent to the PM but retention of localization in proximity to mitochondria (Fig 3A). IEM localization analysis of EGFP‐ORP5ΔPH confirmed that 71% of ORP5ΔPH localized to non‐cortical ER structures (Fig EV4D (arrowheads) and E), and 28% at sites where ER and mitochondria membranes are within 30 nm (Fig EV4D (red arrows) and E). Likewise, EGFP‐ORP8 was found mostly in the reticular ER with a minor localization at ER–mitochondria contacts and the mitochondrial localization was unchanged in EGFP‐ORP8ΔPH mutant (Fig EV4C). We next aimed to better characterize the interaction between ORP5 and PTPIP51 and to identify the domains involved. To address the role of the ORD domain of ORP5 in its subcellular targeting to mitochondria membranes, HeLa cells were transfected with a plasmid expressing EGFP fused to the ORD domain of ORP5 and the intracellular distribution of EGFP‐ORP5ORD was examined by confocal microscopy. In agreement with ORP5 localization to ER–mitochondria contacts, EGFP‐ORP5ORD fusion protein was recruited to the mitochondria, indicating that the ORD domain is sufficient to target a heterologous protein (EGFP) to the mitochondria (Fig 3B). Interestingly, its targeting to the mitochondria was increased in cells co‐expressing PTPIP51.
Figure 3. Recruitment of ORP5 and ORP8 to ER–mitochondria contact sites requires their lipid‐binding domain.
-
AConfocal images of HeLa cells transfected with EGFP‐ORP5, EGFP‐ORP5ΔPH, or EGFP‐ORP5ΔORD and immunostained using anti‐TOM20 antibody to visualize mitochondria. Scale bar, 10 μm.
-
BConfocal images of HeLa cells transfected with the EGFP‐tagged ORD domain of ORP5 (EGFP‐ORD ORP5) and immunostained using anti‐TOM20 antibody (upper panels) or co‐transfected with PTPIP51‐HA and immunostained using HA antibody to detect PTPIP51. Scale bar, 10 μm.
-
C, DConfocal images of HeLa cells transfected with EGFP‐ORP5ΔPH or EGFP‐ORP5ΔORD (C) or EGFP‐ORP5, EGFP‐ORP5L389D or EGFP‐ORP8L425D (D) together with PTPIP51‐HA and immunostained using HA antibody to detect PTPIP51. Scale bar, 10 μm.
-
EEGFP‐ORP5, EGFP‐ORP8, EGFP‐ORP5L389D, EGFP‐ORP8L425D, GFP‐Sec61β, or EGFP alone were immunoprecipitated from lysates of HeLa cells co‐expressing PTPIP51‐HA and analyzed by Western blot with antibodies against GFP and HA.
Source data are available online for this figure.
To further address the targeting role of the ORD domain of ORP5, we extended our localization analysis at non‐cortical ER membranes by confocal imaging to the ORP5 construct lacking the ORD domain (EGFP‐ORP5ΔORD) (Fig 3A). Interestingly, loss of the ORD domain fully localized ORP5 at the PM, indicating that this domain is required for the localization at ER–mitochondria contacts. Co‐expression of ORP5 PH and ORD deletion mutants with PTPIP51‐HA confirmed that the EGFP‐ORP5ΔORD was no longer recruited to mitochondria while the EGFP‐ORP5ΔPH still was (Fig 3C). Co‐immunoprecipitation experiments from HeLa cells transfected with GFP‐Sec61β, EGFP‐ORP5 or EGFP‐ORP5 ORD, and transmembrane (TM) deletion mutants (EGFP‐ORP5ΔORD, EGFP‐ORP5ΔTM) together with PTPIP51‐HA or PTPIP51‐HA lacking the TM domain (PTPIP51‐HA ΔTM) showed that the ORD domain but also the TM domains of ORP5 and PTPIP51 are required for their interaction (Fig EV3D). These data indicate that both proteins should be properly localized at ER–mitochondria contact sites in order for them to interact. It is also possible that their interaction at such sites might not be direct but mediated by another still unidentified mitochondrial determinant.
