

Abstract
The fibric acid derivative, fenofibrate (FF) has been used in the US since 1998 to manage patients with dyslipidemia. Typical changes in serum lipids as a result of FF treatment include clinically important mean reductions of serum triglycerides (TG) by a mean change of −93.7 mg/dL (−39.3%), increases of high density lipoprotein cholesterol (HDLC) by +5.5 mg/dL (+12.4%), and reductions in low density lipoprotein cholesterol (LDLC) by −17.9 mg/dL (−12.3%). The greatest reductions in serum TG are usually observed in subjects with elevated baseline TG including those with the metabolic syndrome (MetS). Although statins remain the mainstay of therapy for most dyslipidemic patients, their combined use with FF would be expected to address residual risk resulting from less than optimal TG and HDLC levels in such patients. Clinical trials examining the cardiovascular benefits of FF alone or combined with statins have produced mixed results. These observations underscore our lack of understanding of which patients may benefit from FF therapy and which do not. Although FF’s basic mechanism of action is known to involve PPAR-α agonist activity resulting in altered transcription of several genes, the actual genetic bases for variability in lipid response is poorly understood. Studies, such as our GOLDN study and others were designed to better understand the genetic determinants of variability in the response to FF treatment and lipid levels. As a result several important genetic determinants of lipid levels have been identified. For example, in the GOLDN study SNPs from different genes were significantly associated with baseline lipid levels before treatment (APOA5-rs662799, rs3135506; APOC3-rs5128, rs2854117, rs4520); APOA4-rs5104; PPARA-rs9626730, rs135543, rs11703495; LPL-rs1801177), after treatment PPARA-rs11708495; LPL-rs1801177, and appeared to modulate overall response to FF treatment (NOS3-rs1799983). In this article, we will review the literature leading up to the contemporary use of FF as an agent to manage patients with dyslipidemia and focus on emerging understanding of the genetic variability in response to FF treatment. On the basis of the available evidence, we conclude that FF is of benefit in the treatment of dyslipidemia, especially among those with MetS. However, more work is needed to specifically identify which individuals derive a benefit from FF administration in terms of clinical outcomes and which do not – particularly in the context of type 2 diabetes.
Keywords: Fenofibrate, clinical trials, lipids, metabolic syndrome, single nucleotide polymorphism, random coefficients model, multivariate analysis
1. BACKGROUND SETTING OF FENOFIBRATE’S USE
Fenofibrate (FF) has been known to positively affect dyslipidemic profiles of most patients by dramatically reducing plasma triglycerides (TG), raising high density lipoprotein cholesterol (HDLC) levels while generally reducing low density lipoprotein cholesterol (LDLC). With the LDLC identified as a primary target in the management of dyslipidemia and the volume of evidence based literature supporting the benefit and safety of HMG CoA reductase inhibitors (statins), the role of fibric acid derivatives and specifically FF has been relatively modest. The contemporary role of fibric acid derivatives and specifically FF, continues to be further refined as clinicians weigh the mixed outcomes from several large clinical trials [1, 2]. In the absence of changes to these guidelines, contemporary practice appears to follow the recommendations by the American Heart Association (AHA) and the National Heart Lung and Blood Institute (NHLBI) for metabolic syndrome (MetS) [3], which identify fibrates and niacin as two options to manage patients whose non-HDLC and/or low HDLC fail to reach targeted levels. The paucity of alternative therapeutic drug entities, which address both TG and HDLC (two MetS components) and non-HDLC targets, supports FF continued use for carefully selected patients.
The clinical diagnosis of MetS and therapeutic management of patients with this condition has been the subject of a special scientific statement by the AHA and NHLBI meeting [3], representing the most commonly followed guide within the US. The current definition of the criteria for the clinical diagnosis of MetS includes the presence of 3 or more of either elevated waist circumference, elevated triglycerides, reduced HDLC, elevated blood pressure, or elevated fasting glucose (see North et al. review paper in this issue for the definition of MetS and Kraja et al. review [4, 5]). In general, the clinical management of patients with the metabolic syndrome focuses on addressing these criteria through both lifestyle modifications and managing metabolic risk factors. Lifestyle changes which address physical inactivity, the atherogenic diet or abdominal obesity each have their positive effects on the metabolic risk factors including the lipid fractions (TG, HDL or non-HDL targets) as well as elevated blood pressure and serum glucose. Consequently, given that two of the 5 criteria are specifically targeted by fibric acid derivatives such as FF, it is natural that drug therapy with FF makes up one of the key therapeutic entities recommended for patients with this syndrome. Given that the prevalence of individuals classified as having MetS within the US is as high as 24–29% [6–8] and increasing and furthermore since obesity and T2DM and/or insulin resistance continues to rise, it is therefore logical that FF’s role be further defined as a logical consideration to address the dyslipidemia associated with each of these phenotypes.
MetS can significantly increase one’s chances for developing threatening diseases such as diabetes mellitus (T2DM) and coronary heart disease (CHD). Individuals with the MetS have 2–4 times greater risk than those who do not have MetS for CHD death [6–8]. Even dyslipidemia in the absence of MetS may qualify a patient for consideration of FF. However, specific recommendations by the National Cholesterol Education Program ATP III (NCEP) panel report [9] identified both FF and niacin as therapeutic options for patients not meeting their non-HDL goal. Non-HDL goals are defined as 30mg/dL greater than an individual’s LDLC goal which may be either 100, 130 or 160 mg/dL based on a count of risk factors (i.e. age, family history, HDLC, smoking status, and systolic blood pressure (SBP) level, 10-year Framingham, Risk, score (http://hp2010.nhlbihin.net/atpiii/calculator.asp) and presence of CHD or CHD risk equivalents (diabetes, stroke, aortic arch atheromatosis, peripheral vascular disease etc.) [9]. Treatment for elevated TG (> 200 mg/dL; normal < 150 mg/dL) based on the NCEP ATP III criteria were considered secondary targets, compared to reaching goals of LDLC, and normalizing HDLC was considered important after reaching goals for LDLC and non-HDLC. Hence, since FF is useful to lower TG and raise HDLC, then it remains an option to consider for those patients not achieving their non-HDL or HDLC goals.
This review is focused on FF role in MetS. Fenofibrate, earlier known also as p-(4-chlorobenzoyl)-phenoxy isobutyrate isopropyl ester, is useful for the treatment of adult patients with very high elevations of serum TG levels and/or high cholesterol levels. About half a century ago, the original fibrate, clofibrate, was developed as a fatty acid analog with clinical studies confirming a marked effect on fatty acids and TG’s metabolism [10, 11]. Later, fibrates were identified to modify the expression of several genes involved in lipoprotein and fatty acid metabolism [12–16]. Such a role was attributed to the fact that FF activated the peroxisome proliferator-activated receptor alpha (PPAR-α) [17].
The determinant role of PPAR-α in mediating many of the effects of fibrates was illustrated by gene knock-out mice with non-functional PPAR-α [18]. Edgar et al. [19] showed that fibrates modify the expression of genes implicated in lipoprotein and fatty acid metabolism via the PPAR-α in liver cells. In addition when FF were used were found to be present in both liver and kidney [20, 21].
Along with dietary advice, several classes of agents have been utilized to help manage patients with dyslipidemia. Of the available classes of agents, HMG CoA reductase inhibitors (statins) have emerged as the most effective, safe and clearly have an overall favorable impact the lipid levels contributing to atherosclerotic cardiovascular disease and cardiovascular outcomes. Given the independent association of elevated cardiovascular risk for those individuals with elevated triglycerides and/or low HDLC- even with modest LDLC levels, the addition of niacin, fish oils or fibrate products remain an option. As a class, fibric acid derivatives, the most common of which include clofibrate, gemfibrozil, bezafibrate, and fenofibrate, have enjoyed a long history of study including their independent or combined use in patients who have markedly high TG or low HDLC. In the US, clofibrate was approved in 1967 with gemfibrozil being introduced in the early 1980s. Although its use in Europe began in the early 1980’s, fenofibrate was approved for use in the US in February 1998 [21]. With the introduction of statins beginning with lovastatin in 1985 the use of fibrates proceeded at a modest pace. With the publications of the Helsinki Heart Study using gemfibrozil in 1987, and the Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT) in 1999, both demonstrating reductions in coronary heart disease events [22, 23], a renewed interest in fibrates ensued. As a chemical entity, FF is well absorbed, but requires conversion via esterases to its active metabolite, fenofibric acid before it will produce its intended pharmacological effect. In an effort to normalize the impact of variability in drug absorption as a result of ingestion in a fed or fasting state, Stamm and Pawan (1998) representing Fournier Laboratories filed Patent No. US6074670, continued to modify the crystalline form of the product. The dosage formulation changes ultimately resulted in an improved bioavailability and therefore necessitated a change in the magnitude of FF per dose from 100 and 300 mg tablets, to 67 mg and 200 mg daily tablets [24]. Guivarc’h et al. [25] and Sauron et al. [26] reported experimental evidence of improved bioavailability of fenofibric acid of drugs in the form of tablet as nanofibrate, in the form of microcoated FF, and in the form of micronized FF and that they eventually had a lack of food effect on their absorption.
