

ABSTRACT
Pyrazinamide (PZA) is a first-line tuberculosis (TB) drug that has been in clinical use for 60 years yet still has an unresolved mechanism of action. Based upon the observation that the minimum concentration of PZA required to inhibit the growth of Mycobacterium tuberculosis is approximately 1,000-fold higher than that of other first-line drugs, we hypothesized that M. tuberculosis expresses factors that mediate intrinsic resistance to PZA. To identify genes associated with intrinsic PZA resistance, a library of transposon-mutagenized Mycobacterium bovis BCG strains was screened for strains showing hypersusceptibility to the active form of PZA, pyrazinoic acid (POA). Disruption of the long-chain fatty acyl coenzyme A (CoA) ligase FadD2 enhanced POA susceptibility by 16-fold on agar medium, and the wild-type level of susceptibility was restored upon expression of fadD2 from an integrating mycobacterial vector. Consistent with the recent observation that POA perturbs mycobacterial CoA metabolism, the fadD2 mutant strain was more vulnerable to POA-mediated CoA depletion than the wild-type strain. Ectopic expression of the M. tuberculosis pyrazinamidase PncA, necessary for conversion of PZA to POA, in the fadD2 transposon insertion mutant conferred at least a 16-fold increase in PZA susceptibility under active growth conditions in liquid culture at neutral pH. Importantly, deletion of fadD2 in M. tuberculosis strain H37Rv also resulted in enhanced susceptibility to POA. These results indicate that FadD2 is associated with intrinsic PZA and POA resistance and provide a proof of concept for the target-based potentiation of PZA activity in M. tuberculosis.
KEYWORDS: Mycobacterium tuberculosis, antimicrobial agents, coenzyme A, fatty acids, metabolism, pyrazinamide
INTRODUCTION
The first-line tuberculosis (TB) drug pyrazinamide (PZA) was discovered over 60 years ago and remains an integral component of chemotherapeutic regimens for TB patients. PZA is extremely effective at sterilizing Mycobacterium tuberculosis in vivo, as evidenced by its ability to reduce the overall duration of treatment for active TB infections from 9 months to 6 months when used during the first 2 months of short-course combination therapy with isoniazid and rifampin (1). Despite the efficacy and continued clinical usage of PZA, its precise mechanism of action remains unclear. It is known that PZA is a prodrug that must be converted to pyrazinoic acid (POA), the presumed active form, by the M. tuberculosis genome-encoded amidase PncA (2, 3). Consequently, the vast majority of PZA-resistant clinical isolates harbor loss-of-function mutations in pncA, which is nonessential for the pathogenesis of M. tuberculosis (4, 5).
Elucidation of the mechanism of action for PZA has been hindered, in part, by the peculiar properties of the action of this drug in vitro. Despite exhibiting excellent bactericidal activity in vivo, PZA is ineffective against M. tuberculosis under standard in vitro growth conditions. While the basis for this disparity remains unclear, the PZA susceptibility of M. tuberculosis can be enhanced by stresses that are known to be encountered during infection, such as low pH (6), nutrient limitation (7, 8), and exposure to hypoxia (9). On the basis of these and other findings, a number of models have been proposed to explain the antimycobacterial action of PZA and POA, including direct inhibition of fatty acid synthase I (10), disruption of the membrane potential and cytoplasmic pH homeostasis via a protonophore activity (11), inhibition of trans-translation (12), and inhibition of the aspartate decarboxylase PanD, involved in pantothenate and coenzyme A (CoA) biosynthesis (13). While the bases for these models provide clues toward understanding PZA and POA susceptibility, fundamental aspects of these models have been challenged by recent reports (7, 14–19). Consistent with a role for PanD in PZA action, it has recently been demonstrated that exogenously supplied intermediates of the CoA biosynthetic pathway can antagonize PZA activity, point mutations in panD are associated with in vitro PZA resistance, and PZA treatment leads to the depletion of intrabacterial levels of pantothenate and CoA (13, 15, 20). However, it is important to note that a strain from which panD was deleted was found to retain susceptibility to PZA when grown with a low concentration of pantetheine (15). Thus, while CoA metabolism is intimately associated with PZA action, PanD is not the principal target for POA (15).
On the basis of the observation that the other first-line TB drugs, isoniazid, rifampin, and ethambutol, are 50- to 4,000-fold more potent than PZA for mediating in vitro growth inhibition of M. tuberculosis (21), it is likely that the bacilli have some degree of intrinsic resistance to PZA that limits the full potential of this drug. The high concentrations of PZA required for treatment efficacy can be problematic, as maintenance of such concentrations in serum and tissues cannot be achieved in many patients (22) and can lead to hepatotoxicity in others (23). Therefore, the identification, characterization, and eventual drug-mediated inhibition of intrinsic PZA resistance mechanisms in M. tuberculosis have the potential to substantially enhance PZA efficacy, reducing clinical doses, side effect severity, and the duration of chemotherapy. In this study, we screened for genes linked to the intrinsic PZA resistance of the M. tuberculosis complex (MTBC) by screening for mutants exhibiting hypersusceptibility to the active form of the drug, POA. We report that the loss of function of the long-chain fatty acyl-CoA ligase FadD2 substantially enhances PZA and POA susceptibility in vitro. These findings suggest that target-based inhibition of functions related to fatty acid metabolism has the potential to dramatically improve the antitubercular activity of PZA.
RESULTS
Screen for mutant strains with hypersusceptibility to POA.
To facilitate the identification of intrinsic PZA resistance mechanisms in the M. tuberculosis complex, M. bovis BCG was transduced with mycobacteriophage phAE180 containing a mariner transposable element (24, 25), and approximately 5,000 mutant strains were subjected to a screen for POA hypersusceptibility. Ten strains that exhibited a 2-fold or greater susceptibility to POA than wild-type M. bovis BCG were isolated on agar medium. The transposon insertion site for the strain exhibiting the greatest degree of hypersusceptibility to POA (a 16-fold reduction in the agar MIC relative to that for wild-type M. bovis BCG) was determined to be located within the 3′ end of the coding DNA sequence (CDS) for FadD2, a long-chain fatty acyl-CoA ligase, resulting in truncation of the C terminus by 35 amino acids. FadD2 is 1 of 36 putative long-chain fatty acyl-CoA ligases encoded by the M. tuberculosis genome (26) and is highly conserved among members of the MTBC and the Mycobacterium genus (see Table S2 in the supplemental material). The M. bovis BCG and M. tuberculosis H37Rv FadD2 amino acid sequences differ at position 112, where the M. bovis BCG enzyme contains a glutamate residue and the M. tuberculosis H37Rv enzyme contains an aspartate residue, but they are otherwise identical (Table S2). On the basis of the locations of the putative substrate-binding and catalytic domains of FadD2, residue 112 is unlikely to be directly involved in catalysis. In addition, the POA hypersusceptibility phenotype conferred by the M. bovis BCG FadD2 loss of function was successfully complemented upon transformation of the fadD2 disruption strain with an integrating mycobacterial expression vector encoding the M. tuberculosis FadD2, suggesting that the M. bovis and M. tuberculosis enzymes are functionally conserved (Table 1; Fig. 1). Furthermore, POA exhibited sustained bactericidal activity against the fadD2 disruption mutant at neutral pH at concentrations as low as 800 μg/ml, in contrast to the 3,200 μg/ml required for similar bactericidal activity against wild-type M. bovis BCG (Fig. 2). Consistent with the recent finding that POA-resistant strains of M. tuberculosis and M. bovis BCG can be isolated at near neutral pH (20, 27), the population that emerged following 3 weeks of incubation with POA maintained the ability to grow when subcultured to fresh medium containing the respective concentration of POA.
