

Abstract
Defects in DNA replication, DNA damage response, and DNA repair compromise genomic stability and promote cancer development. In particular, unrepaired DNA lesions can arrest the progression of the DNA replication machinery during S‐phase, causing replication stress, mutations, and DNA breaks. HUWE1 is a HECT‐type ubiquitin ligase that targets proteins involved in cell fate, survival, and differentiation. Here, we report that HUWE1 is essential for genomic stability, by promoting replication of damaged DNA. We show that HUWE1‐knockout cells are unable to mitigate replication stress, resulting in replication defects and DNA breakage. Importantly, we find that this novel role of HUWE1 requires its interaction with the replication factor PCNA, a master regulator of replication fork restart, at stalled replication forks. Finally, we provide evidence that HUWE1 mono‐ubiquitinates H2AX to promote signaling at stalled forks. Altogether, our work identifies HUWE1 as a novel regulator of the replication stress response.
Keywords: DNA replication, genomic instability, H2AX, HUWE1, PCNA
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
DNA replication is fundamental for genomic integrity. Obstacles to DNA replication, such as unrepaired DNA lesions, generate replication stress by blocking the progression of replicative polymerases. Prolonged stalling of the replication machinery can result in replication fork collapse, generating DNA breaks. This process is a major mechanism for genomic instability, leading to tumorigenesis 1, 2, 3. To avoid this, cells developed DNA damage tolerance mechanisms that restart the stalled forks by bypassing these lesions. Several components of the replication fork are involved in promoting fork restart; central among those is proliferating cell nuclear antigen (PCNA), a homotrimeric ring‐shaped DNA clamp that tethers polymerases to DNA during replication 4. PCNA also serves as a docking platform for proteins that carry out various DNA metabolic processes 4. Most proteins interact with PCNA via the PCNA‐interacting peptide (PIP) motif Qxxhxxaa (where h indicates an aliphatic hydrophobic residue and a indicates an aromatic residue) 5. At stalled replication forks, PCNA becomes mono‐ubiquitinated by the ubiquitin ligase Rad18, promoting recruitment of specialized low‐fidelity polymerases that are able to replicate through DNA lesions—a process termed translesion synthesis (TLS) 6, 7, 8, 9. These polymerases contain not only PIP motifs, but also ubiquitin binding domains, which explains their enhanced affinity for ubiquitinated PCNA 10. PCNA is thus essential for alleviating replication stress.
HUWE1 (also known as ARF‐BP1, HECTH9, MULE, and Lasu1) is a large (482 kDa) evolutionarily conserved E3 ubiquitin ligase of the HECT family 11, 12. HUWE1 plays important roles in regulating cell proliferation, cell death, development, and tumorigenesis. HUWE1 mutations have been found in many cancers including lung, stomach, breast, colorectal, hepatic, and brain carcinomas 13, 14, 15, 16, 17, 18. There is ongoing debate whether HUWE1 plays an oncogenic or tumor suppressive role, with evidence for both activities 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29. HUWE1 regulates cellular homeostasis by maintaining steady‐state levels of p53 13, 30. Moreover, it promotes cell survival and proliferation by ubiquitinating Myc with Lys63‐linked ubiquitin chains, which recruit the coactivator p300 14. HUWE1 was also shown to regulate DNA repair. It was reported that HUWE1 targets for degradation: the checkpoint proteins CDC6 31, TopBP1, and Miz1 20, 32; the base excision repair polymerases β and λ 33, 34, 35; and the homologous recombination factor BRCA1 36. Through these activities, HUWE1 directly inhibits DNA repair.
In contrast, we report here a surprising role for HUWE1 in preserving genomic stability, by promoting tolerance to replication stress. We found that HUWE1 contains a PIP‐box and directly interacts with PCNA, which is essential for replication fork stability and genomic integrity. Moreover, we show that HUWE1 mono‐ubiquitinates H2AX to promote replication stress signaling.
Results
HUWE1 is required for DNA damage tolerance and maintenance of genomic integrity
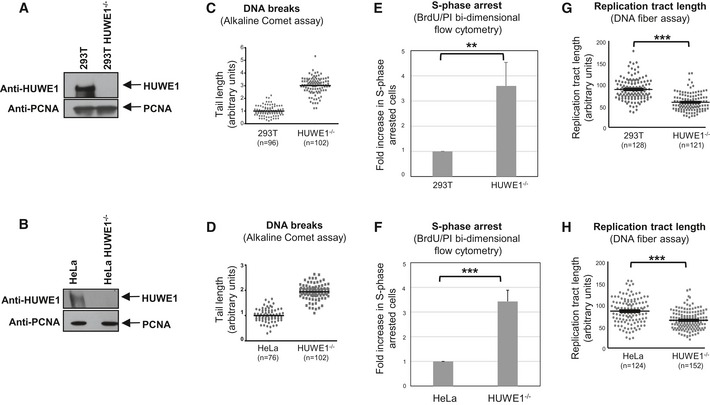
A broad range of substrates have been identified for HUWE1‐mediated ubiquitination. However, mechanistic understanding of the pathways controlled by HUWE1 is still lacking. To address this, we employed the CRISPR/Cas9 technology to knockout HUWE1 in human embryonic kidney 293T cells, HeLa cervical adenocarcinoma cells, and 8988T pancreatic adenocarcinoma cells (Figs 1A and B, and EV1A). Strikingly, HUWE1‐knockout cells showed a significant increase in DNA breaks in the absence of any DNA damage treatment, as measured by the alkaline comet assay (Figs 1C and D, and EV1B). This suggests that there is increased replication stress in the absence of HUWE1. Indeed, cell cycle distribution analyses using BrdU/PI bi‐dimensional flow cytometry indicated increased S‐phase arrest (cells with S‐phase DNA content, but negative for BrdU incorporation), coupled with a reduction in BrdU‐positive cells undergoing DNA synthesis (Figs 1E and F, and EV1C–F). Moreover, using the DNA fiber assay, we found that HUWE1‐knockout cells have shorter replication tracts (Fig 1G and H), indicative of replication stress. Finally, we also employed siRNA (Figs 2A and B, and EV1G) to transiently downregulate HUWE1 in 293T, 8988T, and HeLa cells. Similar to the knockout cells, HUWE1‐knockdown cells showed increased S‐phase arrest, a smaller proportion of BrdU‐positive cells undergoing DNA synthesis, and reduced replication tract length (Figs 2C–F and EV1H and I). These data indicate that HUWE1‐deficient cells are unable to resolve endogenous DNA damage, resulting in DNA replication glitches.
Figure 1. HUWE1‐knockout cells show genomic instability and increased replication stress.
-
A, BWestern blot showing the absence of HUWE1 protein in 293T (A) and HeLa (B) cells subjected to CRISPR/Cas9‐mediated HUWE1 deletion.
-
C, DHUWE1‐knockout 293T (C) and HeLa (D) cells show increased DNA breaks in the absence of exogenous DNA damage treatment. Results from the alkaline comet assay are shown. The “n” numbers of comet tails analyzed (pooled from two independent experiments), as well as the mean, are indicated on the graphs. HUWE1‐knockout HeLa cells did not show increased breakage in the neutral comet assay (Fig EV1B), indicating that the majority of breaks observed in cycling HUWE1‐knockout cells are not double‐strand breaks, but rather single‐strand breaks and other types of lesions.
-
E, FIncreased S‐phase arrest in HUWE1‐knockout 293T (E) and HeLa (F) cells. Cycling cells were incubated with BrdU and subjected to BrdU/PI bi‐dimensional flow cytometry. Representative flow cytometry profiles are presented in Fig EV1D–F. Bars represent the fold increase in the percentage of cells with S‐phase DNA content (between 2N and 4N) but negative for BrdU staining. Bars represent the average of three independent experiments, with error bars showing SD. The P‐values are 0.0091 (E) and 0.0007 (F).
-
G, HThe DNA fiber assay shows reduced replication tract length in HUWE1‐knockout 293T (G) and HeLa (H) cells in the absence of exogenous DNA damage treatment. The “n” numbers of fibers analyzed (pooled from three independent experiments), as well as the mean ± SEM, are indicated on the graphs. P‐values are 9.8 × 10−22 (G) and 1.0 × 10−10 (H).
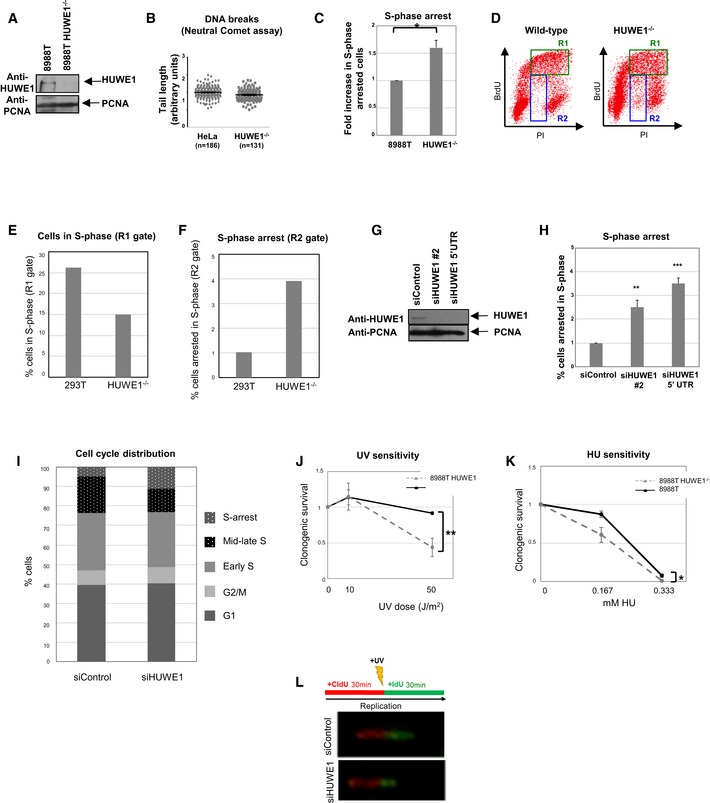
Figure EV1. Phenotypes of HUWE1‐deficient cells.
