

ABSTRACT
Cytomegalovirus (CMV) infection is a significant complication after kidney transplantation. We examined the ability of RG7667, a combination of two monoclonal antibodies, to prevent CMV infection in high-risk kidney transplant recipients in a randomized, double-blind, placebo-controlled trial. CMV-seronegative recipients of a kidney transplant from a CMV-seropositive donor (D+R−) were randomized to receive RG7667 (n = 60) or placebo (n = 60) at the time of transplant and 1, 4, and 8 weeks posttransplant. Patients were monitored for CMV viremia every 1 to 2 weeks posttransplant for 24 weeks. Patients who had seroconverted (D+R+) or withdrawn before dosing were excluded from the analysis (n = 4). CMV viremia occurred in 27 of 59 (45.8%) patients receiving RG7667 and 35 of 57 (61.4%) patients receiving placebo (stratum-adjusted difference, 15.3%; P = 0.100) within 12 weeks posttransplant and in 30 of 59 (50.8%) patients receiving RG7667 and 40 of 57 (70.2%) patients receiving placebo (stratum-adjusted difference, 19.3%; P = 0.040) within 24 weeks posttransplant. Median time to CMV viremia was 139 days in patients receiving RG7667 compared to 46 days in patients receiving placebo (hazard ratio, 0.53; P = 0.009). CMV disease was less common in the RG7667 than placebo group (3.4% versus 15.8%; P = 0.030). Adverse events were generally balanced between treatment groups. In high-risk kidney transplant recipients, RG7667 was well tolerated, numerically reduced the incidence of CMV infection within 12 and 24 weeks posttransplant, delayed time to CMV viremia, and was associated with less CMV disease than the placebo. (This study has been registered at ClinicalTrials.gov under registration no. NCT01753167.)
KEYWORDS: cytomegalovirus, kidney transplantation, monoclonal antibodies
INTRODUCTION
Cytomegalovirus (CMV) is a significant cause of morbidity and mortality in immunocompromised individuals, including solid-organ transplant (SOT) and hematopoietic stem cell transplant recipients (1). While prophylactic antiviral medication has decreased the incidence of CMV infection and disease in the early posttransplant period (2–4), these agents have clinically significant toxicities (5). Furthermore, late-onset CMV disease, which is associated with allograft failure and mortality (6–8), has been increasingly recognized as an important complication (9).
Kidneys are the most commonly transplanted solid organs (10), and CMV-seronegative recipients from a CMV-seropositive donor (D+R−) are the subgroup at highest risk for CMV infection and disease (11). CMV intravenous immunoglobulin (CMV-IVIG) decreases the incidence of CMV disease in D+R− kidney transplant recipients (12) and has been approved for prevention of CMV disease in SOT and kidney transplant recipients by U.S. and Canadian regulatory agencies, respectively (13, 14). However, prophylactic use of CMV-IVIG is uncommon (15, 16), likely due to the availability of oral antivirals and cost (17–19).
Compared to polyclonal antibody preparations and small-molecule antivirals, a monoclonal antibody-based therapy may offer several advantages, including higher target specificity, the potential for greater potency with the ability to administer higher doses of antibody, and lower toxicity (17, 20, 21). RG7667 is a combination of two monoclonal antibodies (MCMV5322A and MCMV3068A) that binds two distinct antigens on the surface of CMV and inhibits its entry into relevant host cells (21). In a phase 1 study, RG7667 was safe and well tolerated in healthy adults (21). This phase 2 trial evaluates the safety and activity of RG7667 for the prevention of CMV infection in D+R− kidney transplant recipients.
(This study was presented in part as an oral presentation at the Interscience Conference on Antimicrobial Agents and Chemotherapy [ICAAC]/International Congress of Chemotherapy and Infection [ICC], San Diego, CA, 17 to 21 September 2015 [22].)
RESULTS
Study population.
Of the 120 patients randomized to receive study drug, 116 patients were included in the modified intention-to-treat (mITT) population (59 RG7667, 57 placebo) (Fig. 1). One hundred eighteen patients were included in the safety population (60 RG7667, 58 placebo), and 98 patients completed the study (48 RG7667, 50 placebo).
FIG 1.
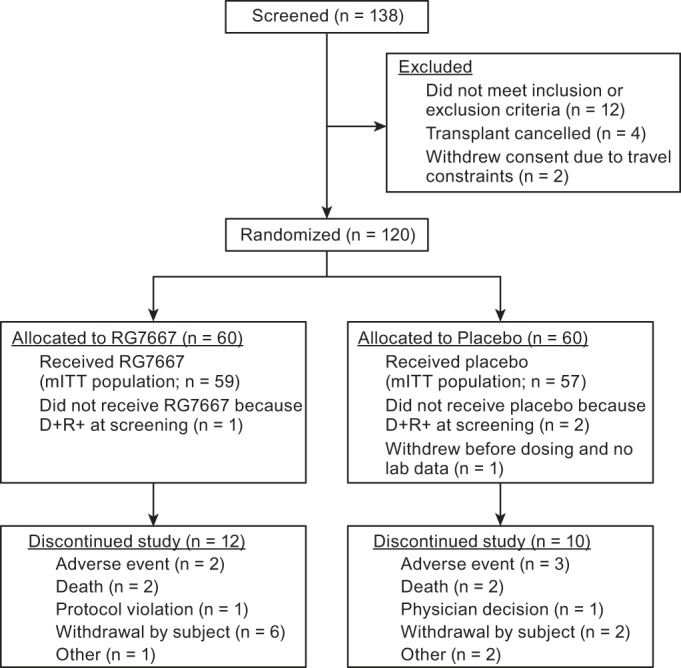
Screening, randomization, and follow-up of patients. D+R+, CMV-seropositive recipient of kidney transplant from CMV-seropositive donor; mITT, modified intention to treat.