Indeed, fenofibric acid delayed release capsules (135 and 45mg) are currently marketed (Trilipix®) by Abbott labs in addition to several contemporary formulations of FF available in the US which include; Tricor® (48 mg or 145 mg of FF tablets), Antara (43 mg or 130 mg of micronized FF), Lipofen® (50 or 150 mg of FF), and Triglide® (50 mg or 160 mg of FF) to mention a few (see Table 1).
Table 1.
Statins, Fenofibrate and a Fibric Acid Marketed in the US
| No | Statin | Brand Name | Manufacturer | Fenofibrate | Brand Name | Manufacturer |
|---|---|---|---|---|---|---|
| 1 | Rosuvastatin | Crestor | AstraZeneca | Tricor | Fenofibrate | Abbott Labs |
| 2 | Atorvastatin | Lipitor | Pfizer | Lipofen | Fenofibrate | Kowa Pharmaceuticals |
| 3 | Simvastatin | Zocor | Merck and Co | Lofibra | Fenofibrate | Teva |
| 4 | Pravastatin | Pravachol | Bristol-Meyers Squibb | Antara | Fenofibrate | Oscient Pharmaceuticals |
| 5 | Fluvastatin | Lescol | Novartis | |||
| 6 | Lovastatin | Mevacor | Merck and Co | Trilipix | Fibric acid | Abbott Labs |
Marketed in the US (Drugs.com, accessed March 21, 2010)
2. FENOFIBRATE MONOTHERAPY
Therapeutic lifestyle changes remain the first consideration before contemplating any drug therapy in the case of managing patients with dyslipidemia. (See details in the review of Djoussé et al. in this issue [27]). As a general rule pharmacotherapy follows lifestyle interventions, if lipid levels are beyond normal values and persist to be abnormal despite life style therapies (see §1). In reviewing nine guidelines for the treatment of patients with low HDLC, Devroey et al. [28] found the majority of them consider low HDLC as a marker of an increased risk for CHD, but only NCEP AT-PIII includes HDLC level in the calculation of coronary risk. Grundy et al. (2005) [3], specifically identifies reduced HDLC as a tertiary target but acknowledges there is not no specific goal to target increases in HDLC. Nonetheless, FF could be considered as an option for raising HDLC. FF has been demonstrated to have a somewhat variable effect raising HDLC in large clinical trials ranging from 1.5–7.4% [29]. For example, a study of 3,090 patients with a previous myocardial infarction or stable angina were treated with bezafibrate (a fibrate drug) 400 mg/day or a placebo and followed for 6.2 years. Bezafibrate raised HDLC 18% and reduced TG 21%. The frequency of the primary end point was 13.6% on bezafibrate versus 15.0% on placebo (p=0.26) [30]. In comparison, FF’s effects in lowering plasma TG levels have been quite variable and clearly depend on baseline levels before therapy. For instance, individuals receiving FF, with highest baseline TG and lowest HDLC seem to demonstrate the greatest benefit in terms of improving their lipid profiles compared to those with only modest elevations in baseline TG and near normal baseline HDLC values [1]. A review of 53 studies enrolling 16,802 subjects showed that FF lowered plasma TG on average by −36%, LDLC by −8%, and increased HDLC by +10% [31]. However, considerable inter-study variability in lipid response exists. In part this is due to the nature of subjects recruited for these studies (having different baseline lipid values) but also as a consequence of other factors which may include the length of study period, baseline lipid values and possibly the use of different formulations and conditions of administration (fasting versus fed). Consequently we conducted our own review of 15 clinical trials utilizing the TriCor® brand of FF which was the formulation used in the GOLDN study which will be discussed later in this article (see §7). In this review, we found the average change, associated with FF treatment was −93.7 mg/dl (−39.3%) for TG, −17.9 mg/dl (−12.3%) for LDLC and +5.5 mg/dl (+12.4%) for HDLC [32–44] (see Fig. 1). Although we recognize that clinicians usually use serum lipid values as their most trusted biomarker of cardiovascular risk and thus measure of drug effect, it is also widely recognized that markers of inflammation are key to the underlying pathophysiology of atherosclerosis. Fenofibrate’s effect of lowered levels of inflammation markers suggests it’s potential to address other biological processes involved in atherogenesis [45]. As a result, the beneficial effects of FF can be easily extended to subjects classified with MetS. Although not systematically evaluated, FF’s use in patients with T2DM seemed to produce mixed results. Specifically, the five year FIELD Study, which evaluated 9,795 subjects with type 2 diabetes who were 50–75 years of age, demonstrated a reduction of 24% of non-fatal myocardial infraction (MI) for those receiving FF-compared to placebo (p=0.01). However this, as well as a lower rate of progression to albuminuria (p=0.002), reduction in need for laser therapy for retinopathy (p=0.0003) and reduction in need for coronary revascularization (p=0.003) conflicted with a non-significant increase in coronary heart disease mortality (p=0.22) [46]. It is possible that such results on mortality were confounded by a larger number of patients allocated to placebo who received statins compared to those allocated to fenofibrate therapy. After four months of treatment LDLC decreased by 12%, TG by 29% and persisted at similar levels after 4 years of treatment. In contrast, HDLC increased by 5% and by the end of the clinical trial its increment leveled to 1% [46, 47]. It is not clear if the increase in mortality in the FIELD study was a result of unequal use of statins between the placebo and FF treated groups or simply the improvement on raised HDLC was too small to benefit from FF as monotherapy. As a result of the FIELD study, clinicians looked to the recently completed ACCORD trial to directly address the impact of adding FF or placebo to baseline statin use in T2DM patients (to be discussed later in §8) and would continue to wait until more still anticipating the role of FF in the treatment of CHD [40, 48–53].
Fig. 1.
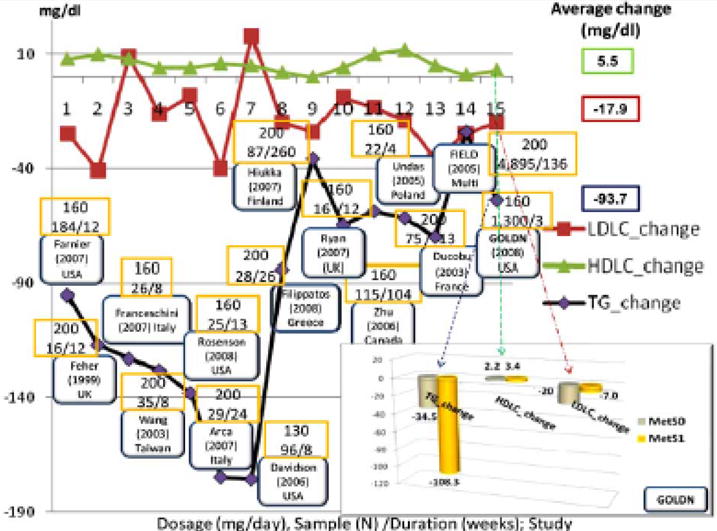
Several clinical trials studying the effects of TriCor® (Fenofibrate) on lipids profile, particularly on LDLC, HDLC and TG. MetS0-non-MetS subjects; MetS1- subjects classified with the NCEP ATPIII MetS. (These clinical trials were classified as part of TriCor® clinical trials based on the information collected from DrugLib.com). In the GOLDN study treatment with FF 160 mg daily for at least 3 weeks decreased plasma triglycerides (TG) from an average 145 mg/dL to 91 mg/dL (−37.2%). TG change was highest for the GOLDN subjects classified with MetS −108.3 mg/dL (−43.2%) than non-MetS subjects −34.5 mg/dL (−31.4%). As expected, the results of the GOLDN study agreed with other studies, where TG was the trait most altered by fenofibrate treatment. LDLC in general had a similar trend as TG, but with a smaller change, whereas the HDLC increased.