-
AWestern blot showing the absence of HUWE1 protein in 8988T cells subjected to CRISPR/Cas9‐mediated HUWE1 deletion.
-
BHUWE1‐knockout HeLa cells show no increase in Neutral Comet tail length. Cells were analyzed under normal growth conditions (no DNA damage treatment). The “n” numbers of comet tails analyzed (pooled from three independent experiments), as well as the mean ± SEM, are indicated on the graphs. We would like to mention that both normal and HUWE1‐knockout cells showed no real “tails” but rather very short “smears”, likely indicating that no DNA breaks are occurring in any of these cells. We measured those tails anyway, and present the result in this figure, in an attempt to provide a quantification of this result.
-
CIncreased S‐phase arrest in HUWE1‐knockout 8988T cells, as measured using the BrdU/PI bi‐dimensional flow cytometry assay. The average of three experiments, with standard deviations, is shown. P‐value is 0.011 (calculated using the t‐test two‐tailed, equal variance).
-
DRepresentative BrdU/PI bi‐dimensional flow cytometry profiles of wild‐type and HUWE1‐knockout 293T cells. Cells in mid‐ and late S‐phase (DNA content >2 N and positive for BrdU) are labeled as R1 region. Cells with S‐phase DNA content (between 2N and 4N) but negative for BrdU staining represent S‐phase‐arrested cells and are indicated as R2 region.
-
E, FQuantifications of R1 and R2 regions from the plot shown in (D), showing the proportions of cells in each region. While only one representative plot is shown, this result was reproduced in three independent experiments.
-
GWestern blot showing that the siRNA oligonucleotides used are efficiently knocking down HUWE1 levels in 293T cells.
-
HIncreased S‐phase arrest in 293T cells following HUWE1 knockdown. The percentage of cells arrested in S‐phase is shown. The average of five experiments, with standard deviations, is presented. The P‐values shown (calculated using the t‐test two‐tailed, equal variance) indicate the statistical significance relative to siControl (0.0024 and 0.0001, respectively).
-
IQuantification of cell cycle distribution of control and HUWE1‐depleted 293T cells, showing the altered distribution of mid‐late S‐phase vs. S‐phase‐arrested cells. Shown are G1 (BrdU‐negative, 2N DNA content); G2/M (BrdU‐negative, 4N DNA content); early S (BrdU‐positive, 2N DNA content); mid‐late S (BrdU‐positive, >2N DNA content); S‐arrest (BrdU‐negative, DNA content between 2N and 4N). The quantification of one representative plot is shown, but this result was reproduced in many independent experiments.
-
J, KUV (J) and HU (K) sensitivity of HUWE1‐knockout 8988T cells. The average of three experiments, with standard deviations, is shown. P‐values (calculated using the t‐test two‐tailed, equal variance) are 0.0029 (J) and 0.0126 (K).
-
LSchematic of the experimental setup, including examples of micrographs, for the DNA fiber experiment.
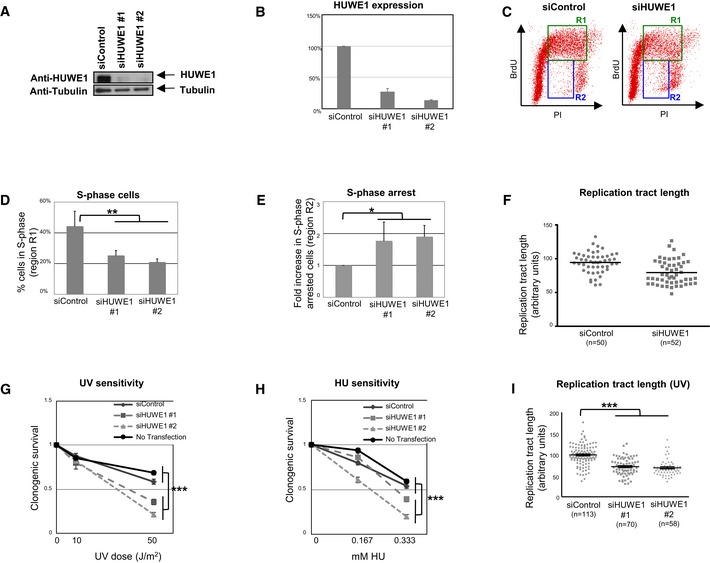
Figure 2. Increased replication stress in HUWE1‐knockdown cells.
-
AHUWE1 levels are efficiently downregulated by two different HUWE1 siRNA oligonucleotides in 8988T cells.
-
BQuantification of HUWE1 expression following siRNA treatment. Band intensity was quantified using ImageJ software and normalized to tubulin loading control. The average of three experiments is shown. Error bars indicate SD.
-
C–ECell cycle analyses by flow cytometry using BrdU/PI double staining show increased replication arrest in HUWE1‐depleted 8988T cells in the absence of exogenous DNA damage treatment. (C) Representative flow cytometry profiles of control and HUWE1‐knockdown cells. R1‐labeled region indicates mid‐ and late S‐phase cells (BrdU‐positive, > 2N DNA content), while R2‐labeled region indicates S‐phase arrested cells (BrdU‐negative, DNA content between 2N and 4N). (D) Quantification of S‐phase cells. Percentage of cells in R1 region is shown. Bars represent the average of three independent experiments. Error bars indicate SD. P‐value is 0.0014. (E) Quantification of S‐phase arrested cells. Bars represent the fold increase in the percentage of cells in R2 region, normalized to siControl‐treated cells. The average of three independent experiments is shown. Error bars indicate SD. P‐value is 0.0135.
-
FDNA fiber experiment showing reduced replication tract length in HUWE1‐depleted HeLa cells in the absence of exogenous DNA damage treatment. The “n” numbers of fibers analyzed (pooled from two independent experiments), as well as the mean, are indicated on the graphs.
-
G, HClonogenic assay showing that HUWE1 knockdown in 8988T cells are sensitive to UV (G) and HU (H). As controls, both cells transfected with non‐targeting siRNA (siControl) and non‐transfected cells were used. Note that the siHUWE1#2 oligonucleotide shows a stronger downregulation of HUWE1 and confers increased sensitivity compared to siHUWE1#1. The average of nine experiments is shown. Error bars represent SEM. P‐values (representing the statistical difference between the samples at the highest dose treatment) are 4.55 × 10−9 (G) and 1.8 × 10−8 (H).
-
IDNA fiber experiments showing decreased replication tract length following UV‐B treatment (30 J/m2) in HUWE1‐depleted HeLa cells. The “n” numbers of fibers analyzed (pooled from three independent experiments), as well as the mean ± SEM, are indicated on the graphs. P‐value is 2.11 × 10−20.
This novel activity of HUWE1 in protecting the replication fork against DNA damage was rather unexpected, since previous studies described HUWE1 as a negative regulator of DNA repair through suppression of HR and BER 33, 34, 35, 36. Therefore, we decided to analyze the impact of HUWE1 on the cellular sensitivity to hydroxyurea and UV radiation, agents that induce replication fork stalling during S‐phase. Clonogenic experiments indicated that HUWE1‐knockdown and knockout cells are hypersensitive to these agents (Figs 2G and H, and EV1J and K). Moreover, DNA fiber assays showed an even stronger reduction of replication tract length in HUWE1‐knockdown cells exposed to UV (Figs 2I and EV1L). Altogether, these results indicate a novel role for HUWE1 in DNA damage tolerance.
HUWE1 interacts with the replication factor PCNA at stalled replication forks
We next investigated whether HUWE1 is directly coupled to the replication machinery. Using the iPOND technique that allows identification of proteins at replication forks 37, 38, we found that HUWE1 is detectable in unchallenged replisomes and accumulates after UV exposure (Fig 3A). Immunofluorescence experiments confirmed that HUWE1 localizes to chromatin foci in response to replication fork stalling induced by HU or UV (Fig 3B). Importantly, these foci show a high degree of overlap with PCNA foci, an essential replication fork component that regulates the restart of stalled replication forks (Figs 3C and EV2A–D).
Figure 3. HUWE1 localizes to replication forks.
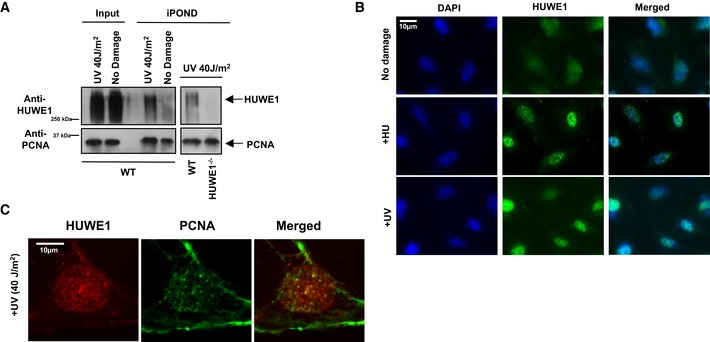
- iPOND experiment showing that HUWE1 localizes to replication forks in 293T cells. Binding to nascent DNA was increased after UV exposure. Because of the difficulties in removing all the cross‐links in high molecular weight proteins, HUWE1 is detected as a high molecular weight smear. A control experiment using the HUWE1‐knockout cells (shown in the right side panel) confirmed the specificity of the HUWE1 signal.