The majority of patients were male (70%) and white (81.7%). Patients randomized to receive RG7667 and placebo were similar in terms of their baseline age, sex, race, ethnicity, weight, height, region, induction immunosuppression, and donor type (Table 1). The RG7667 group had a higher proportion of glomerulonephritis and a lower proportion of polycystic kidney disease as the reason for transplant. There was a higher proportion with two HLA-DR mismatches and a lower proportion with one HLA-DR mismatch in the RG7667 than the placebo group.
TABLE 1.
Participant characteristics
| Characteristica | Value for: |
|
|---|---|---|
| RG7667 (n = 60) | Placebo (n = 60) | |
| Age (yr), mean (SD) | 49.2 (14.3) | 49.5 (12.9) |
| Male sex | 42 (70.0) | 42 (70.0) |
| Race | ||
| White | 49 (81.7) | 49 (81.7) |
| Ethnicity | ||
| Not Hispanic or Latino | 53 (88.3) | 49 (81.7) |
| Height (cm), mean (SD) | 173.1 (12.0) | 173.5 (10.5) |
| Weight (kg), mean (SD) | 80.0 (16.0) | 81.8 (16.2) |
| Reason for transplantation | ||
| Diabetes and/or hypertension | 20 (33.3) | 18 (30.0) |
| Glomerulonephritis | 17 (28.5) | 12 (20.0) |
| Polycystic kidney disease | 8 (13.3) | 13 (21.7) |
| Other/unknown/not specified | 15 (25.4) | 17 (28.3%) |
| Region: United States | 29 (48.3) | 29 (48.3) |
| ATGb and/or alemtuzumab for induction of immunosuppression | 14 (23.3) | 16 (26.7) |
| Donor type | ||
| Deceased | 35 (58.3) | 36 (60.0) |
| Living, related | 16 (26.7) | 13 (21.7) |
| Living, unrelated | 9 (15.0) | 9 (15.0) |
| Missing | 0 | 2 (3.3) |
| HLA-A mismatchc | ||
| 0 | 18 (30.0) | 16 (26.7) |
| 1 | 28 (46.7) | 25 (41.7) |
| 2 | 10 (16.7) | 13 (21.7) |
| Not done or unobtainable | 4 (6.7) | 6 (10.0) |
| HLA-B mismatch | ||
| 0 | 16 (26.7) | 19 (31.7) |
| 1 | 21 (35.0) | 17 (28.3) |
| 2 | 19 (31.7) | 17 (28.3) |
| Not done or unobtainable | 4 (6.7) | 7 (11.7) |
| HLA-DR mismatch | ||
| 0 | 17 (28.3) | 16 (26.7) |
| 1 | 20 (33.3) | 28 (46.7) |
| 2 | 16 (26.7) | 9 (15.0) |
| Not done or unobtainable | 7 (11.7) | 7 (11.7) |
Data are numbers (percent) unless otherwise specified.
ATG, antithymocyte globulin.
HLA, human leukocyte antigen.
Efficacy.
The proportion of patients exhibiting the primary endpoint of CMV viremia within 12 weeks posttransplant was lower in the RG7667 group (27 of 59 [45.8%]) than in the placebo group (35 of 57 [61.4%]) (Table 2). However, the stratum-adjusted difference (15.3%) was not significant at the unadjusted 5% level of significance (95% confidence interval [CI], −2.8% to 32.2%; P = 0.100). The primary endpoint was also observed less frequently in the RG7667 than the placebo group within subgroups stratified by region and induction immunosuppression (see Table S1 in the supplemental material). CMV viremia within 24 weeks posttransplant occurred significantly less frequently in the RG7667 (30 of 59 [50.8%]) than placebo (40 of 57 [70.2%]) group (stratum-adjusted difference, 19.3%; 95% CI, 1.4% to 35.6%; P = 0.040).
TABLE 2.