3. FENOFIBRATE AS COMBINATION THERAPY WITH STATINS
Statins (Table 1) work primarily by inhibiting HMG-CoA reductase, which is a rate-limiting step in endogenous synthesis of cholesterol. They are also known to induce the expression of LDL receptors in the liver, which in turn increase the catabolism of plasma LDLC and lower the plasma concentrations of cholesterol. A meta-analysis of 14 statin trials (n=90,056 randomized subjects) showed a 40% decrease in risk of coronary events over 5 years would be predicted by 29% decrease in LDL cholesterol levels [54]. These trials collectively demonstrated that there was no perceptible increase in risk for cancer or other substantial risk of life threatening complications associated with statin drug therapy. Statins are however, not without important toxicities. It is generally recommended that patients who are prescribed statins have a baseline and follow up measure of serum transaminases such as AST or ALT as a marker of potential liver damage. Furthermore myositis which can manifest as generalized muscle pain and/or weakness may be an indicator of muscle damage such that, if accompanied by an elevation in creatinine kinase, may represent myopathy. Factors associated with the risk of experiencing these side effects include dose of statin used, age of the patient, renal function or use of concomitant medications which inhibit the metabolism or hepatic uptake of the statins [55]. For a more comprehensive history of the data supporting the use of statin drug to manage patients with dyslipidemia, see Steinberg [56] and Davidson and Robinson [57] reviews.
Although the data supporting the use of statins to improve dyslipidemia profiles were accumulating, so too was experience in combination drug therapy with other lipid lowering agents including fibrates [58–62]. Similar precautions exist for combination therapy with statins and FF in that co-administration should proceed cautiously to the elderly based on expected impaired renal function [63]. Failure to identify indicators of potential myositis may lead to more serious side effects including a greater risk in developing rhabdomyolysis, which can be life threatening or lead to kidney failure. Generally speaking, FF is less likely to interfere with CYP based metabolism of most statins compared to gemfibrozil as fenofibrate is less potent of an inhibitor of those critical pathways of metabolism of statin [64]. Moreover, a survey of the reports from the United States Food and Drug Administration from January 1998–March 2002 showed that the combined use of gemfibrozil and a statin resulted in 590 cases of rhabdomyolysis compared with 16 cases with FF and statin therapy per million prescriptions [65]. Another study named SAFARI showed that a combination therapy of simvastatin 20 mg and FF 160 mg in patients (n=411) with combined hyperlipidemia resulted in additional improvement in all lipoprotein parameters measured compared with simvastatin 20 mg monotherapy (n=207) and was well tolerated [66]. Based on these results and that of several other studies, combination therapy of statins and fibrates, appear promising [67].
4. FENOFIBRATE’S EFFECT ON THE METABOLIC SYNDROME AND CARDIOVASCULAR OUTCOMES
Given that FF favorably affects serum TG and HDLC, its use in patients with MetS is logical [9]. Indeed, FF would be expected to address both of these two out of five components of the MetS either as monotherapy or more likely in combination therapy with a statin [68, 69]. Similarly, patients with T2DM often have elevated TG and low HDLC making this high CAD risk population ideal targets for fenofibrate therapy. Several studies have been done which explore FF’s role in such patients and provide an indication of its potential benefits. For example, the Diabetes Atherosclerosis Intervention Study (DIAS) reported a significant reduction in CAD progression in subjects with T2D. In a subpopulation of the overall study, 25 patients assigned to once daily 200 mg FF, compared to 21 patients assigned to placebo demonstrated changes in lipid particle size (larger plasma LDL size) and ratio of total cholesterol to HDLC was lower in the FF group [70]. Also Lemieux et al. [71] studying the effects of FF (n=36) or pravastatin (n=43), found a favorable effect of FF therapy on LDLC peak particle size compared with placebo therapy. Unfortunately there have been a limited number of outcome trials using FF as the study drug. Nonetheless, we should be able to glean some sense of the value of FF from analyzing the findings of outcome trials using other fibrates. For example, the VA-HIT Trial, with a total of 2,531 men with a history of CHD who had low HDLC levels (mean, 32 mg/dl) and low LDLC levels (mean, 111 mg/dl), assigned subjects to receive either 1,200 mg/day of gemfibrozil or placebo. After a 5 year follow-up, treatment with gemfibrozil resulted in a significant 22% reduction in the combined incidence of nonfatal myocardial infarction (MI) and CHD death. A considerable number of these individuals fit the profile of having MetS. Specifically, 50% of individuals had high TG (>150 mg/dl), 75% had low HDLC (< 35 mg/dl), 57% had hypertension, 25% had diabetes and 13% had impaired fasting glucose [72].
Over 80% of The FIELD Study subjects would be later classified with MetS. Analysis of their data showed that MetS components identified higher CVD risk in individuals with type 2 diabetes, so the absolute benefits of FF was likely to be greater when metabolic syndrome features were present [46]. Rosenson et al. [73] reported that 3 months of FF therapy (once daily 160mg/dl) improved lipoproteinemia, oxidative stress, and inflammatory response in subjects with hypertriglyceridemia and MetS.
Rationally, given that FF favorably generally has positive effects on an individual’s lipid profile including LDLC, TG and HDLC, one might expect that the establishment between FF drug therapy and risk for CHD would have been established long ago [74–78]. Moreover, individuals that have T2DM have as much as a two to four fold higher risk of myocardial infarction, stroke, and death from cardiovascular disease compared to those without T2DM. Consequently the opportunity to favorably impact outcomes should have been all that easier to establish. A sub study was conducted on patients with diabetes participating in the VA-HIT Trial (n=627) compared to those without diabetes (n=142). Following 5 years of treatment with gemfibrozil the investigators observed an average reduction of TG levels by 31% and a corresponding increase in HDLC of 6%. More importantly, these changes were associated with a, statistically significant 22% reduction in relative risk of CHD and a 24% reduction in combined outcome of death from CHD, nonfatal myocardial infarction, or stroke [72, 74]. Additional information on the effects of antihyperlipidemics on CHD is provided also in the review article of this issue by de Las Fuentes et al. [79].
5. FENOFIBRATE’S MECHANISM OF ACTION
Given that FF’s mechanism of action is through agonist activity of the PPAR-α gene, it is natural to expect it to have a positive impact on the key components making up MetS (TG and HDLC) and thereby reducing their contributions to macro or microvascular disease. As a PPAR-α agonist, FF in turn modulates expression of target genes implicated in lipoprotein and fatty acid metabolism (Supplemental Fig. 1). PPAR-α binds the retinoid X receptor, forming a complex or heterodimer that interacts with a DNA response element. This interaction modulates the expression of lipoprotein lipase and suppresses the production of lipoprotein lipase inhibitor apolipoprotein C-III. It has also the effect of reducing the availability of free fatty acids for TG synthesis via an increase in β- oxidation of fatty acids and the inhibition of de novo synthesis of fatty acids [80–82]. In addition, production of apolipoprotein B and VLDL is also reduced. Correspondingly, an increase in the synthesis of apolipoprotein A-I and apolipoprotein A-II, the main apolipoproteins in HDLC, together with reduced cholesteryl ester transfer protein–mediated transfer of lipid from HDLC to VLDL contribute to the observed increase in plasma levels of HDLC (Supplemental Fig. 1) [29].
The HDLC concentrations are also increased by fibrates secondary to the reduction in plasma TG concentrations from the increased activity of lipoprotein lipase [80]. Fibrates also appear to stimulate reverse cholesterol transport by modulating macrophage cholesterol efflux, and bile acid synthesis, ultimately enhancing HDL concentrations [83].
6. CLINICAL TRIALS ON FENOFIBRATE
Fig. (1) and Supplemental Table 1 summarize several clinical trial findings on FF effects on TG, HDLC and LDLC. Based on www.clinicaltrials.gov (accessed on March 20, 2010), which represents a service of National Institutes of Health (NIH) in the US for registered clinical trials, we found 99 clinical trials have/are studying FF, and of those 38 focus at FF in combination with statins. Among the major clinical trials in chronological order, are the World Health Organization (WHO) trial (n=7,194 men) which studied clofibrate’s effects on non-fatal MI. The authors reported an increase of 11% more deaths in the clofibrate treated group in a period of 13.2 years which was higher during the treatment period and its origins remain unexplained [84]. The Helsinki Heart Study (n=4,081 men) investigated gemfibrozil’s effects on non-fatal MI/CHD deaths. Tenkanen et al., [85] reported an 18 year follow up on mortality with finding on a decrease of relative risks (RR) 32% of CHD mortality, and for individuals in the original gemfibrozil group with both body mass index and TG level in the highest tertiles had a 71% lower RR of CHD mortality. The VA-HIT studied gemfibrozil’s (n=2,531 men with CHD) effect on non-fatal MI/CHD death. This study reported a 22% reduction on the endpoints [23]. The FIELD study aimed to assess the effect of FF on CVD in patients with diabetes (n=9,795 men and women). Although an improvement in lipids profile, in the original report the end points did not improve versus placebo. Recently, Burgess et al. [86] reported that among The FIELD Study subjects having silent MI, fenofibrate reduced subsequent clinical CVD events by 78%. Also based on The FIELD Study a significant 30% reduction in the need for laser therapy, less albuminuria progression and less non traumatic distal amputations were found, which suggest a benefit in preventing of diabetes [87]. Recently the ACCORD trial has started to analyze and summarize its results (see §8).