- Immunofluorescence experiment showing that HUWE1 localizes to chromatin foci in HeLa cells exposed to replication fork stalling agents HU (2 mM for 16 h) and UV (40 J/m2, analyzed 2 h later).
- Co‐immunofluorescence experiments showing co‐localization of HUWE1 and PCNA in chromatin foci. U2OS cells were analyzed 2 h after exposure to UV (40 J/m2). Quantifications of co‐localization, and additional micrographs are presented in Fig EV2A–D.
Figure EV2. Co‐immunofluorescence experiments showing co‐localization of HUWE1 and PCNA .
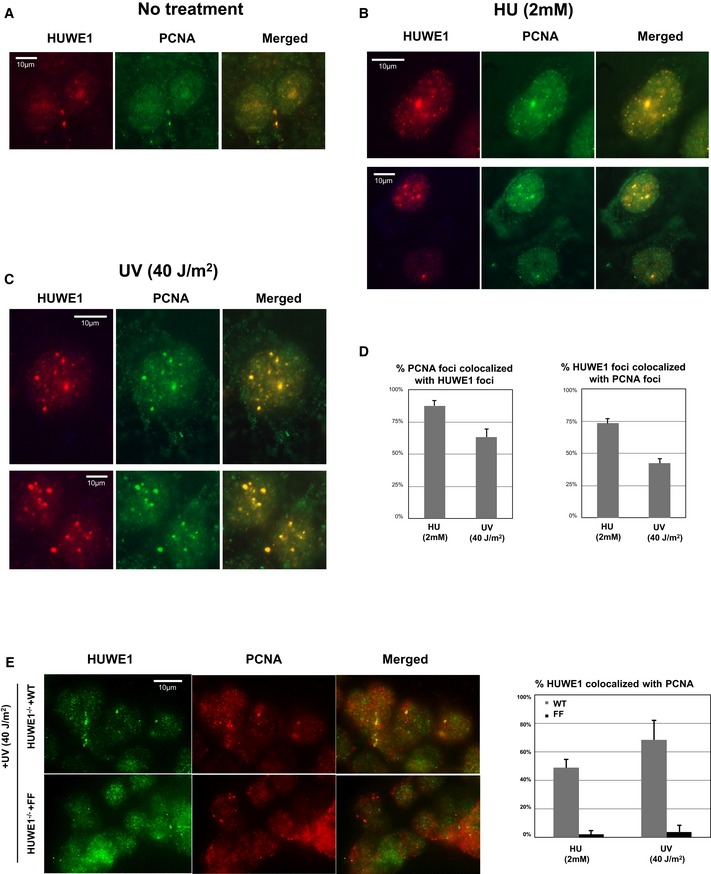
-
A–CRepresentative immunofluorescence micrographs of U2OS cells untreated (A), treated with 2 mM HU for 24 h (B), or exposed to 40 J/m2 UV and analyzed 2 h later (C).
-
DQuantifications showing the percentage of PCNA foci co‐localizing with HUWE1 foci and, reciprocally, the percentage of HUWE1 foci co‐localizing with PCNA foci. Only cells with more than five foci were analyzed. The average of three experiments, with standard errors, is shown. In total, 191 HUWE1 and 156 PCNA foci were counted.
-
EHUWE1‐knockout cells corrected with wild‐type or PIP mutant were subjected to immunofluorescence using HUWE1 and PCNA antibodies. Similar to the endogenous protein, Myc‐HUWE1 wild‐type forms UV‐ and HU‐induced chromatin foci. In contrast, the PIP‐box mutant does not co‐localize with PCNA after damage exposure. Shown are representative micrographs, as well as quantifications of the percentage of PCNA foci co‐localizing with HUWE1 and, reciprocally, the percentage of HUWE1 foci co‐localizing with PCNA, in wild‐type and PIP mutant‐corrected cells. Only cells with more than five foci were analyzed. The average of three experiments, with standard errors, is shown. A total number of foci counted were as follows: for the HU experiment: 162 HUWE1‐WT foci with 144 PCNA foci; 196 HUWE1‐FF foci with 163 PCNA foci. For the UV experiment: 146 HUWE1‐WT foci with 121 PCNA foci; 101 HUWE1‐FF foci with 78 PCNA foci.
This co‐localization suggested that HUWE1 might directly interact with PCNA. HUWE1 is a very long polypeptide (4,374 amino acids), containing a HECT‐type ubiquitin ligase domain in its C‐terminus. When surveying its amino acid sequence, we observed the presence of a putative PCNA‐interacting motif (PIP‐box QPAVEAFF right before the start of the HECT domain (Fig 4A). Reciprocal co‐immunoprecipitation studies of endogenous proteins confirmed that HUWE1 and PCNA interact in 293T, 8988T, and MCF7 cells (Fig 4B–F). This interaction was not reduced in Rad18‐knockout cells, showing that it does not require PCNA ubiquitination (Fig 4G).
Figure 4. HUWE1 interacts with PCNA .
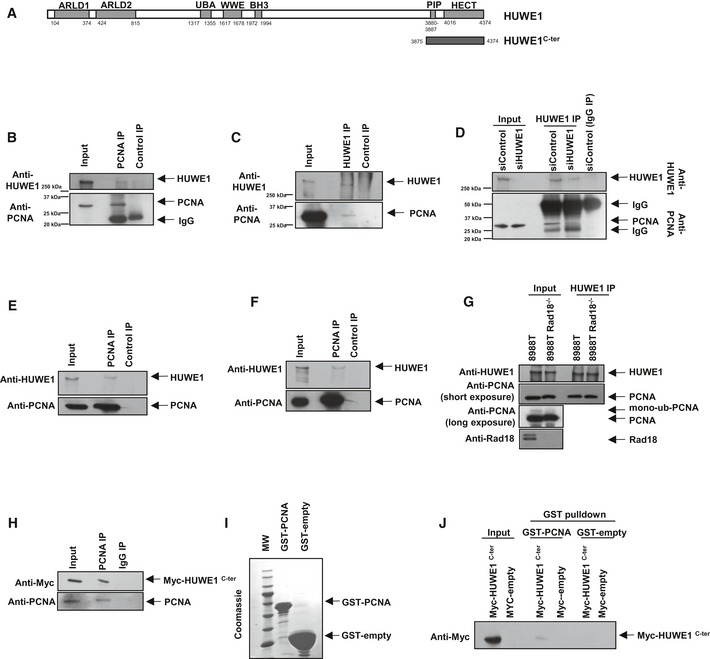
-
ASchematic representation of full‐length HUWE1. Shown is the PIP‐box domain we describe here (aa 3880‐3887). ARLD: Armadillo repeat‐like domain; UBA: ubiquitin‐associated domain; BH3: Bcl2‐homology three domain; HECT: homologous to the E6‐AP carboxyl terminus (catalytic ubiquitin ligase domain). The span of the HUWE1C‐ter (3875‐end) fragment used in subsequent studies is also indicated.
-
B–DInteraction between endogenous HUWE1 and PCNA in 293T cells. (B) Anti‐PCNA immunoprecipitation, showing that HUWE1 co‐precipitates. (C) Reciprocal experiment showing that PCNA co‐precipitates with HUWE1 in anti‐HUWE1 immunoprecipitation. (D) As control, endogenous HUWE1 was depleted by siRNA treatment, and extracts were subjected to anti‐HUWE1 immunoprecipitation. The HUWE1 blot shows that much less HUWE1 is precipitated by anti‐HUWE1 antibodies from HUWE1‐knockdown cells than control, as expected. PCNA is co‐precipitated by anti‐HUWE1 antibodies from control, but not HUWE1‐depleted cells, showing that the interaction is specific and not due to unspecific binding of PCNA to anti‐HUWE1 antibodies.
-
E, FHUWE1 co‐precipitates with PCNA in anti‐PCNA immunoprecipitation from extracts of MCF7 (E) and 8988T (F) cells, showing that the HUWE1–PCNA interaction can be detected in different cell lines.
-
GCo‐immunoprecipitation experiment from control and Rad18‐knockout 293T cells, showing that the HUWE1–PCNA interaction is not affected by loss of PCNA ubiquitination. Rad18‐knockout cells were obtained by CRISPR/Cas9 technology. The Rad18 and PCNA blots show loss of Rad18 and of PCNA ubiquitination, respectively.
-
HMyc‐HUWE1C‐ter fragment was transfected in 293T cells. Anti‐PCNA immunoprecipitation shows that the C‐terminal HUWE1 fragment interacts with PCNA.
-
ICoomassie staining of recombinant GST‐PCNA expressed and purified from bacteria.
-
JGST‐pull downs using GST‐PCNA or GST‐empty as control, and extracts of 293T cells transfected with Myc‐HUWE1C‐ter, showing interaction of this fragment with recombinant PCNA.
To further analyze the HUWE1–PCNA interaction, we cloned a C‐terminal truncation of HUWE1 containing the PIP and HECT domains (HUWE1C‐ter; Fig 4A). This truncation strongly interacted with PCNA in co‐immunoprecipitation and GST‐PCNA pull‐down experiments (Fig 4H–J). To determine the importance of the PIP‐box for this interaction, we next introduced point mutations in conserved residues of the PIP motif. This abolished the ability of HUWE1C‐ter to interact with PCNA in co‐immunoprecipitation and GST‐pull‐down studies (Fig 5A–D). We also obtained a full‐length clone of HUWE1 with a C‐terminal Myc epitope tag and performed Myc‐tag immunoprecipitation from cells transiently transfected with wild‐type or PIP‐box mutant HUWE1. PCNA co‐precipitated with full‐length wild‐type HUWE1, but not the PIP‐box mutant variant (Fig 5E). These results show that HUWE1 directly interacts with PCNA through the PIP‐box motif.