Efficacy endpoints
| Endpointa | Value(s) for: |
|
|---|---|---|
| RG7667 (n = 59) | Placebo (n = 57) | |
| CMV viremia within 12 weeks posttransplant | ||
| n (%) | 27 (45.8) | 35 (61.4) |
| Stratum-adjusted difference, % (95% CI) | 15.3 (−2.8–32.2) | |
| P value | 0.100 | |
| CMV viremia within 24 weeks posttransplant | ||
| n (%) | 30 (50.8) | 40 (70.2) |
| Stratum-adjusted difference, % (95% CI) | 19.3 (1.4–35.6) | |
| P value | 0.040 | |
| Median time to viremia (days) | 139 | 46 |
| HR (95% CI) | 0.53 (0.33–0.86) | |
| P value | 0.009 | |
| Median viral load at initial detection, copies/ml (IU/ml) | 342 (311) | 1051 (956) |
| Median peak viral load, copies/ml (IU/ml) | 2,965 (2,698) | 6,397 (5,821) |
| Receipt of preemptive anti-CMV therapy, n (%) | ||
| Within 12 weeks posttransplant | 29b (49.2) | 36b (63.2) |
| Within 24 weeks posttransplant | 32 (54.2) | 40 (70.2) |
| CMV disease within 24 weeks posttransplant | ||
| n (%) | 2 (3.4) | 9 (15.8) |
| P value | 0.030 | |
CI, confidence interval; HR, hazard ratio.
Some patients in the RG7667-treated (n = 2) and placebo-treated (n = 1) groups received valganciclovir therapy on day 1 before there was any evidence of viremia, in violation of the protocol, and were counted in the group of patients that received preemptive therapy. The medical monitor informed the site investigator of the error, who stopped the prophylactic therapy or withdrew the patient.
The median time to CMV viremia was significantly delayed in patients receiving RG7667 compared to placebo (139 versus 46 days; hazard ratio, 0.53 relative to placebo group; 95% CI, 0.33 to 0.86; P = 0.009) (Table 2 and Fig. 2). Delayed median time to CMV viremia in the RG7667 group compared with the placebo was also observed in subgroups stratified by region and induction immunosuppression (Table S2). Among those with a detectable viral load, the median viral load at initial detection was 342 copies/ml (311 IU/ml) in the RG7667 group and 1,051 copies/ml (956 IU/ml) in the placebo group. The median peak viral load was 2,965 copies/ml (2,698 IU/ml) in the RG7667 group and 6,397 copies/ml (5,821 IU/ml) in the placebo group. Receipt of preemptive anti-CMV therapy within 12 and 24 weeks posttransplant in the RG7667 (29 of 59 [49.2%] and 32 of 59 [54.2%], respectively) and placebo (36 of 57 [63.2%] and 40 of 57 [70.2%], respectively) groups closely mirrored the incidence of CMV viremia (Table 2).
FIG 2.
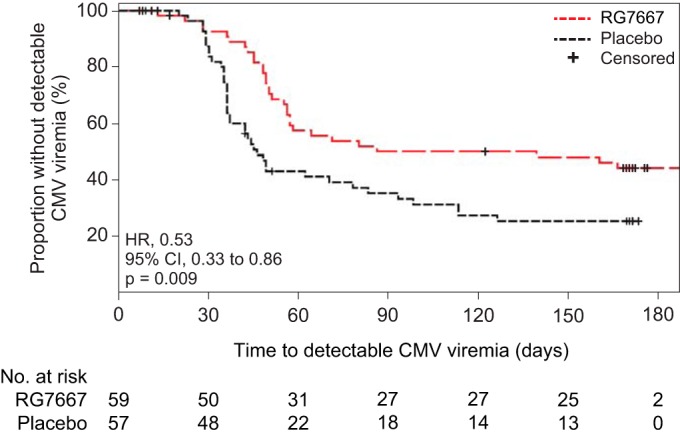
Time to detectable CMV viremia according to study drug group. The proportion of patients without detectable CMV viremia during follow-up according to study group (modified intention-to-treat population) is shown.
A significantly lower proportion of patients in the RG7667 group developed CMV disease within 24 weeks posttransplant than in the placebo group (2 of 59 [3.4%] versus 9 of 57 [15.8%], respectively; P = 0.030) (Table 2). In the RG7667 group, both patients (2 of 59 [3.4%]) experienced an occurrence of CMV syndrome as a late event (onset on days 126 and 145) with no reports of tissue-invasive CMV. In the placebo group, 9 patients experienced 7 occurrences of CMV syndrome and 4 occurrences of tissue-invasive CMV (gastroenteritis, enteritis, or hepatitis). Five of these patients (5 of 57 [8.8%]) experienced CMV syndrome (n = 3, with 3 occurrences) or tissue-invasive CMV (n = 2, with 3 occurrences) as a late event (onset on days 111 to 122).
Safety.
There were 698 treatment-emergent adverse events (TEAEs) in 59 of 60 (98.3%) patients in the RG7667 group and 831 TEAEs in 57 of 58 (98.3%) patients in the placebo group (Table 3). TEAEs were generally balanced between the treatment groups. There was a higher incidence (i.e., difference of at least 10%) of hyperkalemia, pyrexia, leukopenia, and neutropenia in the placebo than RG7667 group; no TEAEs occurred more frequently in the RG7667 than in the placebo group. Kidney transplant rejection occurred less frequently in the RG7667 (3 of 60 [5.0%]) than placebo (9 of 58 [15.5%]) group. Infusion reactions with RG7667 were predominantly mild and not more frequent than with the placebo.