TABLE 1.
Susceptibility of M. bovis BCG and M. tuberculosis strains to first-line TB drugs on agar medium
| Strain | Characteristic | MIC90a (μg/ml) |
|||
|---|---|---|---|---|---|
| POA | RIF | INH | EMB | ||
| M. bovis BCG | Wild type | 1,250–2,500 | 0.16 | 0.125 | 5 |
| M. bovis BCG fadD2::kan/pJT6a | Mutant with a transposon insertion disrupting fadD2 expression | 78 | 0.01 | 0.125 | 2.5 |
| M. bovis BCG fadD2::kan/pJT6a::fadD2 | Complemented fadD2 disruption mutant | 1,250 | 0.08 | 0.125 | 5 |
| M. bovis BCG ΔfadD2/pTIC6a | Mutant with the hyg and sacB marker exchanged for the fadD2 CDS | 39 | ND | ND | ND |
| M. bovis BCG ΔfadD2/pTIC6a::fadD2 | Complemented fadD2 deletion mutant | 2,500 | ND | ND | ND |
| M. tuberculosis H37Rv | Wild type | 625 | ND | ND | ND |
| M. tuberculosis H37Rv ΔfadD2 | Mutant with the hyg and sacB marker exchanged for the fadD2 CDS | 156 | ND | ND | ND |
The MIC90 was defined as the minimum drug concentration necessary for inhibition of at least 90% of the growth relative to the growth on the drug-free control culture. ND, not determined; POA, pyrazinoic acid; RIF, rifampin; INH, isoniazid; EMB, ethambutol.
FIG 1.

Growth kinetics of Mycobacterium bovis BCG strains in the presence of PZA or POA at pH 6.8. M. bovis BCG strains in mid-logarithmic phase were subcultured into 7H9 medium supplemented with OADC (10%), glycerol (0.2%), and tyloxapol (0.05%) at neutral pH (6.8). Strains ectopically expressing the M. tuberculosis PncA in a wild-type background (A) and a fadD2 disruption background (B) were grown in the presence of various PZA concentrations. Wild-type M. bovis BCG (C), the M. bovis BCG fadD2 disruption mutant (D), and the complemented fadD2 disruption mutant (E) were grown in the presence of a range of POA concentrations.
FIG 2.
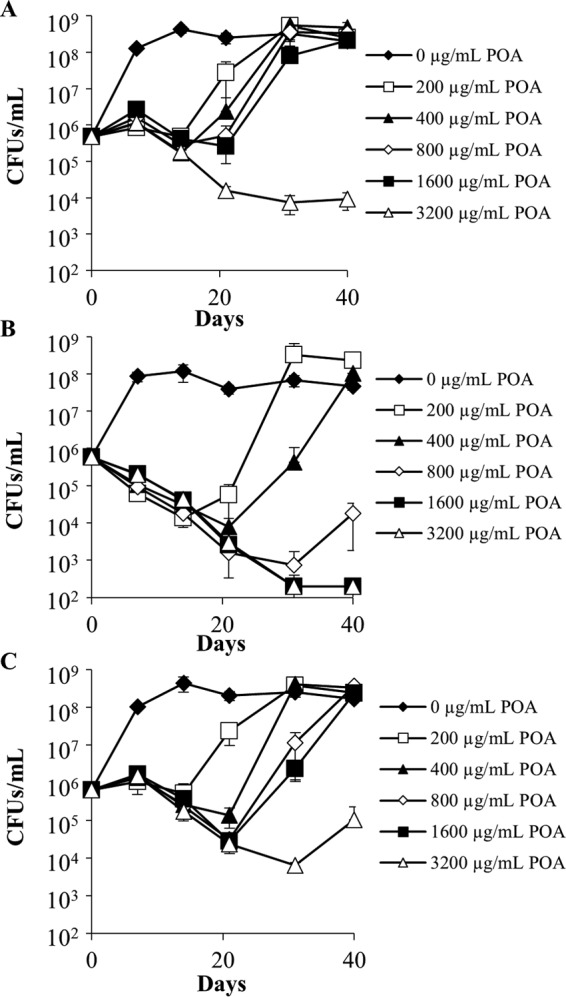
Bactericidal activity of pyrazinoic acid against Mycobacterium bovis BCG strains at pH 6.8. Wild-type M. bovis BCG (A), M. bovis BCG fadD2::kan/pJT6a (B), and M. bovis BCG fadD2::kan/pJT6a::fadD2 (C) cells in mid-logarithmic phase were subcultured into supplemented 7H9 medium at neutral pH (6.8) and treated with a range of POA concentrations. Aliquots were drawn from each culture at the indicated time points, serially diluted, and plated on drug-free supplemented 7H9 agar to permit CFU enumeration. The limit of detection was 200 CFU/ml. Error bars depict the standard deviations of the cell concentrations at a particular time point from three independent experiments.
FadD2 loss of function confers hypersusceptibility to PZA.
In order to assess the PZA susceptibility of the fadD2 disruption mutant, ectopic expression of the M. tuberculosis PncA enzyme was necessary, since M. bovis BCG possesses a missense mutation in PncA that renders the amidase nonfunctional and therefore incapable of converting PZA to POA (3). To address this issue, both wild-type M. bovis BCG and the fadD2 disruption mutant were transformed with an integrating mycobacterial expression vector expressing the M. tuberculosis PncA. As expected, expression of the M. tuberculosis PncA in the wild-type M. bovis BCG background conferred PZA susceptibility at acidic pH (pH 5.8) (MIC90 = 200 μg/ml; Fig. 3) but not at neutral pH (pH 6.8) (MIC90 > 3,200 μg/ml; Fig. 1). This result is consistent with the PZA susceptibility profile observed for wild-type M. tuberculosis, where a lack of susceptibility was observed at neutral pH under standard growth conditions in vitro (MIC90 > 3,200 μg/ml). However, expression of the M. tuberculosis PncA in the fadD2 disruption mutant conferred susceptibility to PZA at neutral pH, as exhibited by the robust growth inhibition observed at PZA concentrations as low as 100 to 200 μg/ml (Fig. 1). The fadD2 disruption mutant also exhibited a 32-fold increase in PZA susceptibility at acidic pH (pH 5.8) and PZA susceptibility at neutral pH on agar medium (Table 2).