Figure 5. The PIP‐box of HUWE1 is essential for its interaction with PCNA .
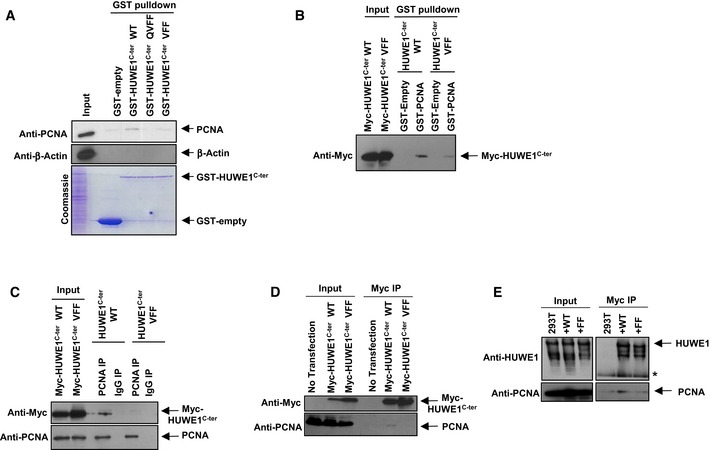
-
A, BGST‐pull downs showing that HUWE1 PIP‐box mutants do not interact with PCNA. (A) Recombinant GST‐tagged HUWE1C‐ter, either wild‐type or PIP‐box mutant variants (QVFF and VFF—the indicated residues, critical for the PIP‐box, were mutated to A), were purified from bacteria (see Coomassie staining) and employed for GST‐pull downs using extracts of 293T cells. PCNA co‐precipitated with wild‐type but not PIP‐box mutants. (B) Reciprocal GST‐pull down using recombinant GST‐PCNA and extracts of 293T cells overexpressing Myc‐HUWE1C‐ter. Wild‐type but not the PIP‐box mutant (VFF) HUWE1 fragment bound to GST‐PCNA.
-
C, DReciprocal co‐immunoprecipitation experiments showing that PIP‐box mutation abolishes PCNA interaction. (C) 293T cells were transfected with Myc‐HUWE1C‐ter variants and subjected to anti‐PCNA (C) or anti‐Myc (D) immunoprecipitation. Only wild‐type but not PIP‐box mutant (VFF) HUWE1 interacted with PCNA in both experimental setups.
-
EMutation of the PIP‐box in full‐length HUWE1 also blocks PCNA interaction. Full‐length Myc‐tagged HUWE1 (wild‐type or PIP mutant) was transfected in 293T cells. Following Myc‐immunoprecipitation, endogenous PCNA was detected in wild‐type but not PIP‐box mutant (FF) complexes.
HUWE1 interaction with PCNA is required for DNA damage tolerance
We next set out to investigate the role of HUWE1 interaction with PCNA. For this, we transfected the HUWE1‐knockout 293T cells described in Fig 1 with full‐length Myc epitope‐tagged HUWE1. We created cells stably expressing wild‐type, PIP‐box mutant (FF), or catalytic mutant (C4341A mutation inactivating the HECT domain) HUWE1 variants. HUWE1 Western blots (Fig 6A) showed that the expression of the exogenous variants stably transfected in the knockout cells is at a similar level to the expression of endogenous HUWE1 in the parental 293T cell line (with the exception of the HECT mutant, which shows reduced expression). As expected, also in this setup, wild‐type but not the PIP‐box mutant HUWE1 interacts with PCNA as shown by Myc‐tag immunoprecipitation experiments (Fig 6B)—further confirming that the HUWE1–PCNA interaction occurs through the PIP‐box. iPOND and immunofluorescence experiments showed that PIP‐box mutant HUWE1 does not efficiently localize to replication forks (Figs 6C and EV2E), indicating that PCNA interaction is required for correct localization of HUWE1.
Figure 6. HUWE1 interaction with PCNA is essential for alleviating replication stress.
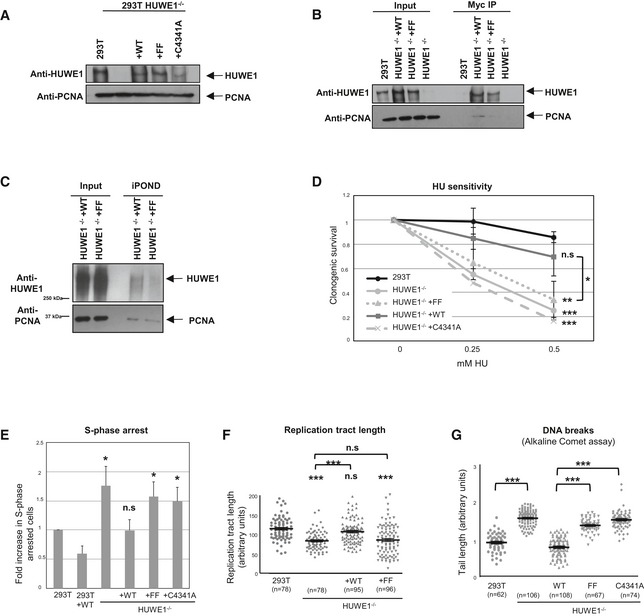
- Stable expression of Myc‐tagged wild‐type, PIP‐box mutant, and catalytically inactive HUWE1 variants in HUWE1‐knockout 293T cells. The Western blot shows that expression of the Myc‐tagged variants stably transfected in the knockout cell lines is similar to the endogenous HUWE1 expression.
- Myc‐tag immunoprecipitation using extracts of the corrected cell lines described above. As expected, PCNA co‐precipitates with wild‐type but not PIP‐box mutant HUWE1 (FF).
- iPOND experiment using HUWE1‐knockout 293T cells, corrected with wild‐type or PIP mutant HUWE1. The PIP mutant shows reduced cross‐linking to nascent DNA, indicating defective recruitment to replication forks.
- Clonogenic assay showing that stable expression of wild‐type HUWE1, but not of the PIP‐box mutant or the catalytic mutant, corrects the HU sensitivity of the HUWE1‐knockout 293T cells. Shown is the average of three independent experiments ± SD. If not indicated otherwise, the P‐values shown specify the statistical significance relative to 293T (for the 0.5 mM HU condition). P‐values are as follows: 0.0003 (293T vs. HUWE1−/−); 0.157 (293T vs. HUWE1−/− +WT); 0.0045 (293T vs. HUWE1−/− +FF); 0.0006 (293T vs. HUWE1−/− +C4341A); 0.047 (HUWE1−/− +WT vs. HUWE1−/− +FF).
- The S‐phase arrest phenotype is also rescued by wild‐type but not PIP mutant or catalytic mutant HUWE1. S‐phase arrested cells were quantified using the BrdU/PI bi‐dimensional flow cytometry assay. Shown is the average of three independent experiments ± SD. The P‐values shown specify the statistical significance relative to 293T (0.018, 0.957, 0.015, and 0.024, respectively). A quantification of the same data, showing the percentage of cells arrested in S‐phase, is shown in Fig EV3A.
- Normal replication tract length, quantified using the DNA fiber assay, is restored by expressing wild‐type, but not PIP mutant HUWE1, in the HUWE1‐knockout 293T cells. The “n” numbers of fibers analyzed (pooled from three independent experiments), as well as the mean ± SEM, are indicated on the graphs. If not indicated otherwise, the P‐values shown specify the statistical significance relative to 293T. P‐values are 1.47 × 10−11 (293T vs. HUWE−/− samples), 0.094 (293T vs. HUWE−/− +WT samples), 1.40 × 10−7 (293T vs. HUWE−/− +FF samples), 1.22 × 10−7 (HUWE−/− vs. HUWE−/− +WT samples), and 0.679 (HUWE−/− vs. HUWE−/− +FF samples).
- Alkaline comet assay showing that wild‐type but not the FF mutant can correct the breakage phenotype of HUWE1‐knockout 293T cells. The “n” numbers of comet tails analyzed (pooled from three independent experiments), as well as the mean ± SEM, are indicated on the graphs. The P‐values indicated are 4.18 × 10−43 (293T vs. HUWE−/− samples), 6.89 × 10−41 (HUWE1−/− +WT vs. FF samples), and 5.97 × 10−50 (HUWE1−/− +WT vs. C4341A samples).
We next investigated whether the PCNA‐HUWE1 interaction is important for relieving replication stress. Wild‐type HUWE1, but not the PIP‐box mutant or the catalytically inactive mutant, could rescue the HU sensitivity of the HUWE1‐knockout cell line (Fig 6D). Moreover, the increased S‐phase arrest, the reduced replication tract length, and the increased DNA breaks phenotypes could also be corrected by wild‐type but not by PIP‐box mutant HUWE1 (Figs 6E–G and EV3A). These results show that HUWE1 interaction with PCNA is essential for DNA damage tolerance and fork progression under replication stress.