TABLE 3.
Summary of TEAEsa
| Event | No. (%) of patients |
|
|---|---|---|
| RG7667 (n = 60) | Placebo (n = 58) | |
| Any TEAE | 59 (98.3) | 57 (98.3) |
| At least one TEAE related to study drug | 12 (20.0) | 14 (24.1) |
| At least one serious TEAE | 30 (50.0) | 35 (60.3) |
| At least one serious TEAE leading to withdrawal from treatment | 2 (3.3) | 2 (3.4) |
| Preferred term | ||
| Kidney transplant rejectionb | 3 (5.0) | 9 (15.5) |
| Hyperkalemia | 7 (11.7) | 13 (22.4) |
| Pyrexia | 5 (8.3) | 11 (19.0) |
| Leukopenia | 4 (6.7) | 14 (24.1) |
| Neutropenia | 2 (3.3) | 9 (15.5) |
Only treatment-emergent adverse event (TEAEs) with a difference of at least 10% between RG7667 and placebo groups are listed.
The preferred terms “kidney transplant rejection” and “transplant rejection” were combined.
During the entire follow-up period, there were 2 deaths in the RG7667 group (due to amebic encephalitis and acute myocardial infarction) and 2 deaths in the placebo group (due to interstitial lung disease and sepsis). No deaths were deemed related to the study drug. In the RG7667 group, 2 of 60 (3.3%) had at least one serious adverse event (SAE) leading to treatment withdrawal (graft loss). In the placebo group, 2 of 58 (3.4%) had at least one SAE leading to treatment withdrawal (worsening interstitial lung disease and kidney dysfunction).
In the RG7667 group, of the 30 patients with CMV viremia during the study, 16 (53.3%) met predefined criteria for resistance testing, of which 4 (25.0%) had a ganciclovir (GCV) resistance mutation. In the placebo group, of the 40 patients with CMV viremia during the study, 26 (65.0%) met predefined criteria for resistance testing, of which 11 (42.3%) had a GCV resistance mutation.
Pharmacokinetics and immunogenicity.
The pharmacokinetic disposition of the constituent antibodies of RG7667 was biphasic (Fig. S1), typical of intravenous monoclonal immunoglobulin G (IgG), and their mean terminal half-lives were consistent with those observed in healthy subjects (21). The proportion of patients with evaluable immunogenicity data who developed antitherapeutic antibodies (ATAs) to MCMV5322A or MCMV3068A after dosing was 14.3% (8 of 56) in the RG7667 group and 6.8% (4 of 59) in the placebo group (see results in the supplemental material). The presence of ATAs was not associated with lower drug exposure (R. Deng, unpublished data).
DISCUSSION
In this phase 2 study, RG7667, a combination of two monoclonal antibodies that inhibits CMV entry into relevant host cells, was administered prophylactically to high-risk (D+R−) kidney transplant recipients. Although RG7667 did not significantly reduce the primary endpoint (i.e., proportion of patients with CMV viremia within 12 weeks posttransplant) compared to placebo, this study demonstrated that RG7667 has anti-CMV activity. Compared with placebo, RG7667 significantly reduced the proportion of patients with CMV viremia within 24 weeks posttransplant, significantly delayed the median time to CMV viremia, and reduced the incidence of CMV disease.
TEAEs were common given the comorbidity burden of the study population, and they were generally balanced between the treatment groups. Notably, kidney transplant rejection, as reported by the investigators, occurred less frequently in the RG7667 than the placebo group. Although biopsy specimens and clinical data were not collected to support the diagnosis of rejection, rejection is a major safety event that would have been reported if it occurred, so we anticipated accurate capture of this outcome. Additionally, GCV resistance was not higher in the RG7667 than the placebo group. Rates of GCV resistance observed in our study may not be comparable to those of other studies due to differences in thresholds for testing, timing of testing, and patient populations. For example, this study exclusively enrolled D+R− patients, who tend to have higher rates of GCV resistance (23).
The proportion of patients with an ATA response was higher in the RG7667 than in the placebo group. These findings were not unexpected, as clinical immunogenicity to recombinant monoclonal antibody therapeutics is not uncommon (24–27). The clinical relevance of this observation is unknown, as there was no measurable effect upon RG7667 pharmacokinetics. Four patients in the placebo group tested negative for ATAs prior to receiving study drug and had at least one positive posttreatment ATA sample. This observation is likely a reflection of the use of two distinct ATA assays (one for each of the two recombinant monoclonal antibodies that make up RG7667), thus increasing the likelihood of positive responses among untreated patients by 2-fold, and the fact that both ATA assays had been developed and validated to be highly sensitive.