7. A CASE STUDY: FENOFIBRATE TREATMENT IN THE GOLDN STUDY AND GENETIC ASSOCIATIONS WITH METABOLIC SYNDROME FACTORS
The Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study is a clinical familial study with two treatment arms, one related to FF treatment and one to a fat-load meal (see Supplemental Methods and Supplemental Fig. (2) for details of the design). Our study focused on examining the effects of FF on the lipid profiles in relation to MetS. In addition, we tested association of candidate single nucleotide polymorphisms (SNPs) of 29 candidate genes with lipids and their change as result of FF treatment. The maximal sampled population consisted of 1,107 white subjects with no missing phenotypes on 10 traits studied (Table 2). Most of the subjects participated in all clinical visits as part of the GOLDN study which, in part recruited from participants of the NHLBI Family Heart Study [88, 89].
Table 2.
Summary of GOLDN Participants’ Characteristics
| Variables | Mean | Median | St. Dev. | Maximum | Minimum | N |
|---|---|---|---|---|---|---|
| Age | 48 | 47 | 16 | 87 | 18 | 1,292 |
| BMI v2 | 28.3 | 27.8 | 5.6 | 52.7 | 16.6 | 1,121 |
| BMI v4 | 28.7 | 28.2 | 5.6 | 52.5 | 16.4 | 792 |
| WAIST | 96.5 | 96 | 16.6 | 222 | 60 | 1,121 |
| WHR v2 | 0.9 | 0.98 | 0.1 | 1.92 | 0.48 | 1,121 |
| GLUC v0* | 98.7 | 95 | 19.4 | 332 | 62 | 1,291 |
| GLUC v2d1 | 101.5 | 98 | 18.7 | 298 | 68 | 1,119 |
| GLUC v4d1 | 99.4 | 96 | 19.1 | 295 | 73 | 860 |
| INS v2d1 | 13.7 | 12 | 8.2 | 82 | 2 | 1,117 |
| INS v4d1 | 13.5 | 11 | 8.4 | 92 | 3 | 857 |
| SBP v2 | 115.2 | 112 | 16.6 | 209 | 70 | 1,121 |
| DBP v2 | 68.1 | 67 | 9.3 | 108 | 41 | 1,121 |
| LDL v0 | 117.8 | 116 | 30.8 | 237 | 46 | 1,290 |
| LDL v1 | 124 | 121 | 31.9 | 245 | 36 | 805 |
| LDL v2d1 | 121.4 | 120 | 31.4 | 236 | 44 | 1,120 |
| LDL v3 | 104.6 | 102 | 31.2 | 216 | 35 | 793 |
| LDL v4d1 | 104.3 | 101 | 31.7 | 209 | 34 | 860 |
| HDL v0 | 48.8 | 47 | 13.3 | 105 | 21 | 1,291 |
| HDL v1 | 46.4 | 44 | 13.2 | 110 | 22 | 804 |
| HDL v2d1 | 47.1 | 45 | 13.1 | 110 | 22 | 1,120 |
| HDL v3 | 49.3 | 47 | 13.4 | 118 | 21 | 793 |
| HDL v4d1 | 49.4 | 47 | 13.6 | 114 | 22 | 860 |
| TG v0 | 133.4 | 108 | 93.9 | 1,380 | 27 | 1,291 |
| TG v1 | 145.5 | 119.5 | 109 | 1,320 | 24 | 804 |
| TG v2d1 | 138.9 | 110 | 115.8 | 2,260 | 23 | 1,119 |
| TG v3 | 91.9 | 75 | 57.1 | 454 | 20 | 788 |
| TG v4d1 | 89.9 | 75 | 54.6 | 455 | 16 | 860 |
v0- visit 0; v1-visit 1; v2- visit 2; v2d1- visit 2, first blood draw; v3- visit 3; v4d1- visit 4, first blood draw.
Previous publications have described a number of polymorphisms in the GOLDN study in relation to lipid levels and their change in response to FF treatment [90–94]. Smith et al. [93], for example, with the GOLDN study data reported 4 SNPs (APOA4_M35, APOC3_3U386, ABCA1_I27943, and LIPC_T224T) responsible for a large portion of the SNP-covariate interaction that predicted the TG change. Different from previous studies, we performed multivariate factor analysis and random coefficients growth curves on the repeated measures. In addition, we assessed the association among a larger set of candidate genes for lipids with two types of responses a) a metabolic syndrome lipid factor from factor analysis for each time point and b) the slopes that represent change before (visits 1 and 2) versus after FF treatment (visits 3 and 4) (see Supplemental Methods). Moreover, the best findings were tested for their association with single traits LDLC, HDLC, and TG.
FF 160 mg once daily for 3–4 weeks decreased plasma triglycerides (TG) from an average 145 mg/dL to 91 mg/dL (−37.2%). TG change, −108.3 mg/dL (−43.2%), was highest in GOLDN subjects classified with MetS based on the National Cholesterol Education Program [9] definition than non-MetS subjects −34.5 mg/dL (−31.4%). Because our association tests are for selected genes, thus we consider the Bonferroni p-value threshold of 0.0004 to secure a family wise of 5% false positives as conservative. Out of 115 SNPs of 29 candidate genes, eighteen selected candidate genes (ABCA1, APOA1, APOA2, APOA4, APOA5, APOB, APOC3, APOE, FABP2, LPL, LIPC, LIPE, MMTP, NOS3, PPARA, PPARG, PPARGC1A, and TCFL2) were classified as members of the MetS gene network [87]. Most of the remaining candidate genes selected were related in some way to lipid metabolism (ABCG5, ABCG8, ADIPOQ, FABP1, LIPG, LRP1, SCARB1, SCD4, WDTC1) and drug metabolism (CYP7A1, PDZK1). A number of SNPs on 11q23 from a cluster of genes APOA5 (rs662799, rs3135506), APOC3 (rs5128, rs2854117, rs4520), APOA4 (rs5104), and from genes PPARA (22q13.31, rs9626730, rs135543, rs11703495) and LPL (8p22, rs1801177) showed significant associations to lipids factors before FF treatment.
Among others, these findings overlap with a linkage region (11q23–11q24) for MetS lipids factor reported in the HyperGEN study [95]. The SNP rs3135506 of APOA5 gene located on 116167617 bps is a coding exon for APOA5 and a promoter for ZNF259 gene. The minor allele of rs3135506 (minor allele frequency (MAF) of about 6% in whites) has been repeatedly associated with increased plasma TG concentrations [96–99]. In our study the rare genotype rs3135506 (C/C) was associated with higher TG levels than the most frequent (G/G) genotype before and after FF treatment. The rs662799 association to lipids-insulin factor in our data although highly significant was problematic, because the rare homozygote genotype was represented by only 1 subject. Hence, a meta p-value of 2.7×10−10 for the same polymorphism reported by Willer et al. [99] in a sample of 8,684 subjects, and a meta p-value of 2.4×10−15 for a sample of 3,248 subjects in association with TG represent a supportive replication evidence.
Although APOA1, APOC3, APOA4, and APOA5 cluster together in 11q23, different haplotypes of APOA5 and APOC3 have been considered to be independently associated with TG levels [100]. The SNP rs4520 is part of a synonymous change in a coding region of APOC3 gene. The genotype (T/T) of less frequent allele (MAF 26.9%) was associated with higher levels of TG before FF treatment. After FF treatment the association of rs4520 was less evident. The SNP rs5104 represents a missense amino-acid change in the APOA4 gene, from the amino acid Asn to Ser when the less frequent genotype (G/G, MAF 13.4%) is present. The G/G genotype for this SNP in our study was associated with higher mean values of TG before the FF treatment.
After FF treatment, PPARA (rs11708495) and LPL (rs1801177) were ranked the top significant associations with factor scores of lipids MetS domain. This shows that PPARA and LPL gene polymorphism association with lipids remain significant even after FF treatment changes in the lipid levels. The association tests on random coefficients of growth curves identified NOS3 (rs1799983) as the top ranking candidate associated with the lipids change (see Supplemental Tables 2–7).