FIG 3.
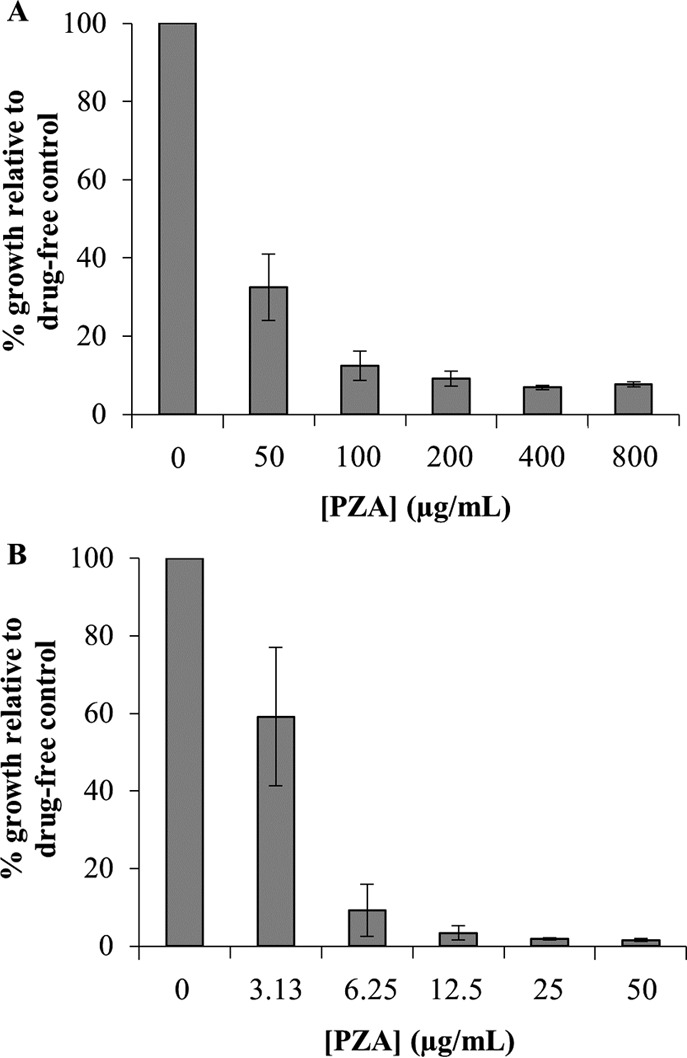
Pyrazinamide susceptibility of Mycobacterium bovis BCG strains ectopically expressing the Mycobacterium tuberculosis PncA at acidic pH. M. bovis BCG/pJT6a::pncA (A) and M. bovis BCG fadD2::kan/pJT6a::pncA (B) were diluted to an OD600 of 0.01 in 7H9 medium supplemented with OADC (10%, vol/vol), glycerol (0.2%, vol/vol), and tyloxapol (0.05%, vol/vol) at pH 5.8. Cells were aliquoted into 30-ml square bottles, and PZA was added to a range of concentrations. Cultures were incubated by shaking at 37°C for 7 days, after which OD600 values were acquired. The average OD600 values for the drug-free cultures for M. bovis BCG/pJT6a::pncA and M. bovis BCG fadD2::kan/pJT6a::pncA after 7 days of incubation were 1.21 ± 0.10 and 2.18 ± 0.18, respectively. The data depicted represent the averages from three independent experiments; error bars depict standard deviations.
TABLE 2.
Susceptibility of Mycobacterium bovis BCG strains expressing the Mycobacterium tuberculosis PncA to PZA on agar medium
| Strain | Characteristic | PZA MIC90a (μg/ml) |
|---|---|---|
| M. bovis BCG/pJT6a::pncA | M. bovis BCG ectopically expressing M. tuberculosis H37Rv PncA | >2,500 |
| M. bovis BCG fadD2::kan/pJT6a::pncA | fadD2 disruption mutant ectopically expressing M. tuberculosis H37Rv PncA | 313 |
The MIC90 was defined as the minimum drug concentration necessary for inhibition of at least 90% of the growth relative to the growth on the drug-free control culture.
FadD2 loss of function confers enhanced susceptibility to rifampin but not to isoniazid or ethambutol.
To determine whether FadD2-mediated intrinsic drug resistance is specific to PZA and POA, susceptibility testing was conducted for the other three first-line TB drugs (isoniazid, rifampin, and ethambutol) on agar medium. The isoniazid and ethambutol MIC90 values for the fadD2 disruption mutant were comparable to those for wild-type M. bovis BCG (Table 1). However, the mutant displayed a 16-fold increase in rifampin susceptibility, and complementation of the fadD2 disruption conferred partial restoration of the wild-type level of rifampin susceptibility (Table 1). In contrast to the PZA and POA hypersusceptibility phenotype of the fadD2 disruption mutant, the enhanced rifampin susceptibility phenotype was apparent only on agar medium and not in liquid culture.
Deletion of fadD2 in M. bovis BCG and M. tuberculosis phenocopies the fadD2 disruption.
To further validate the role of FadD2 in the intrinsic PZA resistance of the MTBC, the M. tuberculosis H37Rv and M. bovis BCG fadD2 CDSs were deleted using the specialized transduction method for allelic exchange (28, 29). Exchange of the fadD2 CDS for a hyg and sacB marker was confirmed by conducting PCR on genomic DNA (gDNA) extracts using primers specific to the chromosomal DNA adjacent to the flanking regions of the fadD2 deletion construct (Table S1). Amplification of the fadD2 locus using wild-type and ΔfadD2 mutant gDNA yielded products that differed by approximately 2 kb in length (Fig. S1). Agar susceptibility testing revealed that the M. tuberculosis H37Rv fadD2 deletion mutant was at least 4-fold more susceptible to POA than the parental strain and the M. bovis BCG fadD2 deletion mutant was at least 32-fold more susceptible to POA than the parental strain. Thus, deletion of the fadD2 CDS has a similar impact on POA susceptibility in both M. tuberculosis and M. bovis BCG (Table 1).
Disruption of FadD2 expression increases susceptibility to certain short-chain fatty acids.