Although RG7667 significantly reduced the proportion of patients with CMV viremia within 24 weeks posttransplant, the proportion with CMV viremia still remained high, necessitating additional antiviral therapy. A number of agents, including small-molecule antivirals and CMV-IVIG, are available to prevent or treat CMV infection and disease (28). Valganciclovir has emerged as the most commonly used CMV prophylaxis in SOT recipients (29), but it has significant toxicities, such as neutropenia (5). Some transplant centers use preemptive antiviral therapy instead of prophylaxis in order to decrease medication toxicities and potentially improve anti-CMV immune responses (23, 28, 30). However, preemptive therapy still results in significant use of antiviral medication (23). Use of preemptive antiviral medication was lower in the RG7667 than in the placebo group, so the addition of RG7667 as an adjunct to existing antiviral therapy could enable lower overall exposure to antiviral medications and their toxicities and warrants further investigation.
During the active dosing period, RG7667 also significantly delayed median time to CMV viremia compared to the placebo. Thus, posttransplant treatment with RG7667 may provide benefits by delaying CMV viremia until a time when immunosuppressant doses are lower and CMV infection may be easier to manage (31, 32). Notably, despite this delay, there did not appear to be deleterious effects as levels of RG7667 decreased. In fact, we observed a smaller proportion of patients with late-onset (i.e., occurring after 3 months) (7) CMV disease in the RG7667 group than with the placebo group during the 24-week study.
Previous evidence from a multicenter randomized controlled trial of CMV-IVIG prophylaxis in the prevention of primary CMV disease in kidney transplant recipients indicated that administration of exogenous antibodies can confer a protective effect, as demonstrated by a 50% reduction in CMV disease in the CMV-IVIG group compared to the placebo (12). While the exact mechanism is unknown, CMV-IVIG is thought to neutralize free CMV virus, thereby inhibiting its entry into host cells, and may enhance antibody-dependent cell-mediated cytotoxicity (ADCC) (33–35). Compared to a polyclonal antibody preparation such as CMV-IVIG, a monoclonal antibody therapy such as RG7667 may have higher target specificity and potency (21). However, a study directly comparing RG7667 to CMV-IVIG would be required in order to draw conclusions about their relative impacts on CMV viremia.
The mechanism of action of RG7667 involves binding to distinct CMV antigens required for cellular entry, which inhibits infection of relevant host cells (21). While in vitro assays may not reflect what occurs in patients, we were unable to detect ADCC or complement-dependent cytotoxicity for either MCMV5322A or MCMV3068A in ARPE-19 epithelial cells or human umbilical vein endothelial cells (HUVECs) infected with CMV (data not shown). In fact, ADCC may even be deleterious to transplant patients if it causes a loss of both infected and noninfected cells as a bystander effect, which could lead to dysfunction in organs infected with CMV. Although a T cell response is critical for long-term protection against late-onset CMV disease (36), our study participants were receiving potent immunosuppression, which would have interfered with our ability to assess T cell response.
Although there was evidence of antiviral activity in this study, RG7667 did not lead to the hypothesized reduction in the primary endpoint of CMV viremia within 12 weeks posttransplant compared to placebo. This may have various causes. First, D+R− patients have a particularly high risk for developing CMV viremia (23). The placebo group had a lower proportion of patients with the primary endpoint than anticipated, resulting in the study being underpowered for the primary endpoint (see the supplemental material). In addition, although RG7667 inhibits CMV entry into epithelial cells, endothelial cells, macrophages, and fibroblasts (21) and would also likely inhibit entry into dendritic cells (37), the formal possibility exists that RG7667 does not neutralize entry into an unidentified cell type. RG7667 may also not inhibit CMV cell-to-cell spread, which may contribute to CMV viremia (38). Even though we observed robust neutralization in vitro using two antibodies, we might have demonstrated greater activity if RG7667 had included a greater number of monoclonal antibodies directed at different targets. Finally, greater concentrations of RG7667 in the target tissues and pharmacologic activity may have been achieved with use of a higher dose, an additional loading dose, more frequent dosing, and a longer treatment duration.
While our study involved D+R− kidney transplant recipients who are at the highest risk for CMV infection and disease (11, 23), we acknowledge that we did not evaluate the use of RG7667 in D+R+ patients who comprise the majority of kidney transplant recipients (39, 40) and who are at risk for superinfection by CMV from a CMV-seropositive donor, particularly in the setting of intense immunosuppression (40). Further study of the utility of RG7667 in a broader population of kidney transplant recipients who may suffer from CMV-related consequences would be of clinical interest.
In summary, this phase 2 study demonstrated that RG7667, a combination of two monoclonal antibodies targeting CMV glycoproteins, has antiviral activity in high-risk kidney transplant recipients. Specifically, RG7667 numerically reduced CMV infection within 12 and 24 weeks posttransplant, delayed time to CMV viremia, and was associated with less CMV disease than placebo. Additionally, RG7667 was well tolerated and had favorable pharmacokinetic and immunogenicity profiles. Further studies of RG7667, incorporating study design modifications such as different dosing parameters, for the prevention and treatment of CMV infection and disease in kidney and other transplant recipients and additional populations, such as pregnant women, are warranted.
MATERIALS AND METHODS
Patients.