Diep et al. [101] and Goya et al. [102] reported that FF increases endothelial Nitric Oxide (NO) availability. They suggested FF reflect beneficial effects on vascular function by increasing NO levels. In our study, use of RCM model identified NOS3 (the endothelial nitric oxide synthase gene) as the most significant gene tested for the association with lipids factor scores with three significant SNPs (rs1799983, rs1800783, rs743507). Of these, rs1799983 was associated with the greatest change in the lipids MetS factor before and after FF treatment. Rs1799983 represents a missense G/T polymorphism with a MAF of 31.9%. Focusing in particular to the association of each trait (LDLC, HDLC, TG and INS) to the most significant SNP (rs1799983), we found the G/G genotype associated with higher values of TG and lower values of HDLC compared to T/T genotype.
The rs1799983 polymorphism in NOS3, is reported to be associated also with significant diastolic blood pressure response to hydrochlorothiazide [103], has been proposed that it may increase the risk of developing diabetic nephropathy [104] and it has been found to be associated to central pulse pressure in the Framingham Heart Study [105]. FF are suggested to have effects on vessel wall for cardiovascular protection by improving endothelial function, besides lipid-lowering effects [106]. Based on the R-squared values identified (Supplemental Tables 2–7), all our top findings had a modest contribution to lipid profiles change. It is not a surprise that identifying PPARA introns in association with changes in the MetS lipids domain coincides with what is already known about FF action. It is important to mention that genes associated with lipids and lipid changes were already identified as part of the MetS gene network [5]. Also PPARA, lipid genes on 11q23 and LPL are part of the PPARA signaling pathway (hsa03320) shown in Supplemental Fig. (1). LPL is also part of the glycerolipid metabolism pathway (hsa00561) and the Alzheimer’s disease (hsa05010) (see KEGG at www.genome.jp). Consequently FF treatment, which causes at least a lowering of TG and LDLC levels and an increment of HDLC, can contribute to ameliorate the progression of MetS.
The GOLDN study showed also that FF treatment substantially alters plasma lipids and can ameliorate MetS profiles (Fig. 1). Our study was based on selected MetS and lipid domains candidate genes. Although the GOLDN study is a large clinical trials using FF, the treatment period of 3–4 weeks does not provide the opportunity to judge the effects of long term FF treatment in terms of clinical outcomes such as morbidity and mortality. We reported in this review several other studies that underlined the benefits of FF use. Recently Blanco-Rivero et al. [107] showed that long term FF treatment in rats induces endothelial dysfunction. Future GWAs and gene expression studies in human and model animals quite possibly may discover a larger network of genes and we will have a better understanding of the systems affected by FF treatment.
8. NEW CHALLENGES
As this article focusing on MetS was being prepared, several new outcome based trials were published on the cardiovascular benefits of various medications in patients with diabetes. Because our article is part of this special issue on MetS, we will merely draw parallels to a few of these studies as they relate to the remaining challenges to our understanding FF’s clinical benefits. The Navigator study followed 9,306 patients with impaired glucose tolerance for 5 years to test the benefits of receiving once daily Valsartan versus placebo. Normally prescribed for patients with hypertension or heart failure, this angiotensin II receptor antagonist had no overall effect on cardiovascular mortality in spite of its benefits in reducing the risk of the incidence of diabetes [108]. So, even though we would expect a medication that corrects hypertension would improve the MetS outcomes, for the end points CHD events, there were no improvements compared to placebo.
Similar findings were observed in the recently completed ACCORD study. This study selected 5,518 patients with T2DM who were treated with open-label simvastatin to receive either masked FF or placebo. In general, the addition of FF to baseline simvastatin therapy failed to demonstrate an improvement in the prospectively selected primary or secondary cardiovascular endpoints which were prospectively monitored (fatal CV events, nonfatal MI, nonfatal stroke etc.). Only in pre-specified subgroup analyses was there a suggested heterogeneity in treatment effect according to sex, with a benefit for men (P=0.01 for interaction), and a possible interaction according to lipid subgroup, favoring those patients with both a high baseline TG level and a low baseline level of HLDC (P=0.057 for interaction) [2]. Despite these findings, and perhaps in light of them, our understanding of precisely which patients benefit from this drug and which do not, remain to be a challenge which may be enlightened by our improved understanding of FF genetic pathway(s) of action. Regardless, it is clear that FF remains an effective therapeutic option for managing the dyslipidemic phenotype relevant to those with MetS and likely select individuals with T2DM as well.
Supplementary Material
Acknowledgments
This work was partially supported by the NIH GOLDN grant R01HL09135701A1.
Footnotes
CONFLICTS OF INTERESTS
None to declare.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Web site along with the published article.
References
- 1.Scott R, Best J, Forder P, Taskinen MR, Simes J, Barter P, Keech A, FIELD Study Investigators Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study: baseline characteristics and short-term effects of fenofibrate. Cardiovasc Diabetol. 2005;4:13. doi: 10.1186/1475-2840-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010 doi: 10.1056/NEJMoa1001282. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F, American Heart Association; National Heart, Lung, and Blood Institute Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 4.Monda KL, North KE, Hunt SC, Rao DC, Province MA, Kraja AT. The genetics of obesity and metabolic syndrome. Endocr Metab Immune Disord Drug Targets. 2010;10:86–108. doi: 10.2174/187153010791213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraja AT, Province MA, Huang P, Jarvis JP, Rice T, Cheverud JM, Rao DC. Trends in metabolic syndrome and gene networks in human and rodent models. Endocr Metab Immune Disord Drug Targets. 2008;8:198–207. doi: 10.2174/187153008785700145. [DOI] [PubMed] [Google Scholar]
- 6.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 7.Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 8.Hoang KC, Ghandehari H, Lopez VA, Barboza MG, Wong ND. Global coronary heart disease risk assessment of individuals with the metabolic syndrome in the U.S. Diabetes Care. 2008;31:1405–1409. doi: 10.2337/dc07-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation and treatment of high blood cholesterol in adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 10.Oliver MF. Further observations on the effect of Atromid and of ethyl chlorophenoxyisobutyrate on serum lipid levels. J Atheroscler Res. 1963;3:427–444. doi: 10.1016/s0368-1319(63)80023-x. [DOI] [PubMed] [Google Scholar]
- 11.Berkowitz D. Effects of chlorophenoxyisobutyrate with and without androsterone on the serum lipids, fat tolerance and uric acid. Metabolism. 1965;14:966–975. doi: 10.1016/0026-0495(65)90112-5. [DOI] [PubMed] [Google Scholar]
- 12.Reddy JK, Goel SK, Nemali MR, Carrino JJ, Laffler TG, Reddy MK, Sperbeck SJ, Osumi T, Hashimoto T, Lalwani ND, Rao MS. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase:3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA. 1986;83:1747–1751. doi: 10.1073/pnas.83.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staels B, Van Tol A, Andreu T, Auwerx J. Fibrates influence the expression of genes involved in lipoprotein metabolism in a tissue selective manner. Arterioscler Thromb. 1992;12:286–294. doi: 10.1161/01.atv.12.3.286. [DOI] [PubMed] [Google Scholar]