To further characterize the phenotypes associated with FadD2 loss of function and gain additional insight into the basis for FadD2-mediated PZA resistance, the susceptibility of the fadD2 disruption mutant strain to fatty acids of various hydrocarbon chain lengths was assessed. Fatty acid susceptibility testing revealed that the fadD2 disruption mutant was between 4- and 8-fold more susceptible than wild-type M. bovis BCG to the short-chain fatty acids propionate (C3:0), butyrate (C4:0), and valerate (C5:0) (Table 3). However, this mutant strain did not exhibit enhanced susceptibility to acetate (C2:0) (Table 3). The fadD2 disruption mutant also exhibited 2-fold increases in susceptibility to the medium-chain fatty acid caprylate (C8:0) and the long-chain fatty acids laurate (C12:0) and oleate (C18:cis-9); however, no change in palmitate (C16:0) susceptibility was observed (Table 3). Complementation of the fadD2 disruption resulted in a partial restoration of the wild-type fatty acid susceptibility profile.
TABLE 3.
Susceptibility of M. bovis BCG strains to various fatty acids
| Fatty acid | MIC90a (mM) |
||
|---|---|---|---|
| BCG | BCG fadD2::kan | BCG fadD2::kan/pJT6a::fadD2 | |
| Acetate (C2:0) | 50 | 50 | 50 |
| Propionate (C3:0) | 50 | 12.5 | 25 |
| Butyrate (C4:0) | 50 | 6.25 | 25 |
| Valerate (C5:0) | 25 | 3.125 | 25 |
| Caprylate (C8:0) | 2 | 1 | 2 |
| Laurate (C12:0) | 0.5 | 0.25 | 0.5 |
| Palmitate (C16:0) | 0.5 | 0.5 | 0.5 |
| Oleate (C18:cis-9) | 1 | 0.5 | 1 |
The MIC90 was defined as the minimum concentration of fatty acid necessary for inhibition of at least 90% of the growth in standard medium without additional fatty acids.
FadD2 enzymatic studies.
To assess the catalytic capabilities of FadD2 and determine if POA undergoes any direct interactions with the enzyme, recombinant histidine-tagged FadD2 was generated in Escherichia coli and purified by nickel affinity chromatography. Enzymatic activity assays were conducted using Ellman's reagent to monitor the consumption of the free thiol group of coenzyme A due to FadD2-catalyzed acyl-CoA ligation (30). The putative long-chain fatty acyl-CoA ligase activity of FadD2 (31) was confirmed using laurate (C12:0), palmitate (C16:0), and oleate (C18:cis-9) as the substrate (Fig. 4). FadD2 exhibited substantially lower levels of activity when it was supplied with the short-chain fatty acids acetate (C2:0), propionate (C3:0), butyrate (C4:0), and valerate (C5:0) and the medium-chain fatty acid caprylate (C8:0) as the substrate (Fig. 4A). Furthermore, no enzymatic activity was observed when POA was supplied as the substrate, and neither POA nor short- or medium-chain fatty acids appeared to interfere with the long-chain fatty acyl-CoA ligase activity of FadD2 (Fig. 4B). A representative kinetic profile of FadD2 with oleate as a substrate is shown in Fig. 4C.
FIG 4.

Enzymatic characterization of the M. tuberculosis FadD2. (A) Fatty acid substrate specificity of FadD2; (B) additional assays conducted to assess the allosteric modulation of FadD2 and use of POA as the substrate; (C) kinetic profile of FadD2 with oleate as the substrate. Recombinant histidine-tagged FadD2 was propagated in E. coli and purified by nickel affinity chromatography. Enzymatic assays were conducted using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB; or Ellman's reagent) to monitor CoA-SH consumption at 412 nm. Specific activities were computed using the previously determined extinction coefficient for DTNB of 13,600 M−1 cm−1. All specific activity values shown are equivalent to the average reaction velocity over the initial 5-min incubation period. Error bars depict the standard deviations from three assays.
Effects of fadD2 disruption on CoA metabolism.
Since pantothenate is a precursor of coenzyme A, a substrate utilized by FadD2 and other fatty acyl-CoA ligases, we sought to determine if pantothenate-mediated antagonism of PZA and POA action was linked to FadD2-dependent intrinsic resistance. It was determined that the fadD2 disruption mutant required 4-fold more pantothenate than wild-type M. bovis BCG to antagonize POA-mediated growth inhibition, and pantothenate antagonism was partially restored to wild-type levels in the complemented strain (Table 4).
TABLE 4.
Minimum concentrations of pantothenate necessary for antagonism of POA activity against Mycobacterium bovis BCG strains
| Strain | Characteristic | Pantothenate MAC10a (μM) |
|---|---|---|
| M. bovis BCG | Wild type | 3.27 |
| M. bovis BCG fadD2::kan/pJT6a | Mutant with a transposon insertion disrupting fadD2 expression | 13.1 |
| M. bovis BCG fadD2::kan/pJT6a::fadD2 | Complemented fadD2 disruption mutant | 6.55 |
MAC10 was defined as the minimum concentration of the antagonistic metabolite necessary to permit at least 10% of the growth exhibited by the drug-free control culture in the presence of drug at two times the MIC90.
Impairment of pantothenate-mediated antagonism in the fadD2 disruption mutant suggests that FadD2 activity may be associated with CoA metabolism. Thus, we measured and compared intracellular CoA levels in M. bovis BCG, the fadD2 mutant strain, and the complemented strain in the absence and presence of POA (Fig. 5). When grown in the absence of POA, the fadD2 mutant strain showed CoA levels that were indistinguishable from those in the wild-type and complemented strains (Fig. 5A). Consistent with recent findings (20), POA treatment led to a significant reduction in CoA levels in the wild-type strain (Fig. 5B). Interestingly, CoA levels in the fadD2 mutant were 20% lower (P < 0.05) than those in the wild-type strain after 48 h of POA treatment. These data support the notion that FadD2 is associated with POA sensitivity through its interaction with CoA metabolism.
FIG 5.