CMV-seronegative patients, at least 18 years old, receiving a first or second kidney transplant from a CMV-seropositive living or deceased donor (D+R−) were enrolled from December 2012 to April 2014 from 39 sites in the United States and European Union. This study was approved by the institutional review board of the study institutions, and all participants provided written informed consent.
Patients were excluded if they were receiving a kidney transplant from a CMV-seronegative donor, were undergoing multiorgan transplant, were suspected of having CMV disease, had received anti-CMV therapy within 30 days before screening or were expected to receive such therapy during the study, had abnormal liver function tests, or had hepatitis B or C or human immunodeficiency virus infection.
Study drug.
RG7667 is a 1:1 ratio of two monoclonal antibodies, briefly described here. MCMV5322A is a human, affinity-matured version of MSL-109 that recognizes CMV glycoprotein H (gH) (21). MCMV3068A is a humanized mouse monoclonal antibody that recognizes the gH/gL/UL128/UL130/UL131 complex (21). MCMV5322A and MCMV3068A (20 mg/ml for each antibody) or their corresponding placebos were instilled in a 1:1 ratio into sterile normal saline to achieve the target dose of 10 mg/kg of body weight of each antibody before infusion.
Study design.
This phase 2, randomized, double-blind, placebo-controlled trial assessed the safety and activity of RG7667, administered prophylactically, for the prevention of CMV infection. Secondary objectives were to characterize the pharmacokinetics and immunogenicity of RG7667.
At the time of transplant, enrolled patients were randomized at a 1:1 ratio using permuted blocks of four to receive a total of four intravenous doses of RG7667 or placebo. Randomization was stratified by region (United States versus European Union) and use of antithymocyte globulin and/or alemtuzumab for induction immunosuppression. The investigators, site clinical staff, and patients were blinded to treatment assignment. Study drug was administered at the time of transplant (within 24 h before to 48 h after transplant; day 1 was the day of the first infusion) and on days 8, 29, and 57 (Fig. 3).
FIG 3.

Study schematic. Randomized patients received study drug on days 1 (within 24 h before to 48 h after transplant), 8, 29, and 57 and were monitored for efficacy and safety outcomes until study completion (day 169).
Patients in both study arms received weekly blood draws during weeks 0 to 12 and at least every other week during weeks 13 to 24 after transplant in order to detect and quantify CMV viral load in plasma samples. Due to differences in diagnostic assays, there is no standard accepted threshold for CMV viremia at which to start preemptive therapy (41); investigators could therefore initiate preemptive antiviral therapy (e.g., valganciclovir, ganciclovir [GCV], foscarnet, or CMV-IVIG) at their discretion when viral load measurements were deemed clinically meaningful. If CMV disease (i.e., CMV syndrome or tissue-invasive CMV disease as detailed in the supplemental material) was suspected, anti-CMV treatment was initiated.
Efficacy assessments.
The primary endpoint was the proportion of patients with CMV viremia (viral load of ≥150 copies/ml [137 IU/ml], the limit of quantification of the assay) within 12 weeks posttransplant. Secondary endpoints included the proportion of patients with CMV viremia within 24 weeks posttransplant, the proportion of patients with CMV disease within 24 weeks posttransplant, time to CMV viremia, viral load at first detection of CMV viremia, peak CMV viral load, and initiation of antiviral therapy within 12 and 24 weeks posttransplant. For endpoint ascertainment, CMV viral load testing was performed by quantitative PCR at a central laboratory on a single platform (COBAS AmpliPrep/COBAS TaqMan CMV test [CAP/CTM CMV test]). CMV viral load testing was also performed on plasma samples at local laboratories to inform decision-making regarding initiation of antiviral therapy.
Safety assessments.
Safety data were assessed from screening until 16 weeks following the last dose of study drug (i.e., approximately 24 weeks posttransplant). Adverse events (AEs) were graded according to the National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI CTCAE; v4.0) (42). Treatment-emergent AEs (TEAEs) were defined as any AEs that occurred after the start of the study drug infusion or were present before but worsened after the start of the study drug infusion. GCV resistance testing was performed as described in the supplemental material.
Pharmacokinetics and immunogenicity assessments.
Enzyme-linked immunosorbent assays were used to determine serum concentrations of MCMV5322A and MCMV3068A and to screen and confirm the presence of antitherapeutic antibodies (ATAs) as described previously (21; see also the supplemental material).
Statistical analyses.
Participant characteristics were summarized for all randomized patients by treatment group using means and standard deviations for continuous variables and frequency counts and percentages for categorical variables.
Efficacy analyses included patients who were D+R− at transplantation with viral load data available for analysis, and patients were assigned to the treatment group to which they were randomized (i.e., modified intention to treat [mITT]). Efficacy endpoints were summarized using frequency counts, percentages, medians, and 95% confidence intervals (CIs). Estimations of the treatment difference and CI between groups were calculated using stratum-adjusted Mantel-Haenszel methodology (43). For time-to-event analyses, Kaplan-Meier methodology and Cox regression were used (supplemental material).