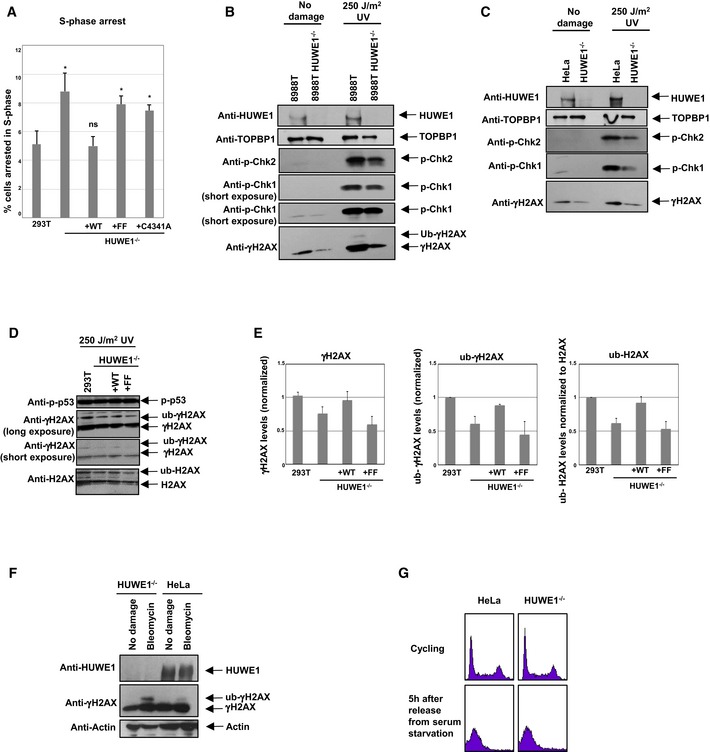
Figure EV3. Impact of HUWE1‐knockout on replication stress signaling and H2AX ubiquitination.
-
ACorrection of the S‐phase arrest phenotype by wild‐type but not PIP mutant HUWE1. The same data as in Fig 6E are shown, but this time the percent of cells in S‐phase is plotted. Shown is the average of three independent experiments ± SD. The P‐values shown (calculated using the t‐test two‐tailed, equal variance), indicate the statistical significance relative to 293T (0.015, 0.865, 0.011, and 0.016, respectively).
-
B, CWestern blots showing reduced γH2AX and phospho‐Chk1 in HUWE1‐knockout 8988T (B) and HeLa (C) cells.
-
DWestern blots showing reduced H2AX modification in HUWE1‐knockout 293T cells, corrected by wild‐type but not PIP mutant HUWE1.
-
EQuantification of γH2AX (against input control), γH2AX ubiquitination (against un‐ubiquitinated γH2AX), and H2AX ubiquitination (against unmodified H2AX). The average of three experiments is shown. Error bars represent standard deviations.
-
FHUWE1 deletion does not reduce H2AX ubiquitination in G1. HeLa cells (wild‐type or HUWE1‐knockout) were arrested in G0 by serum starvation for 24 h, than released in normal media. Cells were analyzed 5 h later, when they reached G1. Cells were either treated with 5 μM bleomycin for the last 2 h of the release or left untreated.
-
GFlow cytometry profile (PI staining) of cells at the harvest time point (5 h after release from serum starvation). The plot indicates that cells are uniformly in G1. For comparison, the normal profile of cycling cells is also presented.
HUWE1 promotes H2AX post‐translational modifications
We next examined whether HUWE1 influences checkpoint signaling at stalled replication forks. Surprisingly, we observed that the levels of mono‐ubiquitinated histone variant H2AX are significantly reduced in HUWE1‐knockout HeLa and 293T cells (Figs 7A and B, and EV3B–E), suggesting that HUWE1 may act as a ubiquitin ligase toward H2AX. Indeed, in vitro ubiquitination reactions showed that HUWE1 can mono‐ubiquitinate H2AX (Fig 7C). H2AX mono‐ubiquitination was shown to promote its phosphorylation 39, 40—an important step in signaling at double‐strand breaks. In line with this, we observed that the level of phosphorylated H2AX (γH2AX) is lower in HUWE1‐knockout cells (Figs 7A and B, and EV3B–E). Wild‐type but not PIP‐box mutant HUWE1 could correct the H2AX mono‐ubiquitination defect of HUWE1‐knockout cells (Figs 7D and EV3D–E). Moreover, HUWE1 knockout did not reduce γH2AX ubiquitination in G1 cells treated with bleomycin—an agent that induces double‐strand breaks (Fig EV3F and G). These results indicate that HUWE1 mono‐ubiquitinates H2AX at stalled replication forks. To further confirm this, we investigated γH2AX recruitment to common fragile sites. These difficult‐to‐replicate DNA regions act as endogenous replication stress elements and are prone to fork collapse and breakage, which can be experimentally exacerbated by treating cells with the replication inhibitor aphidicolin 41. As previously shown 42, γH2AX can be detected by chromatin immunoprecipitation (ChIP) at the fragile site FRA3B in aphidicolin‐treated cells (Fig 7E). This binding was dramatically reduced in HUWE1‐deficient cells (Fig 7E), indicating that HUWE1 is important for γH2AX activity during the replication stress response.
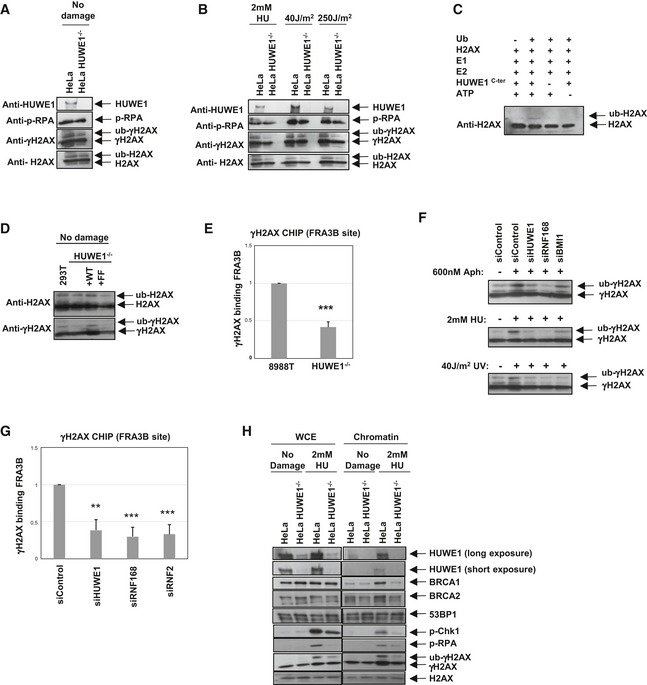
Figure 7. HUWE1 is a novel ubiquitin ligase for H2AX .
-
A, BThe impact of HUWE1 on H2AX phosphorylation and ubiquitination, under normal conditions (A) or upon induction of replication stress (2 mM HU for 18 h, or 2 h after exposure to 40 J/m2 UV). Control or HUWE1‐knockout HeLa cells were analyzed.
-
CIn vitro ubiquitination assay showing that recombinant HUWE1C‐ter can mono‐ubiquitinate H2AX.
-
DWild‐type but not the PIP mutant can correct the defective H2AX ubiquitination in HUWE1‐knockout 293T cells.
-
EChromatin immunoprecipitation showing that γH2AX binding to the common fragile site FRA3B is reduced in HUWE1‐knockout 8988T cells. Cells were treated with 600 nM aphidicolin for 24 h. Binding was quantified relative to input material. Shown is the average of four experiments ± SD. P‐value is 4.4 × 10−6.
-
FWestern blots showing that several H2AX ubiquitin ligases participate in γH2AX ubiquitination following replication stress. HUWE1, RNF168, and BMI1 were knocked‐down in HeLa cells. The efficiency of the knockdown is shown in Fig EV4. Cells were treated with 600 nM aphidicolin for 24 h, 2 mM HU for 24 h, or analyzed 2 h after exposure to 40 J/m2 UV.
-
GChromatin immunoprecipitation from HeLa cells, showing that knockdown of H2AX ubiquitin ligases can reduce γH2AX binding to FRA3B. Cells were treated with 600 nM aphidicolin for 24 h. Binding was quantified relative to input material. Shown is the average of three experiments ± SD. The P‐values shown indicate the statistical significance relative to siControl (0.0016, 0.0005, and 0.0009, respectively).
-
HChromatin fractionation experiments showing that HUWE1‐knockout HeLa cells, treated with 2 mM HU for 24 h, fail to efficiently recruit BRCA1 and BRCA2 proteins to DNA. In contrast, 53BP1 chromatin association is not induced by HU treatment, and not affected by HUWE1 knockout, and thus can serve as loading control. Shown are blots of whole‐cell extract (WCE) samples, representing input material, and of chromatin pellet samples.
At double‐strand breaks, H2AX can be ubiquitinated by several ubiquitin ligases including RNF168 and RNF2/BMI1 40, 43. Similar to HUWE1 depletion, knockdown of RNF168, RNF2, or BMI1 resulted in decreased γH2AX ubiquitination (Figs 7F and EV4), and γH2AX binding to FRA3B (Fig 7G) following aphidicolin treatment. Thus, efficient H2AX ubiquitination during the response to replication stress is achieved through the activity of multiple ubiquitin ligases.
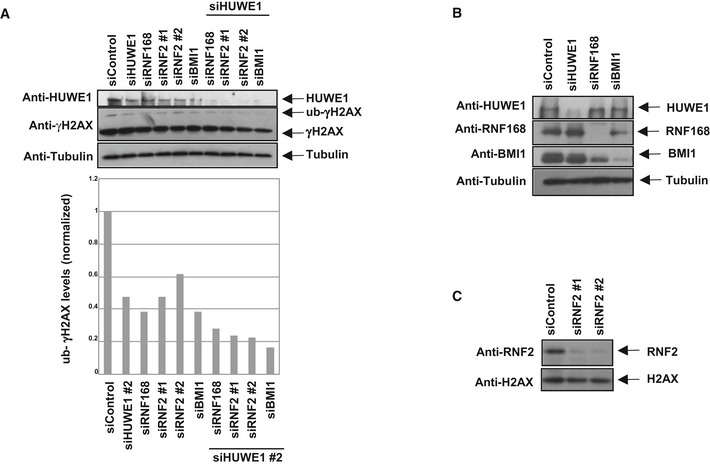
Figure EV4. Co‐depletion of HUWE1 and other H2AX ubiquitin ligases enhances the H2AX ubiquitination defect.