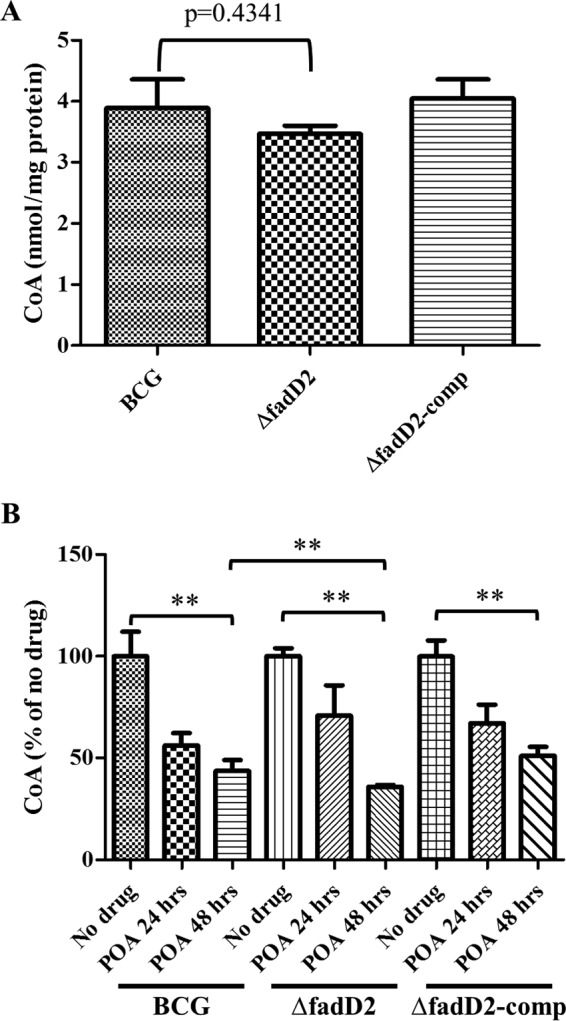
Intracellular CoA levels of Mycobacterium bovis BCG strains. (A) Intracellular CoA levels were measured from wild-type M. bovis BCG (BCG), M. bovis BCG fadD2::hyg (ΔfadD2), and M. bovis BCG fadD2::hyg/pTIC6a::fadD2 (ΔfadD2-comp) that were grown in supplemented 7H9 medium at neutral pH (pH 6.8) until early logarithmic phase (OD600 = 0.2 to 0.3). (B) Intracellular CoA levels were measured 24 h (POA 24 hrs) and 48 h (POA 48 hrs) after 1 mg/ml POA was added to M. bovis BCG strains grown at early logarithmic phase. Intracellular CoA levels were measured as described in Materials and Methods. CoA levels were normalized by the protein concentrations. These results represent the means and standard deviations from three biological replicates. P values of pairwise comparisons (marked by the ends of the brackets) were calculated by using the Student t test. **, P < 0.05.
DISCUSSION
Potentiating the activity of existing antitubercular agents is a major facet of the effort to improve TB chemotherapeutic regimens, prevent the emergent spread of drug-resistant TB, and ultimately eradicate M. tuberculosis. In this study, we demonstrate that the long-chain fatty acyl-CoA ligase FadD2 is a mediator of intrinsic PZA and POA resistance in members of the MTBC and that FadD2 loss of function confers hypersusceptibility to PZA and POA. While the precise mechanism of FadD2-mediated PZA intrinsic resistance remains unclear, our findings demonstrate a central role for CoA and lipid metabolism.
We found that FadD2 loss of function conferred dramatic increases in susceptibility to both PZA and POA. Significantly, a FadD2 loss-of-function transposon mutant was found to be fully susceptible to PZA at neutral pH under normal growth conditions, in contrast to the high degree of PZA tolerance of wild-type bacilli observed under these same conditions. This finding provides further validation of the recent report that the PZA mechanism of action is acid independent (7) and suggests that the FadD2 pathway is a contributor to the lack of PZA efficacy observed at neutral pH against wild-type M. tuberculosis under standard in vitro growth conditions (6).
Interestingly, we also found that a FadD2 loss of function rendered M. bovis BCG 16-fold more susceptible to rifampin. Although the enhanced rifampin susceptibility of the fadD2 mutant was observed only on agar medium and not in liquid culture, this observation suggests that FadD2 activity is associated with a multidrug tolerance mechanism. Mechanistically, perturbations in lipid metabolism afforded by disruption of fadD2 expression may necessitate some degree of metabolic reprogramming at the transcriptional level, and rifampin treatment would hinder the ability of the cell to conduct this shift in gene expression.
The most parsimonious explanation for FadD2-mediated PZA resistance is that the enzyme directly acts upon POA and covalently modifies it, perhaps via formation of an adduct with coenzyme A, such that the drug is inactive against the bacterium. Although chimeric acyl-CoA ligases capable of utilizing both aromatic acids and fatty acids as the substrates have been described (32), our assessments of FadD2 substrate specificity and catalytic capabilities indicate that POA does not directly interact with the enzyme either as a substrate or as an allosteric modulator. Another possibility for the mechanistic basis of this phenomenon is that in a wild-type background, POA inhibits 1 of the other 35 FadD enzymes in the MTBC proteome for which FadD2 provides an alternate fatty acid detoxification pathway. Thus, when FadD2 functionality is lost during POA treatment, the bacterium has no mechanism for the detoxification of cytotoxic fatty acids. We found that a FadD2 loss of function conferred hypersusceptibility to the short-chain fatty acids propionate, butyrate, and valerate but not to acetate or the long-chain fatty acids that are the primary substrates of FadD2. This observation does not necessarily preclude the possibility of fatty acid cytotoxicity being linked to POA action, however, since a loss of FadD2 activity could result in the accumulation of long-chain fatty acids that inhibit the utilization of short-chain fatty acids via a negative-feedback mechanism. Nevertheless, despite the remarkable amount of redundancy in the MTBC proteome with regard to long-chain fatty acyl-CoA ligases, the numerous striking phenotypic effects associated with a FadD2 loss of function indicate that this specific enzyme possesses a particularly important role in drug resistance and fatty acid tolerance.
Another potential explanation for the phenotypes associated with the FadD2 loss of function is that FadD2 activity is required for the maintenance of a sufficient intracellular long-chain fatty acyl-CoA pool for downstream metabolic processes, such as β-oxidation, triacylglycerol synthesis, or membrane lipid synthesis, and that functional lipid metabolism is required for persistence during PZA treatment. Our findings suggest that the precise PZA mechanism of action may be associated with lipid metabolism, which is consistent with the previous report that PZA treatment leads to significant reductions in whole-cell fatty acid synthesis (10). Additional experimentation will be required to elucidate the mechanistic basis for FadD2-mediated PZA resistance.
The identification of FadD2 as a novel player in intrinsic PZA resistance suggests that drug-mediated inhibition of FadD2 or other components of this pathway could dramatically enhance PZA efficacy. On the basis of our in vitro findings, the FadD2 loss of function and PZA treatment exhibit synergy in their ability to provide robust inhibition of bacterial growth and enhance bactericidal activity. If FadD2 inhibition in vivo does indeed confer PZA hypersusceptibility, lower doses of PZA could be administered, in turn reducing adverse drug reactions, expediting the sterilization of infection, and mitigating the proliferation of drug-resistant bacilli. Since lower doses of PZA would presumably be required for activity, it is possible that the coadministration of a FadD2 inhibitor with a normal dose of PZA could overcome the resistance resulting from the pncA loss of function by relying on host amidase activity for PZA bioactivation (33). Further, the potentiation of susceptibility could subvert pncA-linked resistance by enabling the efficacious use of POA in lieu of PZA. Although additional studies will be essential to determine whether FadD2 mediates PZA resistance in vivo, our findings suggest that cotargeting of this pathway during antitubercular chemotherapy has the potential to improve treatment outcomes.