Safety analyses included randomized patients who had received at least one dose of study drug, with patients assigned to the treatment group associated with the regimen received. Safety data were summarized using frequency counts and percentages. Assessment of kidney transplant rejection was a safety outcome of particular interest and relevance and was made based on safety data (combining the preferred terms “kidney transplant rejection” and “transplant rejection”) reported by the investigators. Pharmacokinetic assessment was performed for serum concentration-time data using standard noncompartmental methods and actual blood sampling times. ATA data were summarized by treatment.
Data analysis was performed using SAS version 9.1 (SAS Institute, Inc., Cary, NC) and Phoenix WinNonlin version 6.2 (Certara, L.P., Princeton, NJ) for the pharmacokinetic analysis. An Internal Monitoring Committee (IMC) examined unblinded data to facilitate ongoing monitoring of safety and tolerability. The study was also monitored by a Scientific Oversight Committee that included the IMC members and two external subject matter experts who remained blinded to treatment allocation.
Supplementary Material
ACKNOWLEDGMENTS
We thank the patients for their participation in this study. We thank the RG7667 investigators: from Belgium, Emine Broeders, Hopital Erasme; Dirk Kuypers, Universitair Ziekenhuis Gent; and Patrick Peeters, Universitair Ziekenhuis Gent; from France, Gilles Blancho, CHU de Nantes; Luc Frimat, Hopitaux De Brabois; Nassim Kamar, Hopital Rangueil; Yvon Lebranchu, CHU de Tours; Christophe Legendre, Hopital Necker; and Pierre-Gilles Merville, Hopital Pellegrin-CHU de Bordeaux; from Germany, Ingeborg Hauser, Klinik Johann Wolfgang von Goethe Uni; Christian Hugo, Universitätsklinikum “Carl Gustav Carus”; Claudia Sommerer, Uniklinikum Heidelberg; and Oliver Witzke, Universitätsklinikum Essen; from Norway, Anders Hartmann, Oslo Universitetssykehus HF; from Spain, Gabriel Bernal Blanco, CEIC del Hospital Virgen del Rocío; Carme Facundo Molas, Fundació Puigvert; Josep Maria Grinyó Boira, Hospital Universitari de Bellvitge; Fransesc Moreso Mateos, Hospital Universitari Vall d'Hebron; and Federico Oppenheimer Salinas, Hospital Clinic i Provincial de Barcelona; from Sweden, Lars Bäckman, Akademiska Sjukhuse; Marie Felldin, Sahlgrenska Universitetssjukhuset; and Lars Wennberg, Karolinska University Hospital; from the United Kingdom, Mark Harber, Royal Free Hospital; and Rachel Hilton, Guy's and St. Thomas' NHS Foundation Trust, Guy's Hospital; from the United States, Yousri Barri, Baylor University Medical Center; Suphamai Bunnapradist, University of California Los Angeles; Laurence Chan, University of Colorado Health Sciences Center; David Cohen, Columbia University; Matthew Cooper, Georgetown University Hospital; Rowena Delos Santos, Washington University School of Medicine/Barnes Jewish Hospital; Shirish Huprikar, Icahn School of Medicine at Mount Sinai; Abhijit (Ajit) Limaye, University of Washington Medical Center; Gautham Mogilishetty, University of Cincinnati College of Medicine; Jack Moore, MedStar Washington Hosp Center; Laura Mulloy, Georgia Regents Medical Center; Oleh Pankewycz, Eric County Medical Center; Ram Peddi, California Pacific Medical Center; Adele Rike-Shields, University of Cincinnati College of Medicine; Steven Steinberg, California Institute of Renal Research; and Flavio Vincenti, University of California San Francisco.
We also thank Aide Castro, Patrick McLeroth, the Genentech Internal Monitoring Committee (Carole Ho, Andrew Erdman, and Tracy Burgess), the external members of the Scientific Oversight Committee (David Snydman and Daniele Lilleri), and Daniel Sheinson for his programming assistance. Manuscript preparation and editorial assistance were provided by Bryan Hains, Sarah Adai, and Deborah Solymar (Genentech, Inc.).
J.H.I., A.K.M., and P.G. have received research funding from Genentech, Inc. A.P. is a consultant to Genentech, Inc., Novartis, and Alexion Pharmaceuticals, Inc. D.R.S. is a consultant to Genentech, Inc., Chimerix, Inc., Merck & Co., Inc., and Millennium Pharmaceuticals, Inc., and has received research support from Merck & Co., Inc., Tetraphase Pharmaceuticals, Actelion Pharmaceuticals US, Inc., and Summit Pharmaceuticals International. B.F. is currently an employee of Gilead Sciences, Inc. A.E.F. is currently an employee of GRAIL, Inc. N.S.S. is currently an employee of BioMarin Pharmaceutical, Inc. X.C.L. is currently an employee of Immune-Onc Therapeutics, Inc. J.M., T.B., M.A.D., B.E., B.F., A.E.F., M.M., R.D., C.M.R., L.A.G., N.S.S. X.C.L., and J.A.T. are/were (at the time of this study) employees of Genentech, Inc. (a member of the Roche Group), and shareholders of Roche.