To test whether ORP5/ORP8 recruitment to ER–mitochondria contact sites is dependent on their lipid transfer activity, we made use of the EGFP‐ORP5L389D and EGFP‐ORP8L425D where the conserved leucine critical for binding phosphatidylserine (PS) is mutated to an aspartic acid 12, 20. Confocal microscopy analysis of HeLa cells transfected with EGFP‐ORP5/ORP8 or EGFP‐ORP5L389D/ORP8L425D together with PTPIP51‐HA showed that the PS binding mutants localized exclusively to cortical ER (Fig 3D). Accordingly, co‐immunoprecipitation experiments confirmed the loss of interaction between EGFP‐ORP5L389D or EGFP‐ORP8L425D and PTPIP51 (Fig 3E). These results indicate that the PS binding by ORP5/ORP8 ORD domains is required for their targeting to ER–mitochondria contact sites and interaction with PTPIP51. Although the underlying mechanisms are still unclear, one possibility is that the loss of ORP5/ORP8 localization at ER–mitochondria contacts might depend on a predominance of the binding to PI4P at ER–PM contacts when the ORD domains of ORP5/ORP8 are unable to bind PS. Another attractive possibility is that ORP5 targeting at ER–mitochondria contact site requires presence of PS within their ORD domain or its transfer to the mitochondria.
ORP5 and ORP8 knockdown affect mitochondrial morphology and function
To address the role of ORP5 and ORP8 at ER–mitochondria contact sites, we depleted HeLa cells of ORP5 or ORP8 using short interfering RNA (siRNA), and robust suppression of both ORP5 and ORP8 was confirmed by Western blotting (Fig 4A). A morphological analysis by EM in control and ORP5‐ and ORP8‐silenced cells expressing HRP‐KDEL (Fig 4B) revealed that the overall ER structure as well as the abundance of ER–PM and ER–mitochondria membrane contacts was not altered by ORP5 and ORP8 knockdown. Interestingly, in ORP5‐ and ORP8‐silenced cells, we found presence of mitochondria with aberrant morphology. These mitochondria, which were still engaged in contacts with the ER, displayed less and disorganized internal cristae that visually resulted in a more translucent lumen as compared to normal mitochondria in siRNA control cells (Fig 4B, insets). A parallel morphological analysis by conventional EM where no proteins were overexpressed, that is, HRP‐KDEL, to better preserve the ultrastructure of internal compartments, gave similar observations (Fig 4D). Quantifications revealed that 37% in siORP5 and 28% in siORP8 of the mitochondria presented aberrant morphology versus 6% in control cells (Fig 4E). This shows that depletion of ORP5/ORP8 leads to defect in mitochondria morphology.
Figure 4. Depletion of ORP5 affects mitochondria morphology and function.
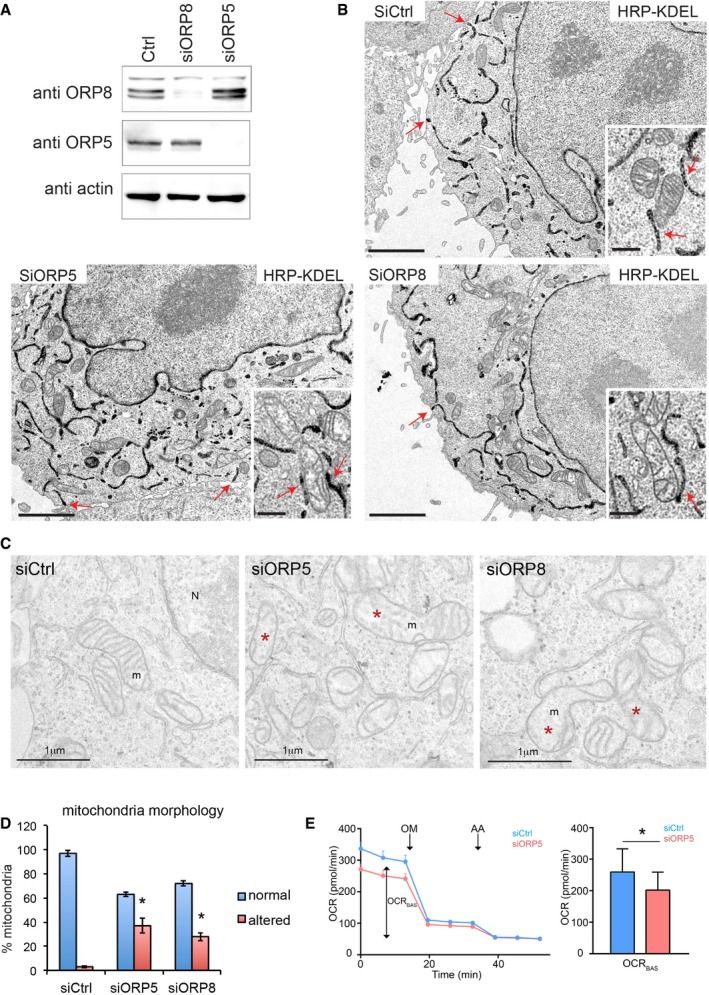
- ORP5, ORP8, and actin levels of HRP‐KDEL‐expressing HeLa cells treated with Ctrl siRNAs (siCtrl) or siRNAs against ORP5 (siORP5) or ORP8 (siORP8).