MATERIALS AND METHODS
Bacterial strains and growth media.
Transposon mutagenesis, a screen for POA hypersusceptibility, and subsequent characterization of the phenotypes associated with the FadD2 loss of function were conducted in M. bovis BCG Pasteur. Mycobacteriophages were propagated in Mycobacterium smegmatis mc2155. Slow-growing mycobacterial strains (M. bovis BCG and M. tuberculosis H37Rv) were cultivated in Middlebrook 7H9 medium supplemented with oleate-albumin-dextrose-catalase (OADC; 10%, vol/vol), glycerol (0.2%, vol/vol), and tyloxapol (0.05%, vol/vol) or on Middlebrook 7H9 or 7H10 agar plates supplemented with OADC (10%, vol/vol) and glycerol (0.2%, vol/vol) unless otherwise indicated. OADC was prepared to the following specifications: sodium oleate (1.89 mM), bovine serum albumin (50 g/liter), dextrose (20 g/liter), catalase (30 mg/liter), and sodium chloride (8.5 g/liter). All experiments involving M. tuberculosis H37Rv and derivatives were conducted in a biosafety level 3 laboratory following institutionally approved protocols and containment requirements. Fast-growing mycobacterial strains (M. smegmatis) were grown in Middlebrook 7H9 medium supplemented with dextrose (0.2%, vol/vol) and tyloxapol (0.05%, vol/vol) or on Middlebrook 7H10 agar supplemented with dextrose (0.2%, vol/vol). Kanamycin and/or hygromycin was included in the growth medium at 50 μg/ml and 150 μg/ml, respectively, when appropriate for selection. E. coli DH5α and HB101 were used for plasmid and phasmid propagation. E. coli DH5α λpir was employed for transposon insertion locus determination. Recombinant M. tuberculosis FadD2 was expressed in E. coli BL21/pGRO7. All E. coli strains were grown in LB broth or on LB agar. Kanamycin, hygromycin, and/or chloramphenicol was included in the growth medium at 50 μg/ml, 150 μg/ml, and 25 μg/ml, respectively, when appropriate for selection.
Drug and metabolite stock solutions.
PZA, isoniazid, and ethambutol were dissolved in dimethyl sulfoxide. Rifampin was dissolved in methanol, and chloramphenicol was dissolved in ethanol. POA stocks were prepared by dissolving POA in distilled H2O (dH2O) via addition of NaOH, adjusting the pH to 7.0, and filter sterilizing the solution. Calcium pantothenate, sodium acetate, sodium butyrate, sodium caprylate, and sodium laurate were dissolved in dH2O and filter sterilized. Sodium propionate and sodium valerate stock solutions were prepared by dissolving the free acids in dH2O by addition of NaOH, adjusting the pH to 7.0, and filter sterilizing the solutions. Palmitic acid was dissolved in warm 50% (vol/vol) ethanol with addition of NaOH. Oleic acid was dissolved in dH2O by addition of NaOH and filter sterilized.
Transposon mutagenesis and screen for POA hypersusceptibility.
Mycobacteriophage containing a mariner transposable element (phAE180) was amplified in M. smegmatis mc2155 and used to transduce M. bovis BCG as previously described (24, 25). Transduced bacilli were plated on supplemented 7H10 agar containing kanamycin. A screen for POA hypersusceptibility was conducted by patching transposon mutant colonies (approximately 5,000 in total) onto supplemented 7H10 agar plates at neutral pH containing no drug or 1,250 μg/ml POA. Strains exhibiting growth on the no-drug control plate but not on the POA plate were selected for POA susceptibility testing. Transposon insertion sites were determined as previously described (25). In brief, genomic DNA was extracted from M. bovis BCG transposon insertion mutants, digested with BssHII, ligated, and used to transform E. coli DH5α λpir. Plasmids were maintained under kanamycin selection. Purified plasmids were subjected to Sanger sequencing using the KanSeq_Rev primer (see Table S1 in the supplemental material), which is oriented outward from the 3′ end of the mariner kanamycin resistance cassette such that the adjacent chromosomal DNA was sequenced. The resulting sequence were aligned with the sequence of the M. bovis BCG genome (GenBank accession number NC_002945.3) to determine insertion sites.
Cloning (complementation, restoration of PncA function, deletion).
Complementation of fadD2 disruption and restoration of pncA function in M. bovis BCG were achieved by transforming the appropriate strains with an integrating mycobacterial expression vector expressing fadD2 or M. tuberculosis H37Rv pncA. The fadD2 CDS was amplified from M. tuberculosis mc27000 genomic DNA by PCR with primers containing HindIII and NheI restriction sites at the 5′ and 3′ ends of the gene, respectively (see Table S1 in the supplemental material). The PCR product and pJT6a (pTIC6a with a hygromycin resistance cassette instead of a kanamycin resistance cassette) were digested with HindIII and NheI and ligated. The pncA CDS was excised from pUMN002::pncA (7) by an HindIII/NheI double digest and ligated into HindIII- and NheI-digested pJT6a. Plasmids were propagated in E. coli and maintained with hygromycin selection. fadD2 was deleted using the specialized transduction method for allelic exchange (28, 29). Briefly, chromosomal regions flanking the fadD2 CDS were amplified by PCR (Table S1), digested, and ligated with the hyg and sacB marker and oriE fragments such that the fadD2 flanking regions now flanked the hyg and sacB marker. Both the resulting plasmid and phAE159 were digested with PacI and ligated. The ligation product was packaged in vitro using a MaxPlax bacteriophage lambda packaging extract, and the resulting phage was used to transduce E. coli HB101. The phasmids were purified and subsequently electroporated into M. smegmatis mc2155 for mycobacteriophage propagation. Once a titer of at least 1010 PFU/ml was attained, phage was used to transduce M. bovis BCG and M. tuberculosis H37Rv. Transduced bacilli were subjected to hygromycin selection on supplemented 7H9 medium.
MIC assays.
For liquid culture MICs, cells in mid-logarithmic phase were diluted to an optical density at 600 nm (OD600) of 0.01 in supplemented 7H9 medium. Diluted cells were aliquoted into 30-ml square bottles, and drug was added to a range of concentrations. Cultures were incubated with shaking (100 rpm) at 37°C for the time period specified for respective experiments, after which OD600 values were acquired to permit MIC determination. The liquid MIC90 was defined as the minimum drug concentration necessary for inhibition of at least 90% of the growth of the drug-free control culture. Drug susceptibility on agar medium was assessed using a modified version of the agar proportion method (34). Cells in mid-logarithmic phase were serially diluted and plated on 7H9 agar plates (7H9 broth [4.7 g/liter], agar [15 g/liter], OADC [10%, vol/vol], glycerol [0.2%, vol/vol], tyloxapol [0.05%, vol/vol]) containing a range of drug concentrations. The plates were incubated for 28 days at 37°C under atmospheric CO2 levels. MICs were determined on the basis of the overall cellular biomass present on a plate with a particular drug concentration. The biomass from a single dilution spot on each plate was resuspended in liquid medium, and OD600 values were acquired for quantification. The agar MIC90 was defined as the minimum drug concentration necessary for inhibition of at least 90% of the biomass formation of the no-drug control.