-
ARepresentative Western blot experiment in HeLa cells. A quantification of this experiment is also shown. This result was replicated in three independent experiments.
-
B, CWestern blots showing the efficient depletion of H2AX ubiquitin ligases.
We next wondered how H2AX ubiquitination by HUWE1 might promote replication fork stability. DNA fiber experiments indicated that knockdown of H2AX results in decreased replication tract length (Fig EV5). Indeed, it has been proposed that coating of damaged DNA by γH2AX promotes the assembly of repair complexes 44, 45. Among other repair factors, the recombination proteins BRCA1 and BRCA2 are known to promote restart of stalled replication forks 46, 47. Chromatin fractionation experiments showed reduced chromatin recruitment of BRCA1 and BRCA2 in HUWE1‐knockout cells following induction of replication stress by HU (Fig 7H). These results indicate that HUWE1‐mediated ubiquitination of H2AX promotes recruitment of repair proteins to restart stalled replication forks.
Figure EV5. H2AX knockdown reduces replication tract length.
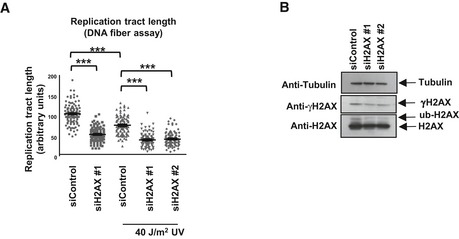
- DNA fiber assay using HeLa cells with H2AX knockdown. Cells were either treated with 40 J/m2 UV (analyzed 2 h later) or left untreated. The “n” numbers of samples are 101, 101, 106, 92, and 100, respectively (pooled from three independent experiments). P‐values (calculated using the t‐test two‐tailed, equal variance) are 1.19 × 10−40 (siControl vs. siH2AX, no damage samples); 5.6 × 10−15 (siControl, no damage vs. UV samples); 1.1 × 10−20 (siControl vs. siH2AX #1, UV samples); 9.01 × 10−32 (siControl vs. siH2AX #2, UV samples).
- Western blot of HeLa cells showing the impact of H2AX siRNA oligonucleotides.
Discussion
HUWE1 is required for genomic integrity and DNA damage tolerance
HUWE1 has been reported to affect a number of genome stability mechanisms. HUWE1 degrades the pre‐replication complex member Cdc6 after exposure to DNA damaging agents, possibly to suppress firing of late or dormant replication origins 31, 48. HUWE1 also controls base excision repair by initiating the degradation of DNA polymerase Polβ, and its knockdown was reported to result in slightly increased rates of repair of hydrogen peroxide‐induced DNA damage 33, 35. Finally, HUWE1 was shown to degrade BRCA1, and HUWE1 knockdown in breast cancer MCF10F cells resulted in increased resistance to double‐strand break‐inducing agents such as ionizing radiation 36. In contrast, our results clearly indicate that HUWE1 knockdown or knockout confers sensitivity to replication fork stalling agents HU and UV (Figs 1 and 2). Moreover, our DNA fiber experiments showed a reduction in progression of replication forks both with and without exogenous DNA damage treatment (Figs 1 and 2). Finally, analysis of cell cycle progression by co‐staining for BrdU incorporation and DNA content (Figs 1 and 2) identified a specific increase in the population of cells arrested in S‐phase. This profile is consistent with increased replication stress in HUWE1‐depleted cells. These results argue that HUWE1 plays an important role in the response to replication stalling, which is separate from the previously described activities in degrading DNA repair proteins. The reduced fiber tract length in HUWE1‐deficient cells would suggest that these cells spend a longer time in S‐phase. However, the number of cells with S‐phase DNA content is similar (but a larger proportion of those are BrdU‐negative). These findings may suggest an additional function of HUWE1 in regulating entry into S‐phase.
HUWE1 is a novel PCNA binding partner
Proliferating cell nuclear antigen is a homotrimeric ring‐shaped sliding clamp that plays a critical role in DNA replication and repair 4. Here, we report for the first time that HUWE1 associates with PCNA via the PIP‐box QPAVEAFF and is associated with replication forks. The co‐localization of PCNA with HUWE1 after DNA damage treatment may indicate that HUWE1 is specifically recruited to stalled forks, but it may also simply mark the sites of repair DNA synthesis. PIP‐box mutations abolished this localization and could not correct the replication stress phenotypes of HUWE1‐knockout cells, indicating that PCNA interaction is essential for this novel activity of HUWE1. HUWE1‐knockout cells also have a growth defect (not shown), again illustrating the broad, pleiotropic impact of HUWE1. While we could correct the HU sensitivity phenotype by expression of exogenous Myc‐HUWE1 (Fig 6), the growth defect was in fact not corrected—potentially indicating a role for transcriptional regulation of HUWE1 expression.
At stalled replication forks, PCNA mono‐ubiquitination results in the recruitment of specialized low‐fidelity TLS polymerases 8, 10. Because of our findings that the E3 ligase HUWE1 interacts with PCNA and that HUWE1 knockdown results in sensitivity to replication fork stalling agents HU and UV, we initially speculated that HUWE1 might mono‐ubiquitinate PCNA to promote DNA damage tolerance and lesion bypass. However, despite our intense efforts, we did not detect a reproducible reduction in PCNA mono‐ubiquitination in HUWE1‐knockout cells.
HUWE1 promotes H2AX modifications at stalled forks
The role of H2AX at sites of double‐strand breaks has been extensively studied 49. H2AX phosphorylation by ATM initiates the damage signaling process by recruiting MDC1, one of the earliest proteins in the signaling cascade. H2AX is also ubiquitinated, which promotes recruitment of repair proteins. Blocking H2AX mono‐ubiquitination also results in reduced γH2AX signal at double‐strand breaks, but how H2AX ubiquitination promotes its phosphorylation is still unclear 39, 40. It was recently shown that γH2AX also accumulates at stalled replication forks, in a process that depends on the ATR kinase, the counterpart of ATM for signaling replication stress 38, 50, 51, 52. The exact role of γH2AX at stalled forks is much less understood compared to its activity at double‐strand break sites. Interestingly, MDC1 was shown recently to be recruited to stalled forks, where it interacts with TopBP1, a known ATR activator, suggesting that γH2AX may be important for amplification of the checkpoint signal at stalled forks 53.
Surprisingly, HUWE1‐knockout cells had reduced γH2AX levels (Fig 7) even though they showed increased replication stress (Figs 1 and 2). This suggests that H2AX phosphorylation at stalled replication forks is impaired in the absence of HUWE1. HUWE1 was previously shown to ubiquitinate H2A during spermatogenesis 54, 55. We noticed that HUWE1‐knockout cells have reduced H2AX mono‐ubiquitination (Fig 7). Importantly, recombinant HUWE1 could mono‐ubiquitinate H2AX in vitro. These results indicate that HUWE1 is a novel ubiquitin ligase for H2AX. The reduced γH2AX levels in HUWE1‐knockout cells may thus be due to the ubiquitination defect, echoing the H2AX ubiquitination–phosphorylation link previously described at double‐strand breaks. Since we could correct the H2AX ubiquitination defect using wild‐type but not PIP mutant HUWE1 (Fig 7), and we observed no defect in H2AX ubiquitination in HUWE1‐knockout cells during G1 (Fig EV3F), we hypothesize that HUWE1 is responsible for H2AX ubiquitination at stalled forks. However, it is possible that HUWE1 also acts on H2AX under other conditions as well. Interestingly, a recent publication reported a role for HUWE1 in regulating levels of H2AX through multi‐ubiquitination‐mediated degradation under steady state vs. ionizing radiation exposure 56. We have not obtained any evidence for multi‐ubiquitination in our in vitro ubiquitination assay and did not observe differences in H2AX levels in our HUWE1‐knockout cells under the replication stress conditions that our studies focused on. Further complicating this picture, we found that multiple E3 ligases are involved in replication stress‐induced H2AX ubiquitination (Figs 7 and EV4). In line with this, we noticed that, in 293T cells, the γH2AX deficiency is a transient phenotype: after several passages, HUWE1 knockout cells tend to recover normal γH2AX levels (not shown). It is likely that backup pathways become activated to restore DNA damage signaling. Future studies aiming at understanding the complex interplay between the activities regulating the activity of H2AX in the response to DNA damage and replication stress are thus very important.
Replication stress is a major threat to genomic integrity, as evidenced by the hypersusceptibility to breakage of common fragile sites. Aphidicolin‐induced γH2AX binding to fragile sites is severely reduced by HUWE1 depletion (Fig 7), showing that HUWE1 is directly involved in protecting replication forks. This is further highlighted by our findings that HUWE1 promotes the recruitment of repair proteins such as BRCA1 and BRCA2 to chromatin under replication stress conditions (Fig 7H). Homologous recombination and other DNA repair mechanisms have been shown to participate in restarting stalled replication forks 3, 57, 58, 59. In particular, the involvement of BRCA1 and BRCA2 in promoting replication fork stability has been well documented 46, 47. Our results thus indicate a model wherein, following replication fork stalling, HUWE1 promotes ubiquitination and phosphorylation of H2AX, which in turn recruits repair complexes to repair and/or restart the damaged fork.