- Electron micrographs of HRP‐KDEL‐expressing HeLa cells treated with Ctrl siRNAs or siRNAs against ORP5 or ORP8. Red arrows indicate ER–PM contact sites. Scale bar, 2 μm. Insets show representative mitochondria and red arrows in the insets indicate ER–mitochondria contacts. Inset scale bar, 500 nm.
- Representative EM micrographs showing the morphology of mitochondria in siCtrl‐, siORP5‐, and siORP8‐treated cells. Scale bar, 1 μm. Red asterisks indicate mitochondria with altered morphology.
- Quantifications of the number of mitochondria with altered morphology in the indicated siRNA conditions. % altered mitochondria ± SEM, n = 25 cells, and 632–710 mitochondria. *P < 0.001 compared to siCtrl.
- Mitochondrial oxygen consumption rate (OCR) measured in Ctrl and ORP5 siRNA‐transfected cells. OCR trace was obtained by sequential measurement of basal OCR (OCRBAS), OCR after the addition of oligomycin (OM), and OCR after addition of antimycin A (AA). Note the reduced basal OCR (OCRBAS) compared to ctrl siRNA cells. Error bars denote ± SEM. Data are the mean of 4 independent repeats (n = 4), *P < 0.05.
Source data are available online for this figure.
Mitochondrial morphology is essential to fulfill their multiple functions and maintain mitochondria bioenergetics. Having established the presence of ORP5 in the MAMs (see Fig 1I) and showed that knockdown of ORP5 leads to altered mitochondrial morphology, we next analyzed if these changes had a functional effect on mitochondrial oxygen consumption, a crucial process in the production of ATP. To this end, we monitored mitochondrial oxygen consumption rate (OCR), in control and ORP5 siRNA‐transfected cells. We found that ORP5‐silenced cells showed a reduction in basal OCR (OCRBAS) of ± 23% compared to ctrl siRNA cells (Fig 4D). Having shown the stronger MAM localization of ORP5, it might still be possible that ORP8 could partially compensate the reduced OCRBAS. Additionally, the reduction in OCRBAS might be exacerbated under metabolic stress conditions.
Mitochondria lipid composition is important to maintain mitochondrial morphology and to stabilize the conformation of respiratory complexes 15, 21. Taking in account the role of ORP5 and ORP8 in transferring PS at ER–PM contact sites 12 and their localization at ER–mitochondria contact sites and defect in mitochondria morphology when depleted, an interesting possibility is that ORP5 and ORP8 mediate PS transport also at ER–mitochondria contact sites.
In summary, we have demonstrated that the mammalian ORP5 and ORP8 proteins localize to ER–mitochondrial MCS, in addition to ER–PM contact sites. Their targeting to ER–mitochondria MCS and interaction with the mitochondrial PTPIP51 requires a functional lipid‐binding/transfer ORD domain. We also show a novel function for ORP5/ORP8 in preserving mitochondria morphology and respiratory activity.
Materials and Methods
Cell culture, siRNA, and transfection
HeLa cells were cultured in DMEM supplemented with 10% FBS at 37°C and 5% CO2. Transfection of plasmids and siRNA oligos was carried out with Lipofectamine 2000 and Oligofectamine (Life Technologies) according to manufacturer's instructions.
Fluorescence microscopy
Immunofluorescence analysis and confocal and TIRF microscopy were carried out as described in the Appendix Supplementary Methods.
Electron microscopy
Conventional EM, including HRP cytochemistry, and immunogold labeling on ultrathin cryosections were performed according to standard procedures 18 (see also Appendix Supplementary Methods). EM sections were observed under a FEI Tecnai 12 microscope equipped with a CCD (SiS 1kx1k keenView) camera.
Biochemical analysis
For immunoprecipitation of EGFP‐tagged ORPs or HA‐tagged PTPIP51, cell lysates were incubated with Chromoteck GFP‐trap agarose beads (Allele Biotech) or anti‐HA agarose beads (Pierce), respectively, and solubilized bead‐bound material was processed for SDS–PAGE and immunoblotting (see also Appendix Supplementary Methods).
Mass spectrometry analysis
MS analysis was carried out by the proteomics/mass spectrometry platform in IJM (http://www.ijm.fr/plateformes/spectrometrie-de-masse). Full experimental procedures are found in the Appendix Supplementary Methods.
Cell fractionation
Crude mitochondria, mitochondria, and mitochondria‐associated membranes fractions were isolated as described 22 (see also Appendix Supplementary Methods).