Growth kinetics experiments.
Cells in mid-logarithmic phase were diluted to an OD600 of 0.01 in supplemented 7H9 medium at neutral pH. Diluted cells were aliquoted into 30-ml square bottles, and drug was added to a range of concentrations. The cultures were incubated with shaking (100 rpm) at 37°C, and OD600 values were acquired at various time points to generate growth curves.
Minimum antagonistic concentration assays.
Cells in mid-logarithmic phase were diluted to an OD600 of 0.01 in supplemented 7H9 medium at neutral pH containing a POA concentration 2-fold greater than the MIC90 for each strain. Diluted cells were aliquoted into 30-ml square bottles, and calcium pantothenate was added to a range of concentrations. A pantothenate-only culture and a culture lacking both pantothenate and POA were prepared as controls. Cultures were incubated for 7 days, after which OD600 values were acquired. The MAC10 was defined as the minimum concentration of the antagonistic metabolite (pantothenate) necessary to permit at least 10% of the growth exhibited by the drug-free control culture in the presence of drug at two times the MIC90.
Assessment of pyrazinoic acid bactericidal activity.
Cells in mid-logarithmic phase were diluted to an OD600 of 0.01 in supplemented 7H9 medium at neutral pH and aliquoted into 30-ml square bottles. Drug was added to a range of concentrations. Upon initial culture inoculation and at various time points thereafter, aliquots were drawn from each culture, serially diluted, and plated on drug-free supplemented 7H9 agar to permit CFU enumeration.
FadD2 overexpression and purification.
The M. tuberculosis H37Rv fadD2 CDS was amplified by PCR with primers containing NheI and HindIII restriction sites at the 5′ and 3′ ends of the gene, respectively (Table S1), digested with NheI and HindIII, and ligated into NheI- and HindIII-digested pET28b such that the pET28b-encoded histidine tag was attached to the N terminus of FadD2. pET28b::fadD2 was transformed into E. coli DH5α for propagation, purified, and subsequently transformed into E. coli BL21/pGRO7 for protein overexpression. E. coli BL21/pGRO7/pET28b::fadD2 was grown in batch culture to mid-logarithmic phase under kanamycin and chloramphenicol selection for plasmid maintenance and in the presence of arabinose (500 μg/ml) to induce GroEL expression. IPTG (isopropyl-β-d-thiogalactopyranoside) was then added to 1 mM to induce FadD2 expression, and the culture was incubated by shaking (200 rpm) at 37°C for 3 h. Cells were pelleted via centrifugation, and the pellets were stored at −20°C. For lysis, the pellets were resuspended in lysis buffer, treated with lysozyme at 1 mg/ml for 30 min on ice, and sonicated on ice. FadD2 was purified by nickel affinity chromatography using Ni-nitrilotriacetic acid agarose for the column matrix (Qiagen).
Enzymatic assays.
The activity of purified FadD2 was assessed using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB; or Ellman's reagent) to spectrophotometrically monitor the consumption of the thiol group of coenzyme A via FadD2-catalyzed fatty acyl-CoA ligation. DTNB binds free thiol to produce a color change quantifiable at 412 nm. Thus, a loss of signal at 412 nm is indicative of FadD2 activity. The assay protocol was adapted from the protocols described by Kang et al. (35) and Guo et al. (36). Briefly, the reaction components (buffer [35], 10 mM ATP, 5 μg protein, and fatty acid substrate [concentration, between 12.5 and 400 μM]) were combined in a 90-μl volume (100-μl final volume after addition of coenzyme A). Control reaction mixtures lacking protein were also prepared. The reaction mixtures were prewarmed at 37°C for 2 min, after which 10 μl of 5 mM coenzyme A (final concentration, 0.5 mM) was added for reaction initiation. The reactions were allowed to proceed for 5 min, followed by a 5-min heat inactivation period at 80°C. After the reaction mixtures had cooled to room temperature, 70 μl of each reaction mixture was transferred to a cuvette containing 600 μl 0.4 mM DTNB in potassium phosphate buffer (pH 7.0). A412 values were acquired after a 5-min color development period. The previously published extinction coefficient of DTNB (13,600 M−1 cm−1) was used to compute specific activity (30).
Intracellular CoA measurement.
Intracellular CoA measurements were performed as previously described with slight modifications (20). Briefly, M. bovis BCG strains were grown until early logarithmic phase (OD600 = 0.2 to 0.3) and collected by centrifugation at 4,000 rpm (4°C) for 20 min. The cell pellet was washed with ice-cold phosphate-buffered saline (pH 7.4) and then resuspended in the same buffer. Metabolites were extracted by bead beating (Beads Blaster 24; Benchmark Scientific) at 6,000 rpm four times for 20 s each time. The cell extract was centrifuged at 13,000 rpm for 10 min. The supernatant was then filtered through a 0.22-μm-pore-size syringe filter. The protein concentration in each sample was measured by using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Large proteins were then removed from the remaining sample by use of a 3-kDa Nanosep spin column (Pall Corporation). CoA levels were measured using a CoA assay kit (catalog number MAK034; Sigma-Aldrich). CoA levels were normalized by the respective protein concentration.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant R01 AI123146 and institutional start-up funds from the University of Minnesota Department of Microbiology and Immunology to A.D.B. B.C.R. was supported by an American Society for Microbiology undergraduate research fellowship and the Undergraduate Research Opportunities Program at the University of Minnesota. N.A.D. was supported by NIH institutional training grant T32 HL07741.
We thank William R. Jacobs, Jr., for providing M. smegmatis mc2155, M. bovis BCG, M. tuberculosis H37Rv, phAE159, and phAE180; Joshua M. Thiede for constructing the pJT6a vector; and Shannon Lynn Kordus for her assistance with FadD2 enzymology.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02130-16.