Materials and Methods
Cell culture and protein techniques
Human HeLa (ATCC# CCL2), 293T (ATCC# 3216), U2OS (ATCC# HTB‐96), MCF7 (ATCC# HTB‐22), and 8988T 60 cells were grown in DMEM (Lonza) supplemented with 15% fetal calf serum. For gene knockouts, the commercially available CRISPR/Cas9 KO plasmids were used (Santa Cruz Biotechnology sc‐404890 for HUWE1, sc‐406099 for Rad18). Single transfected cells were sorted into 96‐well plates, and resulting colonies were screened by Western blot. Gene knockout was confirmed by genomic sequencing. Cell extracts, co‐immunoprecipitation assays, chromatin fractionation, and GST‐pull‐down experiments were performed as previously described 60, 61, 62, 63. In vitro ubiquitination was done as previously described for HUWE1 13, using commercially available recombinant UBE1 E1 and UBCH7 E2 (Boston Biochem). Antibodies used in this study are as follows: HUWE1 (Novus NB100‐652 and Bethyl A300‐486A), PCNA (Abcam ab29), BrdU and IdU (BD 347580), CldU (Abcam ab6326), tubulin (Genetex gt114), Rad18 (Novus NB100‐61063), c‐Myc (Santa Cruz Biotechnology sc‐40), actin (Genetex gt5512), γH2AX (Santa Cruz Biotechnology sc‐101696), H2AX (Novus NB100‐638), p‐RPA (Bethyl A300‐245A), RNF168 (Millipore 06‐1130), RNF2 (Abcam ab101273), BMI1 (Abcam ab126783), BRCA1 (Santa Cruz Biotechnology sc‐642), BRCA2 (Calbiochem OP95), 53BP1 (Bethyl A300‐272A), p‐Chk1 (S317) (Cell Signaling Technology 2344S), p‐Chk2 (T68) (Cell Signaling Technology 2661S), TOPBP1 (Novus NB100‐217), and p‐p53 (S15) (Cell Signaling Technology 9284P).
Immunofluorescence
Cells were first incubated in pre‐extraction buffer (25 mM HEPES pH 7.5, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM glucose, 0.5% Triton X‐100) for 5 min on ice and then fixed in 4% formaldehyde for 10 min at room temperature. Cells were permeabilized with 0.2% Triton X‐100 for 5 min at room temperature. Slides were blocked with 2% BSA in PBS with 0.2% Tween and incubated with primary antibodies (diluted in blocking buffer) overnight at 4°C. Next, slides were washed three times in blocking buffer and incubated with secondary antibodies Alexa Fluor 568 and 488 (Invitrogen A11036 and A11001) for 1 h at room temperature.
Plasmids and siRNA
The cDNA for HUWE1C‐ter (3875‐end) was obtained by gene synthesis (GeneArt, Invitrogen). The cDNA for full‐length Myc‐tagged HUWE1 was purchased from Origene (Cat. No. RC215250). The cDNA for H2AX was obtained from PlasmID (clone ID HsCD00415019). For transient transfection, cDNA fragments were cloned in pCMV‐Myc (Clonetech) with a GFP tag. For bacteria expression, HUWE1C‐ter was cloned into pGEX‐6P1 (GE Healthcare). Stable cell lines were obtained by selection with 10 mg/ml geneticin. Plasmid and siRNA transfections were done using Lipofectamine LTX and Lipofectamine RNAiMAX (Invitrogen), respectively. For gene knockdown, cells were transfected with siRNA twice in consecutive days. The siRNA targeting sequences used are as follows:
siHUWE1#1: CATGAGACATCAGCCCACCCTTAAAA
siHUWE1#2: CACACCAGCAATGGCTGCCAGAATT (unless indicated otherwise in the figures, this was the targeting sequence used for HUWE1)
siHUWE1 5′UTR: AGCCTGACCTGAGTGGGTTAGTGAT
siRNF2 #1: GAGGCAATAACAGATGGCTTAGAAA
siRNF2 #2: TCCAGTAATGGATGGTGCTAGTGAA
siBMI1: AATGGAAGTGGACCATTCCTTCTCC
siRNF168: ACAGGACAGGTTATTGGCATTACAA
siH2AX #1: CGCGACAACAAGAAGACGCGAATCA
siH2AX #2: GCGACAACAAGAAGACGCGAATCAT
BrdU/PI bi‐dimensional flow cytometry
As previously described 61, 64, cells were incubated with 20 μM BrdU for 30 min and then harvested by trypsinization and fixed in 70% ethanol overnight. Cells were incubated in 0.1 M HCl/0.5% Triton X‐100 for 10 min on ice. Samples were spun down, resuspended in water, boiled for 10 min, and placed on ice for 10 min. Samples were then incubated with primary antibody (Anti‐BrdU, BD 347580) and subsequently with secondary antibody Alexa Fluor 488 (Invitrogen A11001) for 30 min each. Before flow cytometry analysis, cells were resuspended in PBS containing 20 μg/ml RNase and 5 μg/ml propidium iodide.
DNA fiber assay
Cells were incubated with 20 μM CIdU for 30 min. Cells were washed with PBS (and irradiated with 30 J/m2 UV‐B if indicated). Fresh media containing 20 μM IdU were added for another 30 min. Cells were lysed in 0.5% SDS, 200 mM Tris–HCl pH 7.4, and 50 mM EDTA. Slides were fixed with methanol:acetic acid (3:1) for 5 min, washed with 2.4 N HCl, and blocked in 5% BSA in PBS. Slides were incubated with primary antibodies (Abcam 6326 for detecting CIdU; BD 347580 for detecting IdU), washed three times with PBS, incubated with secondary antibodies Alexa Fluor 568 and 488 (Invitrogen A11031 and A21208), washed three times with PBS, and mounted.
Other functional assays
Clonogenic assays were performed as described 61, 64. The alkaline comet assay was performed using the CometAssay Kit (Trevigen 4250) according to manufacturer's instructions. Chromatin immunoprecipitation for FRA3B site was performed as previously described 62.
iPOND
Cells (approx. 108 for each condition) were incubated with 10 μM EdU (5‐ethynil‐2’‐deoxyuridine; Invitrogen) for 20 min, fixed in 1% formaldehyde for 25 min, and scraped in PBS with 0.01% Triton X‐100. Cells were then permeabilized in PBS with 0.25% Triton X‐100 for 20 min and incubated in click reaction buffer (10 mM sodium ascorbate, 2 mM CuSO4, 10 μM azide‐PEG11‐biotin, 0.01% Triton X‐100, 0.5% BSA, in PBS) for 90 min at room temperature. Next, cell pellets were resuspended in RIPA buffer, sonicated, and cleared by high‐speed centrifugation. Nascent DNA was captured by incubation of the cleared extracts with Streptavidin T1 Dynabeads (Invitrogen) for 60 min. The beads were then boiled for 30 min in Laemmli buffer containing 100 mM DTT to elute the proteins and reverse the cross‐links.
Statistical analyses
For all functional assays, the statistical analysis performed was the t‐test (two‐tailed, equal variance), using PRISM software. Statistical significance is indicated for each graph (ns = not significant, for P > 0.05; * for P < 0.05; ** for P < 0.01; *** for P < 0.001). The exact P‐values are indicated in the figure legends. All Western blots, co‐immunoprecipitations, and FACS analyses are representative of at least three independent experiments.
Author contributions
KNC, VAJS, RF, and GLM designed the experiments. KNC, CMN, DC, YIK, MRD‐D, VAJS, SD, and GLM performed the experiments. KNC, MRD‐D, VAJS, and GLM analyzed the data. KNC and GLM wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We would like to thank Sanziana Rotariu and Hejuan He for early contributions to this work, and Alan D'Andrea, James Broach, Stefan Jentsch, Wafik El‐Deiry, David Cortez, Wojciech Piwko, Kyungjae Myung, David Kozono, Sergei Grigoryev, Kristin Eckert, Thomas Spratt, Faoud Ishmael, Laura Carrel, and Gregory Yochum for materials, support, and advice. This work was supported by: NIH 1R01ES026184, Department of Defense CA140303, St. Baldrick Foundation, V Foundation, Concern Foundation, Gittlen Foundation (to GLM); American Cancer Society ACS‐IRG‐13‐043‐01 (to CMN); American Cancer Society ACS‐IRG‐57‐001‐53, Lung Cancer Colorado Fund, and United Against Lung Cancer (to SD).