- 14.Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, Auwerx J. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl Coenzyme A oxidase. J Clin Invest. 1995;95:705–712. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5% flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992;11:433–439. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferatoractivated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA. 1993;90:2160–2164. doi: 10.1073/pnas.90.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gebel T, Arand M, Oesch F. Induction of the peroxisome proliferator activated receptor by fenofibrate in rat liver. FEBS Lett. 1992;309:37–40. doi: 10.1016/0014-5793(92)80734-x. [DOI] [PubMed] [Google Scholar]
- 18.Lee SST, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez F. Targeted disruption of the a isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar AD, Tomkiewicz C, Costet P, Legendre C, Aggerbeck M, Bouguet J, Staels B, Guyomard C, Pineau T, Barouki R. Fenofibrate modifies transaminase gene expression via a peroxisome proliferator activated receptor -dependent pathway. Toxicology Lett. 1998;98:13–23. doi: 10.1016/s0378-4274(98)00042-3. [DOI] [PubMed] [Google Scholar]
- 20.Chapman MJ. Pharmacology of fenofibrate. Am J Med. 1987;83:21–25. doi: 10.1016/0002-9343(87)90867-9. [DOI] [PubMed] [Google Scholar]
- 21.Guay DRP. Micronized fenofibrate: a new fibric acid hypolipidemic agent. Ann Pharmacother. 1999;33:1083–1103. doi: 10.1345/aph.18432. [DOI] [PubMed] [Google Scholar]
- 22.Huttunen J, Manninen V, Manttari M, Koskinen P, Romo M, Tenkanen L, Heinonen O, Frick M. The Helsinki Heart Study: central findings and clinical implications. Ann Med. 1991;23:155–159. doi: 10.3109/07853899109148041. [DOI] [PubMed] [Google Scholar]
- 23.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 24.Stamm A, Pawan S. Patent US6074670 Fenofibrate pharmaceutical composition having high bioavailability and method for preparing it. 1998
- 25.Guivarc’h PH, Vachon MG, Fordyce D. A new fenofibrate formulation: results of six single-dose, clinical studies of bioavailability under fed and fasting conditions. Clin Ther. 2004;26:1456–1469. doi: 10.1016/j.clinthera.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Sauron R, Wilkins M, Jessent V, Dubois A, Maillot C, Weil A. Absence of a food effect with a 145 mg nanoparticle fenofibrate tablet formulation. Int J Clin Pharmacol Ther. 2006;44:64–70. doi: 10.5414/cpp44064. [DOI] [PubMed] [Google Scholar]
- 27.Djoussé L, Padilla H, Nelson TL, Gaziano JM, Mukamal KJ. Diet and metabolic syndrome. Endocr Metab Immune Disord Drug Targets. 2010;10:124–137. doi: 10.2174/187153010791213056. [DOI] [PubMed] [Google Scholar]
- 28.Devroey D, Vantomme K, Betz W, Vandevoorde J, Kartounian J. A review of the treatment guidelines on the management of low levels of high-density lipoprotein cholesterol. Cardiology. 2004;102:61–66. doi: 10.1159/000077906. [DOI] [PubMed] [Google Scholar]
- 29.Keating GM, Croom KF. Fenofibrate: a review of its use in primary dyslipidaemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs. 2007;67:121–153. doi: 10.2165/00003495-200767010-00013. [DOI] [PubMed] [Google Scholar]
- 30.Goldenberg I, Benderly M, Goldbourt U. BIP Study Group. Secondary prevention with bezafibrate therapy for the treatment of dyslipidemia: an extended follow-up of the BIP trial. J Am Coll Cardiol. 2008;51:459–465. doi: 10.1016/j.jacc.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 31.Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45:185–197. doi: 10.1016/j.jacc.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Farnier M, Roth E, Gil-Extremera B, Mendez GF, Macdonell G, Hamlin C, Perevozskaya I, Davies MJ, Kush D, Mitchel YB, Ezetimibe/Simvastatin + Fenofibrate Study Group Efficacy and safety of the coadministration of ezetimibe/simvastatin with fenofibrate in patients with mixed hyperlipidemia. Am Heart J. 2007;153:335.e1–e8. doi: 10.1016/j.ahj.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Feher MD, Caslake M, Foxton J, Cox A, Packard CJ. Atherogenic lipoprotein phenotype in type 2 diabetes: reversal with micronised fenofibrate. Diabetes Metab Res Rev. 1999;15:395–399. doi: 10.1002/(sici)1520-7560(199911/12)15:6<395::aid-dmrr65>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 34.Franceschini G, Calabresi L, Colombo C, Favari E, Bernini F, Sirtori CR. Effects of fenofibrate and simvastatin on HDL-related biomarkers in low-HDL patients. Atherosclerosis. 2007;195:385–391. doi: 10.1016/j.atherosclerosis.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Wang TD, Chen WJ, Lin JW, Cheng CC, Chen MF, Lee YT. Efficacy of fenofibrate and simvastatin on endothelial function and inflammatory markers in patients with combined hyperlipidemia: relations with baseline lipid profiles. Atherosclerosis. 2003;170:315–323. doi: 10.1016/s0021-9150(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 36.Rosenson RS. Fenofibrate reduces lipoprotein associated phospholipase A2 mass and oxidative lipids in hypertriglyceridemic subjects with the metabolic syndrome. Am Heart J. 2008;155:499.e9–e16. doi: 10.1016/j.ahj.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Arca M, Montali A, Pigna G, Antonini R, Antonini TM, Luigi P, Fraioli A, Mastrantoni M, Maddaloni M, Letizia C. Comparison of atorvastatin versus fenofibrate in reaching lipid targets and influencing biomarkers of endothelial damage in patients with familial combined hyperlipidemia. Metabolism. 2007;56:1534–1541. doi: 10.1016/j.metabol.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 38.Davidson MH, Bays HE, Stein E, Maki KC, Shalwitz RA, Doyle R, TRIMS Investigators Effects of fenofibrate on atherogenic dyslipidemia in hypertriglyceridemic subjects. Clin Cardiol. 2006;29:268–273. doi: 10.1002/clc.4960290609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filippatos TD, Liberopoulos EN, Kostapanos M, Gazi IF, Papavasiliou EC, Kiortsis DN, Tselepis AD, Elisaf MS. The effects of orlistat and fenofibrate, alone or in combination, on high-density lipoprotein subfractions and pre-beta1-HDL levels in obese patients with metabolic syndrome. Diabetes Obes Metab. 2008;10:476–483. doi: 10.1111/j.1463-1326.2007.00733.x. [DOI] [PubMed] [Google Scholar]
- 40.Hiukka A, Leinonen E, Jauhiainen M, Sundvall J, Ehnholm C, Keech AC, Taskinen MR. Long-term effects of fenofibrate on VLDL and HDL subspecies in participants with type 2 diabetes mellitus. Diabetologia. 2007;50:2067–2075. doi: 10.1007/s00125-007-0751-8. [DOI] [PubMed] [Google Scholar]
- 41.Ryan KE, McCance DR, Powell L, McMahon R, Trimble ER. Fenofibrate and pioglitazone improve endothelial function and reduce arterial stiffness in obese glucose tolerant men. Atherosclerosis. 2007;194:e123–e130. doi: 10.1016/j.atherosclerosis.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Zhu S, Su G, Meng QH. Inhibitory effects of micronized fenofibrate on carotid atherosclerosis in patients with essential hypertension. Clin Chem. 2006;52:2036–2042. doi: 10.1373/clinchem.2006.074724. [DOI] [PubMed] [Google Scholar]
- 43.Undas A, Celinska-Löwenhoff M, Domagala TB, Iwaniec T, Dropinski J, Löwenhoff T, Szczeklik A. Early antithrombotic and anti-inflammatory effects of simvastatin versus fenofibrate in patients with hypercholesterolemia. Thromb Haemost. 2005;94:193–199. doi: 10.1160/TH05-01-0067. [DOI] [PubMed] [Google Scholar]
- 44.Ducobu J, VanHaelst L, Salomon H. Comparison of micronized fenofibrate and pravastatin in patients with primary hyperlipidemia. J Cardiovasc Pharmacol. 2003;41:60–67. doi: 10.1097/00005344-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 45.Vega GL, Cater NB, Hadizadeh DR, 3rd, Meguro S, Grundy SM. Free fatty acid metabolism during fenofibrate treatment of the metabolic syndrome. Clin Pharmacol Ther. 2003;74:236–244. doi: 10.1016/S0009-9236(03)00170-X. [DOI] [PubMed] [Google Scholar]