REFERENCES
- 1.Ormerod LP, Horsfield N. 1987. Short-course antituberculous chemotherapy for pulmonary and pleural disease: 5 years' experience in clinical practice. Br J Dis Chest 81:268–271. doi: 10.1016/0007-0971(87)90160-4. [DOI] [PubMed] [Google Scholar]
- 2.Konno K, Feldmann FM, McDermott W. 1967. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis 95:461–469. [DOI] [PubMed] [Google Scholar]
- 3.Scorpio A, Zhang Y. 1996. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med 2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 4.Scorpio A, Lindholm-Levy P, Heifets L, Gilman R, Siddiqi S, Cynamon M, Zhang Y. 1997. Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 41:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilchèze C, Weinrick B, Wong K-W, Chen B, Jacobs WR Jr. 2010. NAD(+) auxotrophy is bacteriocidal for the tubercle bacilli. Mol Microbiol 76:365–377. doi: 10.1111/j.1365-2958.2010.07099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDermott W, Tompsett R. 1954. Activation of pyrazinamide and nicotinamide in acidic environments in vitro. Am Rev Tuberc 70:748–754. [DOI] [PubMed] [Google Scholar]
- 7.Peterson ND, Rosen BC, Dillon NA, Baughn AD. 2015. Uncoupling environmental pH and intrabacterial acidification from pyrazinamide susceptibility in Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:7320–7326. doi: 10.1128/AAC.00967-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Q, Chen Z-F, Li Y-Y, Zhang Y, Ren Y, Fu Z, Xu S-Q. 2007. Nutrient-starved incubation conditions enhance pyrazinamide activity against Mycobacterium tuberculosis. Chemotherapy 53:338–343. doi: 10.1159/000107723. [DOI] [PubMed] [Google Scholar]
- 9.Wade MM, Zhang Y. 2004. Anaerobic incubation conditions enhance pyrazinamide activity against Mycobacterium tuberculosis. J Med Microbiol 53:769–773. doi: 10.1099/jmm.0.45639-0. [DOI] [PubMed] [Google Scholar]
- 10.Zimhony O, Cox JS, Welch JT, Vilchèze C, Jacobs WRJ. 2000. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med 6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. 2003. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother 52:790–795. doi: 10.1093/jac/dkg446. [DOI] [PubMed] [Google Scholar]
- 12.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi W, Chen J, Feng J, Cui P, Zhang S, Weng X, Zhang W, Zhang Y. 2014. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg Microbes Infect 3:e58. doi: 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boshoff HI, Mizrahi V, Barry CE III. 2002. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J Bacteriol 184:2167–2172. doi: 10.1128/JB.184.8.2167-2172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dillon NA, Peterson ND, Rosen BC, Baughn AD. 2014. Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob Agents Chemother 58:7258–7263. doi: 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemens SP, Sharpe CA, Cynamon MH. 1996. Activity of pyrazinamide in a murine model against Mycobacterium tuberculosis isolates with various levels of in vitro susceptibility. Antimicrob Agents Chemother 40:14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Personne Y, Parish T. 2014. Mycobacterium tuberculosis possesses an unusual tmRNA rescue system. Tuberculosis 94:34–42. doi: 10.1016/j.tube.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Speirs RJ, Welch JT, Cynamon MH. 1995. Activity of n-propyl pyrazinoate against pyrazinamide-resistant Mycobacterium tuberculosis: investigations into mechanism of action of and mechanism of resistance to pyrazinamide. Antimicrob Agents Chemother 39:1269–1271. doi: 10.1128/AAC.39.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baughn AD, Deng J, Vilcheze C, Riestra A, Welch JT, Jacobs WR Jr, Zimhony O. 2010. Mutually exclusive genotypes for pyrazinamide and 5-chloropyrazinamide resistance reveal a potential resistance-proofing strategy. Antimicrob Agents Chemother 54:5323–5328. doi: 10.1128/AAC.00529-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopal P, Yee M, Sarathy J, Low JL, Sarathy JP, Kaya F, Dartois V, Gengenbacher M, Dick T. 2016. Pyrazinamide resistance is caused by two distinct mechanisms: prevention of coenzyme A depletion and loss of virulence factor synthesis. ACS Infect Dis 2:616–626. doi: 10.1021/acsinfecdis.6b00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. 2014. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother 69:2420–2425. doi: 10.1093/jac/dku136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang KC, Leung CC, Yew WW, Lau TY, Tam CM. 2008. Hepatotoxicity of pyrazinamide. Am J Respir Crit Care Med 177:1391–1396. doi: 10.1164/rccm.200802-355OC. [DOI] [PubMed] [Google Scholar]
- 24.Kriakov J, Lee SH, Jacobs WR. 2003. Identification of a regulated alkaline phosphatase, a cell surface-associated lipoprotein, in Mycobacterium smegmatis. J Bacteriol 185:4983–4991. doi: 10.1128/JB.185.16.4983-4991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci U S A 96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. 2004. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 27.Lanoix J-P, Tasneen R, O'Brien P, Sarathy J, Safi H, Pinn M, Alland D, Dartois V, Nuermberger E. 2016. High systemic exposure of pyrazinoic acid has limited antituberculosis activity in murine and rabbit models of tuberculosis. Antimicrob Agents Chemother 60:4197–4205. doi: 10.1128/AAC.03085-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 29.Jain P, Hsu T, Arai M, Biermann K, Thaler DS, Nguyen A, González PA, Tufariello JM, Kriakov J, Chen B, Larsen MH, Jacobs WR Jr. 2014. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. mBio 5:e01245-14. doi: 10.1128/mBio.01245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellman G. 1959. Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 31.Nambi S, Gupta K, Bhattacharyya M, Ramakrishnan P, Ravikumar V, Siddiqui N, Thomas AT, Visweswariah SS. 2013. Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J Biol Chem 288:14114–14124. doi: 10.1074/jbc.M113.463992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koetsier MJ, Jekel PA, van den Berg MA, Bovenberg RAL, Janssen DB. 2009. Characterization of a phenylacetate-CoA ligase from Penicillium chrysogenum. Biochem J 417:467–476. doi: 10.1042/BJ20081257. [DOI] [PubMed] [Google Scholar]
- 33.Via LE, Savic R, Weiner DM, Zimmerman MD, Prideaux B, Irwin SM, Lyon E, O'Brien P, Gopal P, Eum S, Lee M, Lanoix J-P, Dutta NK, Shim T, Cho JS, Kim W, Karakousis PC, Lenaerts A, Nuermberger E, Barry CE, Dartois V. 2015. Host-mediated bioactivation of pyrazinamide: implications for efficacy, resistance, and therapeutic alternatives. ACS Infect Dis 1:203–214. doi: 10.1021/id500028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Middlebrook G, Cohn ML. 1958. Bacteriology of tuberculosis: laboratory methods. Am J Public Health Nations Health 48:844–853. doi: 10.2105/AJPH.48.7.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang Y, Zarzycki-Siek J, Walton CB, Norris MH, Hoang TT. 2010. Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS One 5:e13557. doi: 10.1371/journal.pone.0013557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo F, Ortega-Pierres G, Argüello-García R, Zhang H, Zhu G. 2015. Giardia fatty acyl-CoA synthetases as potential drug targets. Front Microbiol 6:753. doi: 10.3389/fmicb.2015.00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.