EMBO Reports (2016) 17: 874–886
See also: KE Coleman & TT Huang (June 2016)
References
- 1. Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papamichos‐Chronakis M, Peterson CL (2013) Chromatin and the genome integrity network. Nat Rev Genet 14: 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaillard H, Garcia‐Muse T, Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15: 276–289 [DOI] [PubMed] [Google Scholar]
- 4. Moldovan GL, Pfander B, Jentsch S (2007) PCNA, the maestro of the replication fork. Cell 129: 665–679 [DOI] [PubMed] [Google Scholar]
- 5. Maga G, Hubscher U (2003) Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci 116: 3051–3060 [DOI] [PubMed] [Google Scholar]
- 6. Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73: 134–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6‐dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- 8. Guo C, Kosarek‐Stancel JN, Tang TS, Friedberg EC (2009) Y‐family DNA polymerases in mammalian cells. Cell Mol Life Sci 66: 2363–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang DJ, Cimprich KA (2009) DNA damage tolerance: when it's OK to make mistakes. Nat Chem Biol 5: 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, et al (2005) Ubiquitin‐binding domains in Y‐family polymerases regulate translesion synthesis. Science 310: 1821–1824 [DOI] [PubMed] [Google Scholar]
- 11. Bernassola F, Karin M, Ciechanover A, Melino G (2008) The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14: 10–21 [DOI] [PubMed] [Google Scholar]
- 12. Chen D, Brooks CL, Gu W (2006) ARF‐BP1 as a potential therapeutic target. Br J Cancer 94: 1555–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen D, Kon N, Li M, Zhang W, Qin J, Gu W (2005) ARF‐BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121: 1071–1083 [DOI] [PubMed] [Google Scholar]
- 14. Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al (2005) The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123: 409–421 [DOI] [PubMed] [Google Scholar]
- 15. Yoon SY, Lee Y, Kim JH, Chung AS, Joo JH, Kim CN, Kim NS, Choe IS, Kim JW (2005) Over‐expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem Biophys Res Commun 326: 7–17 [DOI] [PubMed] [Google Scholar]
- 16. Confalonieri S, Quarto M, Goisis G, Nuciforo P, Donzelli M, Jodice G, Pelosi G, Viale G, Pece S, Di Fiore PP (2009) Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 28: 2959–2968 [DOI] [PubMed] [Google Scholar]
- 17. Liu YX, Zhang SF, Ji YH, Guo SJ, Wang GF, Zhang GW (2012) Whole‐exome sequencing identifies mutated PCK2 and HUWE1 associated with carcinoma cell proliferation in a hepatocellular carcinoma patient. Oncol Lett 4: 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A (2008) The HECT‐domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N‐Myc oncoprotein. Nat Cell Biol 10: 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, et al (2013) A Network of Substrates of the E3 Ubiquitin Ligases MDM2 and HUWE1 Control Apoptosis Independently of p53. Sci Signal 6: ra32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L, Silvester J, Snow B, Harris IS, Sasaki M, Li WY, et al (2013) Mule/Huwe1/Arf‐BP1 suppresses Ras‐driven tumorigenesis by preventing c‐Myc/Miz1‐mediated down‐regulation of p21 and p15. Genes Dev 27: 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pervin S, Tran A, Tran L, Urman R, Braga M, Chaudhuri G, Singh R (2011) Reduced association of anti‐apoptotic protein Mcl‐1 with E3 ligase Mule increases the stability of Mcl‐1 in breast cancer cells. Br J Cancer 105: 428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qi CF, Zhang R, Sun J, Li Z, Shin DM, Wang H, Kovalchuk AL, Sakai T, Xiong H, Kon N, et al (2013) Homeostatic defects in B cells deficient in the E3 ubiquitin ligase ARF‐BP1 are restored by enhanced expression of MYC. Leuk Res 37: 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maltseva DV, Khaustova NA, Fedotov NN, Matveeva EO, Lebedev AE, Shkurnikov MU, Galatenko VV, Schumacher U, Tonevitsky AG (2013) High‐throughput identification of reference genes for research and clinical RT‐qPCR analysis of breast cancer samples. J Clin Bioinforma 3: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Groot RE, Ganji RS, Bernatik O, Lloyd‐Lewis B, Seipel K, Šedová K, Zdráhal Z, Dhople VM, Dale TC, Korswagen HC, et al (2014) Huwe1‐mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci Signal 7: ra26 [DOI] [PubMed] [Google Scholar]
- 25. Jang ER, Shi P, Bryant J, Chen J, Dukhande V, Gentry MS, Jang H, Jeoung M, Galperin E (2014) HUWE1 is a molecular link controlling RAF‐1 activity supported by the Shoc2 scaffold. Mol Cell Biol 34: 3579–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peter S, Bultinck J, Myant K, Jaenicke LA, Walz S, Müller J, Gmachl M, Treu M, Boehmelt G, Ade CP, et al (2014) Tumor cell‐specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol Med 6: 1525–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schaub FX, Cleveland JL (2014) Tipping the MYC‐MIZ1 balance: targeting the HUWE1 ubiquitin ligase selectively blocks MYC‐activated genes. EMBO Mol Med 6: 1509–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hao Z, Duncan GS, Su YW, Li WY, Silvester J, Hong C, You H, Brenner D, Gorrini C, Haight J, et al (2012) The E3 ubiquitin ligase Mule acts through the ATM‐p53 axis to maintain B lymphocyte homeostasis. J Exp Med 209: 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vaughan L, Tan CT, Chapman A, Nonaka D, Mack NA, Smith D, Booton R, Hurlstone AF, Malliri A (2015) HUWE1 ubiquitylates and degrades the RAC activator TIAM1 promoting cell‐cell adhesion disassembly, migration, and invasion. Cell Rep 10: 88–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brooks CL, Gu W (2006) p53 ubiquitination: Mdm2 and beyond. Mol Cell 21: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall JR, Kow E, Nevis KR, Lu CK, Luce KS, Zhong Q, Cook JG (2007) Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol Biol Cell 18: 3340–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herold S, Hock A, Herkert B, Berns K, Mullenders J, Beijersbergen R, Bernards R, Eilers M (2008) Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J 27: 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV, Kessler BM, Sharma RA, McKenna WG, Dianov GL (2009) Ubiquitin ligase ARF‐BP1/Mule modulates base excision repair. EMBO J 28: 3207–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khoronenkova SV, Dianov GL (2011) The emerging role of Mule and ARF in the regulation of base excision repair. FEBS Lett 585: 2831–2835 [DOI] [PubMed] [Google Scholar]
- 35. Markkanen E, van Loon B, Ferrari E, Parsons JL, Dianov GL, Hübscher U (2012) Regulation of oxidative DNA damage repair by DNA polymerase λ and MutYH by cross‐talk of phosphorylation and ubiquitination. Proc Natl Acad Sci USA 109: 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Lu G, Li L, Yi J, Yan K, Wang Y, Zhu B, Kuang J, Lin M, Zhang S, et al (2014) HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin‐proteasome pathway. Biochem Biophys Res Commun 444: 549–554 [DOI] [PubMed] [Google Scholar]
- 37. Dungrawala H, Cortez D (2015) Purification of proteins on newly synthesized DNA using iPOND. Methods Mol Biol 1228: 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu CY, Kang HY, Yang WL, Wu J, Jeong YS, Wang J, Chan CH, Lee SW, Zhang X, Lamothe B, et al (2011) Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J Biol Chem 286: 30806–30815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pan MR, Peng G, Hung WC, Lin SY (2011) Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem 286: 28599–28607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ozeri‐Galai E, Bester AC, Kerem B (2012) The complex basis underlying common fragile site instability in cancer. Trends Genet 28: 295–302 [DOI] [PubMed] [Google Scholar]
- 42. Lu X, Parvathaneni S, Hara T, Lal A, Sharma S (2013) Replication stress induces specific enrichment of RECQ1 at common fragile sites FRA3B and FRA16D. Mol Cancer 12: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK (2012) RNF168 ubiquitinates K13‐15 on H2A/H2AX to drive DNA damage signaling. Cell 150: 1182–1195 [DOI] [PubMed] [Google Scholar]
- 44. Hoeller D, Dikic I (2009) Targeting the ubiquitin system in cancer therapy. Nature 458: 438–444 [DOI] [PubMed] [Google Scholar]
- 45. Peterson CL, Cote J (2004) Cellular machineries for chromosomal DNA repair. Genes Dev 18: 602–616 [DOI] [PubMed] [Google Scholar]
- 46. Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman‐Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al (2014) BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun 5: 5496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall JR, Lee HO, Bunker BD, Dorn ES, Rogers GC, Duronio RJ, Cook JG (2008) Cdt1 and Cdc6 are destabilized by rereplication‐induced DNA damage. J Biol Chem 283: 25356–25363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR‐dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- 51. Ewald B, Sampath D, Plunkett W (2007) H2AX phosphorylation marks gemcitabine‐induced stalled replication forks and their collapse upon S‐phase checkpoint abrogation. Mol Cancer Ther 6: 1239–1248 [DOI] [PubMed] [Google Scholar]
- 52. Gagou ME, Zuazua‐Villar P, Meuth M (2010) Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 21: 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J, Gong Z, Chen J (2011) MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J Cell Biol 193: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu Z, Oughtred R, Wing SS (2005) Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol Cell Biol 25: 2819–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Z, Miao D, Xia Q, Hermo L, Wing SS (2007) Regulated expression of the ubiquitin protein ligase, E3(Histone)/LASU1/Mule/ARF‐BP1/HUWE1, during spermatogenesis. Dev Dyn 236: 2889–2898 [DOI] [PubMed] [Google Scholar]
- 56. Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H, et al (2015) ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient gammaH2AX foci formation. Cell Rep 13: 2728–2740 [DOI] [PubMed] [Google Scholar]
- 57. McGlynn P, Lloyd RG (2002) Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol 3: 859–870 [DOI] [PubMed] [Google Scholar]
- 58. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yeeles JT, Poli J, Marians KJ, Pasero P (2013) Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5: a012815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Connor KW, Dejsuphong D, Park E, Nicolae CM, Kimmelman AC, D'Andrea AD, Moldovan GL (2013) PARI overexpression promotes genomic instability and pancreatic tumorigenesis. Cancer Res 73: 2529–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nicolae CM, Aho ER, Vlahos AH, Choe KN, De S, Karras GI, Moldovan GL (2014) The ADP‐ribosyltransferase PARP10/ARTD10 interacts with proliferating cell nuclear antigen (PCNA) and is required for DNA damage tolerance. J Biol Chem 289: 13627–13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nicolae CM, Aho ER, Choe KN, Constantin D, Hu HJ, Lee D, Myung K, Moldovan GL (2015) A novel role for the mono‐ADP‐ribosyltransferase PARP14/ARTD8 in promoting homologous recombination and protecting against replication stress. Nucleic Acids Res 43: 3143–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D'Andrea AD (2012) Inhibition of homologous recombination by the PCNA‐interacting protein PARI. Mol Cell 45: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI (2013) Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet 9: e1003542 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File