- 46.Scott R, O’Brien R, Fulcher G, Pardy C, D’Emden M, Tse D, Taskinen MR, Ehnholm C, Keech A. Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care. 2009;32:493–498. doi: 10.2337/dc08-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, Forder P, Pillai A, Davis T, Glasziou P, Drury P, Kesäniemi YA, Sullivan D, Hunt D, Colman P, d’Emden M, Whiting M, Ehnholm C, Laakso M, FIELD study investigators Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomized controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 48.Vergès B. Fenofibrate therapy and cardiovascular protection in diabetes: recommendations after FIELD. Curr Opin Lipidol. 2006;17:653–658. doi: 10.1097/01.mol.0000252612.21602.e3. [DOI] [PubMed] [Google Scholar]
- 49.Backes JM, Gibson CA, Ruisinger JF, Moriarty PM. Fibrates: what have we learned in the past 40 years? Pharmacotherapy. 2007;27:412–424. doi: 10.1592/phco.27.3.412. [DOI] [PubMed] [Google Scholar]
- 50.Balfour JA, McTavish D, Heel RC. Fenofibrate, a review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidemia. Drugs. 1990;40:260–290. doi: 10.2165/00003495-199040020-00007. [DOI] [PubMed] [Google Scholar]
- 51.Haffner SM, Ashraf T. Predicting risk reduction of coronary disease in patients who are glucose intolerant: a comparison of treatment with fenofibrate and other lipid-modifying agents. Manag Care Interface. 2000;13:52–58. [PubMed] [Google Scholar]
- 52.UK HDL-C Consensus Group. Role of fibrates in reducing coronary risk: a UK consensus. Curr Med Res Opin. 2004;20:241–247. doi: 10.1185/030079903125002892. [DOI] [PubMed] [Google Scholar]
- 53.Evans M, Roberts A, Davies S, Rees A. Medical lipid-regulating therapy: current evidence, ongoing trials and future developments. Drugs. 2004;64:1181–1196. doi: 10.2165/00003495-200464110-00003. [DOI] [PubMed] [Google Scholar]
- 54.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, Cholesterol Treatment Trialists’ (CTT) Collaborators Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 55.McKenney JM, Davidson MH, Jacobson TA, Guyton JR, National Lipid Association Statin Safety Task Force Liver Expert Panel Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97(8A):89C–94C. doi: 10.1016/j.amjcard.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 56.Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J Lipid Res. 2006;47:1339–1351. doi: 10.1194/jlr.R600009-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Davidson MH, Robinson JG. Lipid-lowering effects of statins: a comparative review. Expert Opin Pharmacother. 2006;7:1701–1714. doi: 10.1517/14656566.7.13.1701. [DOI] [PubMed] [Google Scholar]
- 58.Wang TD, Chen WJ, Lin JW, Cheng CC, Chen MF, Lee YT. Efficacy of fenofibrate and simvastatin on endothelial function and inflammatory markers in patients with combined hyperlipidemia: relations with baseline lipid profiles. Atherosclerosis. 2003;170:315–323. doi: 10.1016/s0021-9150(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 59.Derosa G, Cicero AE, Bertone G, Piccinni MN, Ciccarelli L, Roggeri DE. Comparison of fluvastatin + fenofibrate combination therapy and fluvastatin monotherapy in the treatment of combined hyperlipidemia, type 2 diabetes mellitus, and coronary heart disease: a 12-month, randomized, double-blind, controlled trial. Clin Ther. 2004;26:1599–1607. doi: 10.1016/j.clinthera.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 60.Farnier M, Freeman MW, Macdonell G, Perevozskaya I, Davies MJ, Mitchel YB, Gumbiner B, Ezetimibe Study Group Efficacy and safety of the coadministration of ezetimibe with fenofibrate in patients with mixed hyperlipidaemia. Eur Heart J. 2005;26:897–905. doi: 10.1093/eurheartj/ehi231. [DOI] [PubMed] [Google Scholar]
- 61.Jones PH, Bays HE, Davidson MH, Kelly MT, Buttler SM, Setze CM, Sleep DJ, Stolzenbach JC. Evaluation of a new formulation of fenofibric acid, ABT-335, co-administered with statins : study design and rationale of a phase III clinical programme. Clin Drug Investig. 2008;28:625–634. doi: 10.2165/00044011-200828100-00003. [DOI] [PubMed] [Google Scholar]
- 62.Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d’Emden MC, Crimet DC, O’Connell RL, Colman PG, FIELD study investigators (2007) Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomized controlled trial. Lancet. 370:1687–1697. doi: 10.1016/S0140-6736(07)61607-9. 2007. [DOI] [PubMed] [Google Scholar]
- 63.Tomlinson B, Chan P, Lan W. How well tolerated are lipid-lowering drugs? Drugs Aging. 2001;18:665–683. doi: 10.2165/00002512-200118090-00003. [DOI] [PubMed] [Google Scholar]
- 64.Bellosta S, Paoletti R, Corsini A. Safety of statins: Focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109(23 Suppl 1):III50–III57. doi: 10.1161/01.CIR.0000131519.15067.1f. [DOI] [PubMed] [Google Scholar]
- 65.Jones PH, Davidson MH. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am J Cardiol. 2005;95:120–122. doi: 10.1016/j.amjcard.2004.08.076. [DOI] [PubMed] [Google Scholar]
- 66.Grundy SM, Vega GL, Yuan Z, Battisti WP, Brady WE, Palmisano J. Effectiveness and tolerability of simvastatin plus fenofibrate for combined hyperlipidemia (the SAFARI trial) Am J Cardiol. 2005;95:462–468. doi: 10.1016/j.amjcard.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 67.Fiévet C, Staels B. Combination therapy of statins and fibrates in the management of cardiovascular risk. Curr Opin Lipidol. 2009;20:505–511. doi: 10.1097/MOL.0b013e328332e9ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davidson MH. Statin/fibrate combination in patients with metabolic syndrome or diabetes: evaluating the risks of pharmacokinetic drug interactions. Expert Opin Drug Saf. 2006;5:145–156. doi: 10.1517/14740338.5.1.145. [DOI] [PubMed] [Google Scholar]
- 69.Gil-Extremera B, Mendez G, Zakson M, Meehan A, Shah A, Lin J, Mitchel Y. Efficacy and safety of ezetimibe/simvastatin co- administered with fenofibrate in mixed hyperlipidemic patients with metabolic syndrome. Metab Syndr Relat Disord. 2007;5:305–314. doi: 10.1089/met.2007.0011. [DOI] [PubMed] [Google Scholar]
- 70.Vakkilainen J, Steiner G, Ansquer JC, Perttunen-Nio H, Taskinen MR. Fenofibrate lowers plasma triglycerides and increases LDL particle diameter in subjects with type 2 diabetes. Diabetes Care. 2002;25:627–628. doi: 10.2337/diacare.25.3.627. [DOI] [PubMed] [Google Scholar]
- 71.Lemieux I, Laperrière L, Dzavik V, Tremblay G, Bourgeois J, Després JP. A 16-week fenofibrate treatment increases LDL particle size in type IIA dyslipidemic patients. Atherosclerosis. 2002;162:363–371. doi: 10.1016/s0021-9150(01)00711-0. [DOI] [PubMed] [Google Scholar]
- 72.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J, for The Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 73.Rosenson RS, Wolff DA, Huskin AL, Helenowski IB, Rademaker AW. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes Care. 2007;30:1945–1951. doi: 10.2337/dc07-0015. [DOI] [PubMed] [Google Scholar]
- 74.Rubins HB, Robins SJ, Collins D, Nelson DB, Elam MB, Schaefer EJ, Faas FH, Anderson JW, for the VA-HIT Study Group Diabetes, Plasma Insulin, and Cardiovascular Disease: Subgroup Analysis From the Department of Veterans Affairs High-Density Lipoprotein Intervention Trial (VA-HIT) Arch Intern Med. 2002;162:2597–2604. doi: 10.1001/archinte.162.22.2597. [DOI] [PubMed] [Google Scholar]
- 75.Williams RR, Skolnick M, Carmelli D, Maness AT, Hunt SC, Hasstedt S, Reiber GE, Jones RK. Utah pedigree studies: design and preliminary data for premature male CHD deaths. Prog Clin Biol Res. 1979;32:711–729. [PubMed] [Google Scholar]
- 76.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 77.Tuomilehto J. Reducing coronary heart disease associated with type 2 diabetes: lifestyle intervention and treatment of dyslipidaemia. Diabetes Res Clin Pract. 2003;61:S27–34. doi: 10.1016/s0168-8227(03)00125-6. [DOI] [PubMed] [Google Scholar]
- 78.Wierzbicki AS. Fibrates in the treatment of cardiovascular risk and atherogenic dyslipidaemia. Curr Opin Cardiol. 2009;24:372–379. doi: 10.1097/HCO.0b013e32832c0b3d. [DOI] [PubMed] [Google Scholar]