Mitochondrial respiration assay
Oxygen consumption rate (OCR) was measured utilizing the extracellular flux analyzer XFp (Seahorse Bioscience Inc.) (see also Appendix Supplementary Methods).
Statistical analysis
FIJI Image Analysis software was used for quantification of fluorescent signals. iTEM software (Olympus) was used for quantification on EM sections. Statistical analysis, including those of electron microscopy data, was made using Student's t‐test for independent samples.
Author contributions
FG conceived and supervised the work. RG, AH, AvV, PA, and FG performed the experiments. RG, AH, and FG analyzed the data. RG, CLJ, and FG wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Jean‐Marc Verbavatz for its critical input in this work. We also thank Patrick Fuchs, Laurence Walch, Alenka Copic, Amelie Bacle, Thierry Galli, and David Tareste for discussion; France Lam, Remi LeBorgne, and Cesar La Torre Garray for help and consultation with microscopy experiments; Karine Eudes for technical help; Sebastien Leon and Benoit Palancade for sharing reagents. We thank Jean‐Michel Camadro and Thibault Leger (IJM proteomics platform) for proteomics analysis and acknowledge the ImagoSeine core facility of the Institut Jacques Monod, member of IBiSA and the France‐BioImaging (ANR‐10‐INBS‐04) infrastructure. Marie Curie CIG (PCIG14‐GA‐2013‐631202‐ERPM) to FG, ANR Jeune Chercheur (ANR‐14‐CE11‐0015) to FG, FWO (G0584.12N, GA01111N) and KU Leuven (C16/15/073) to PA supported this work, and ANR Jeune Chercheur (ANR‐14‐CE11‐0015) to FG supported salaries to RG and AH.
EMBO Reports (2016) 17: 800–810
References
- 1. Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B (2013) Organization and function of membrane contact sites. Biochim Biophys Acta 1833: 2526–2541 [DOI] [PubMed] [Google Scholar]
- 2. Tatsuta T, Scharwey M, Langer T (2014) Mitochondrial lipid trafficking. Trends Cell Biol 24: 44–52 [DOI] [PubMed] [Google Scholar]
- 3. Lahiri S, Toulmay A, Prinz WA (2015) Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol 33: 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olkkonen VM, Li S (2013) Oxysterol‐binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog Lipid Res 52: 529–538 [DOI] [PubMed] [Google Scholar]
- 5. de Saint‐Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G (2011) Osh4p exchanges sterols for phosphatidylinositol 4‐phosphate between lipid bilayers. J Cell Biol 195: 965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G, Antonny B (2013) A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155: 830–843 [DOI] [PubMed] [Google Scholar]
- 7. Raychaudhuri S, Prinz WA (2010) The diverse functions of oxysterol‐binding proteins. Annu Rev Cell Dev Biol 26: 157–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tong J, Yang H, Yang H, Eom SH, Im YJ (2013) Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol‐binding proteins. Structure 21: 1203–1213 [DOI] [PubMed] [Google Scholar]
- 9. Moser von Filseck J, Vanni S, Mesmin B, Antonny B, Drin G (2015) A phosphatidylinositol‐4‐phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat Commun 6: 6671 [DOI] [PubMed] [Google Scholar]
- 10. Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H (2011) A role for oxysterol‐binding protein‐related protein 5 in endosomal cholesterol trafficking. J Cell Biol 192: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W, Drin G (2015) INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4‐phosphate. Science 349: 432–436 [DOI] [PubMed] [Google Scholar]
- 12. Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P (2015) INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5‐ and ORP8‐mediated ER‐plasma membrane contacts. Science 349: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shiao YJ, Lupo G, Vance JE (1995) Evidence that phosphatidylserine is imported into mitochondria via a mitochondria‐associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J Biol Chem 270: 11190–11198 [DOI] [PubMed] [Google Scholar]
- 14. Vance JE (2014) MAM (mitochondria‐associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841: 595–609 [DOI] [PubMed] [Google Scholar]
- 15. Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE (2005) Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem 280: 40032–40040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML (2012) Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem 287: 17589–17597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM (2008) The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol Biol Cell 19: 5143–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giordano F, Saheki Y, Idevall‐Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P (2013) PI(4,5)P2‐Dependent and Ca(2+)‐Regulated ER‐PM Interactions Mediated by the Extended Synaptotagmins. Cell 153: 1494–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay‐Barrena G, Lin WL, Xu YF, Lewis J et al (2014) ER‐mitochondria associations are regulated by the VAPB‐PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: 3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin AC (2013) Interactome map uncovers phosphatidylserine transport by oxysterol‐binding proteins. Nature 501: 257–261 [DOI] [PubMed] [Google Scholar]
- 21. Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE (2013) Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem 288: 4158–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: 1582–1590 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4