- 79.de Las Fuentes L, de Simone G, Arnett DK, Dávila-Román VG. Molecular Determinants of the Cardiometabolic Phenotype. Endocr Metab Immune Disord Drug Targets. 2010;10:109–123. doi: 10.2174/187153010791213119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barbier O, Torra IP, Duguay Y, Blanquart C, Fruchart JC, Glineur C, Staels B. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:717–726. doi: 10.1161/01.atv.0000015598.86369.04. [DOI] [PubMed] [Google Scholar]
- 81.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 83.Grundy SM, Vega GL. Fibric acids: effects on lipids and lipoprotein metabolism. Am J Med. 1987;83:9–20. doi: 10.1016/0002-9343(87)90866-7. [DOI] [PubMed] [Google Scholar]
- 84.WHO cooperative trial on primary prevention of ischaemic heart disease with clofibrate to lower serum cholesterol: final mortality follow-up. Report of the Committee of Principal Investigators. Lancet. 1984;2:600–604. [PubMed] [Google Scholar]
- 85.Tenkanen L, Mänttäri M, Kovanen PT, Virkkunen H, Manninen V. Gemfibrozil in the treatment of dyslipidemia: an 18-year mortality follow-up of the Helsinki Heart Study. Arch Intern Med. 2006;166:743–748. doi: 10.1001/archinte.166.7.743. [DOI] [PubMed] [Google Scholar]
- 86.Burgess DC, Hunt D, Li L, Zannino D, Williamson E, Davis TM, Laakso M, Kesäniemi YA, Zhang J, Sy RW, Lehto S, Mann S, Keech AC. Incidence and predictors of silent myocardial infarction in type 2 diabetes and the effect of fenofibrate: an analysis from the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Eur Heart J. 2010;31:92–99. doi: 10.1093/eurheartj/ehp377. [DOI] [PubMed] [Google Scholar]
- 87.Ansquer JC, Foucher C, Aubonnet P, Le Malicot K. Fibrates and microvascular complications in diabetes–insight from the FIELD study. Curr Pharm Des. 2009;15:537–552. doi: 10.2174/138161209787315701. [DOI] [PubMed] [Google Scholar]
- 88.Higgins M, Province M, Heiss G, Eckfeldt J, Ellison RC, Folsom AR, Rao DC, Sprafka JM, Williams R. NHLBI Family Heart Study: objectives and design. Am J Epidemiol. 1996;143:1219–1228. doi: 10.1093/oxfordjournals.aje.a008709. [DOI] [PubMed] [Google Scholar]
- 89.Arnett DK, Miller MB, Coon H, Ellison RC, North KE, Province M, Leppert M, Eckfeldt JH. Genome-wide linkage analysis replicates susceptibility locus for fasting plasma triglycerides: NHLBI Family Heart Study. Hum Genet. 2004;115:468–474. doi: 10.1007/s00439-004-1182-y. [DOI] [PubMed] [Google Scholar]
- 90.Rasmussen-Torvik LJ, Pankow JS, Peacock JM, Borecki IB, Hixson JE, Tsai MY, Kabagambe EK, Arnett DK. Suggestion for linkage of chromosome 1p35.2 and 3q28 to plasma adiponectin concentrations in the GOLDN Study. BMC Med Genet. 2009;10:39. doi: 10.1186/1471-2350-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y, Ordovas JM, Gao G, Province M, Straka RJ, Tsai MY, Lai CQ, Zhang K, Borecki I, Hixson JE, Allison DB, Arnett DK. The SCARB1 gene is associated with lipid response to dietary and pharmacological interventions. J Hum Genet. 2008;53:709–717. doi: 10.1007/s10038-008-0302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shen J, Arnett DK, Parnell LD, Peacock JM, Lai CQ, Hixson JE, Tsai MY, Province MA, Straka RJ, Ordovas JM. Association of common C-reactive protein (CRP) gene polymorphisms with baseline plasma CRP levels and fenofibrate response: the GOLDN study. Diabetes Care. 2008;31:910–915. doi: 10.2337/dc07-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith JA, Arnett DK, Kelly RJ, Ordovas JM, Sun YV, Hopkins PN, Hixson JE, Straka RJ, Peacock JM, Kardia SL. The genetic architecture of fasting plasma triglyceride response to fenofibrate treatment. Eur J Hum Genet. 2008;16:603–613. doi: 10.1038/sj.ejhg.5202003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lai CQ, Arnett DK, Corella D, Straka RJ, Tsai MY, Peacock JM, Adiconis X, Parnell LD, Hixson JE, Province MA, Ordovas JM. Fenofibrate effect on triglyceride and postprandial response of apolipoprotein A5 variants: the GOLDN study. Arterioscler Thromb Vasc Biol. 2007;27:1417–1425. doi: 10.1161/ATVBAHA.107.140103. [DOI] [PubMed] [Google Scholar]
- 95.Kraja AT, Hunt SC, Pankow JS, Myers RH, Heiss G, Lewis CE, Rao DC, Province MA. Quantitative trait loci for metabolic syndrome in the hypertension genetic epidemiology network study. Obes Res. 2005;13:1885–1890. doi: 10.1038/oby.2005.231. [DOI] [PubMed] [Google Scholar]
- 96.Ahituv N, Akiyama J, Chapman-Helleboid A, Fruchart J, Pennacchio LA. In vivo characterization of human APOA5 haplotypes. Genomics. 2007;90:674–679. doi: 10.1016/j.ygeno.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 97.Lai CQ, Demissie S, Cupples LA, Zhu Y, Adiconis X, Parnell LD, Corella D, Ordovas JM. Influence of the APOA5 locus on plasma triglyceride, lipoprotein subclasses, and CVD risk in the Framingham Heart Study. J Lipid Res. 2004;45:2096–2105. doi: 10.1194/jlr.M400192-JLR200. [DOI] [PubMed] [Google Scholar]
- 98.Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 99.Qi L, Liu S, Rifai N, Hunter D, Hu FB. Associations of the apolipoprotein A1/C3/A4/A5 gene cluster with triglyceride and HDL cholesterol levels in women with type 2 diabetes. Atherosclerosis. 2007;192:204–210. doi: 10.1016/j.atherosclerosis.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 100.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM, Strait J, Duren WL, Maschio A, Busonero F, Mulas A, Albai G, Swift AJ, Morken MA, Narisu N, Bennett D, Parish S, Shen H, Galan P, Meneton P, Hercberg S, Zelenika D, Chen WM, Li Y, Scott LJ, Scheet PA, Sundvall J, Watanabe RM, Nagaraja R, Ebrahim S, Lawlor DA, Ben-Shlomo Y, Davey-Smith G, Shuldiner AR, Collins R, Bergman RN, Uda M, Tuomilehto J, Cao A, Collins FS, Lakatta E, Lathrop GM, Boehnke M, Schlessinger D, Mohlke KL, Abecasis GR. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diep QN, Benkirane K, Amiri F, Cohn JS, Endemann D, Schiffrin EL. PPAR alpha activator fenofibrate inhibits myocardial inflammation and fibrosis in angiotensin II-infused rats. J Mol Cell Cardiol. 2004;36:295–304. doi: 10.1016/j.yjmcc.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Goya K, Sumitani S, Xu X, Kitamura T, Yamamoto H, Kurebayashi S, Saito H, Kouhara H, Kasayama S, Kawase I. Peroxisome proliferator-activated receptor alpha agonists increase nitric oxide synthase expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:658–663. doi: 10.1161/01.ATV.0000118682.58708.78. [DOI] [PubMed] [Google Scholar]
- 103.Maitland-van der Zee AH, Turner ST, Schwartz GL, Chapman AB, Klungel OH, Boerwinkle E. A multilocus approach to the antihypertensive pharmacogenetics of hydrochlorothiazide. Pharmacogenet Genomics. 2005;15:287–293. doi: 10.1097/01213011-200505000-00003. [DOI] [PubMed] [Google Scholar]
- 104.Möllsten A, Wessman M, Svensson M, Forsblom C, Parkkonen M, Brismar K, Groop PH, Dahlquist G. Glu298Asp and NOS4ab polymorphisms in diabetic nephropathy. Ann Med. 2006;38:522–528. doi: 10.1080/07853890600969213. [DOI] [PubMed] [Google Scholar]
- 105.Mitchell GF, Guo CY, Kathiresan S, Vasan RS, Larson MG, Vita JA, Keyes MJ, Vyas M, Newton-Cheh C, Musone SL, Camargo AL, Drake JA, Levy D, O’Donnell CJ, Hirschhorn JN, Benjamin EJ. Vascular stiffness and genetic variation at the endothelial nitric oxide synthase locus: the Framingham Heart study. Hypertension. 2007;49:1285–1290. doi: 10.1161/HYPERTENSIONAHA.106.085266. [DOI] [PubMed] [Google Scholar]
- 106.Otsuki M, Goya K, Kasayama S. Vascular endothelium as a target of beraprost sodium and fenofibrate for antiatherosclerotic therapy in type 2 diabetes mellitus. Vasc Health Risk Manag. 2005;1:209–215. [PMC free article] [PubMed] [Google Scholar]
- 107.Blanco-Rivero J, Márquez-Rodas I, Xavier FE, Aras-López R, Arroyo-Villa I, Ferrer M, Balfagón G. Long-term fenofibrate treatment impairs endothelium-dependent dilation to acetylcholine by altering the cyclooxygenase pathway. Cardiovasc Res. 2007;75:398–407. doi: 10.1016/j.cardiores.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 108.The NAVIGATOR Study Group. Effect of Valsartan on the Incidence of Diabetes and Cardiovascular Events. N Engl J Med. 2010 doi: 10.1056/NEJMoa1001121. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 109.Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53:983–997. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.