This work was supported by Genentech, Inc. (South San Francisco, California). Genentech, Inc., was involved in the study design, data interpretation, and the decision to submit for publication in conjunction with the authors.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01794-16.
REFERENCES
- 1.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Gane E, Saliba F, Valdecasas GJ, O'Grady J, Pescovitz MD, Lyman S, Robinson CA. 1997. Randomised trial of efficacy and safety of oral ganciclovir in the prevention of cytomegalovirus disease in liver-transplant recipients. The Oral Ganciclovir International Transplantation Study Group. Lancet 350:1729–1733. [DOI] [PubMed] [Google Scholar]
- 3.Lowance D, Neumayer HH, Legendre CM, Squifflet JP, Kovarik J, Brennan PJ, Norman D, Mendez R, Keating MR, Coggon GL, Crisp A, Lee IC. 1999. Valacyclovir for the prevention of cytomegalovirus disease after renal transplantation. International Valacyclovir Cytomegalovirus Prophylaxis Transplantation Study Group. N Engl J Med 340:1462–1470. [DOI] [PubMed] [Google Scholar]
- 4.Goodrich JM, Bowden RA, Fisher L, Keller C, Schoch G, Meyers JD. 1993. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann Intern Med 118:173–178. doi: 10.7326/0003-4819-118-3-199302010-00003. [DOI] [PubMed] [Google Scholar]
- 5.Hodson EM, Ladhani M, Webster AC, Strippoli GF, Craig JC. 2013. Antiviral medications for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst Rev 2:CD003774. [DOI] [PubMed] [Google Scholar]
- 6.Limaye AP, Bakthavatsalam R, Kim HW, Kuhr CS, Halldorson JB, Healey PJ, Boeckh M. 2004. Late-onset cytomegalovirus disease in liver transplant recipients despite antiviral prophylaxis. Transplantation 78:1390–1396. doi: 10.1097/01.TP.0000145989.22373.03. [DOI] [PubMed] [Google Scholar]
- 7.Arthurs SK, Eid AJ, Pedersen RA, Kremers WK, Cosio FG, Patel R, Razonable RR. 2008. Delayed-onset primary cytomegalovirus disease and the risk of allograft failure and mortality after kidney transplantation. Clin Infect Dis 46:840–846. doi: 10.1086/528718. [DOI] [PubMed] [Google Scholar]
- 8.Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang ML, Myerson D, Stevens-Ayers T, Flowers ME, Cunningham T, Corey L. 2003. Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood 101:407–414. doi: 10.1182/blood-2002-03-0993. [DOI] [PubMed] [Google Scholar]
- 9.Singh N. 2006. Cytomegalovirus infection in solid organ transplant recipients: new challenges and their implications for preventive strategies. J Clin Virol 35:474–477. doi: 10.1016/j.jcv.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Department of Health and Human Services Health Resources and Services Administration. 2014. United States Organ Transplantation. OPTN/SRTR. Annual Data Report 2012. U.S. Department of Health and Human Services Health Resources and Services Administration, Washington, DC. [Google Scholar]
- 11.De Keyzer K, Van Laecke S, Peeters P, Vanholder R. 2011. Human cytomegalovirus and kidney transplantation: a clinician's update. Am J Kidney Dis 58:118–126. doi: 10.1053/j.ajkd.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Snydman DR, Werner BG, Heinze-Lacey B, Berardi VP, Tilney NL, Kirkman RL, Milford EL, Cho SI, Bush HL Jr, Levey AS, Strom TB, Carpenter CB, Levey RH, Harmon WE, Zimmerman CE Jr, Shapiro ME, Steinman T, LoGerfo F, Idelson B, Schröter GPJ, Levin MJ, McIver J, Leszczynski J, Grady GF. 1987. Use of cytomegalovirus immune globulin to prevent cytomegalovirus disease in renal-transplant recipients. N Engl J Med 317:1049–1054. doi: 10.1056/NEJM198710223171703. [DOI] [PubMed] [Google Scholar]
- 13.U.S. Food and Drug Administration. March 2007. Cytomegalovirus immune globulin intravenous. http://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/UCM197962.pdf Accessed 22 October 2014.
- 14.CSL Behring Canada. 2014. Product monograph: Cytogam cytomegalovirus immune globulin intravenous (human). http://www.cslbehring.ca/docs/534/245/2015-03-02_182259_E_Cytogam.pdf Accessed 7 December 2014.
- 15.Fisher RA, Kistler KD, Ulsh P, Bergman GE, Morris J. 2012. The association between cytomegalovirus immune globulin and long-term recipient and graft survival following liver transplantation. Transpl Infect Dis 14:121–131. doi: 10.1111/j.1399-3062.2011.00664.x. [DOI] [PubMed] [Google Scholar]
- 16.Snydman DR, Kistler KD, Ulsh P, Bergman GE, Vensak J, Morris J. 2011. The impact of CMV prevention on long-term recipient and graft survival in heart transplant recipients: analysis of the Scientific Registry of Transplant Recipients (SRTR) database. Clin Transplant 25:E455–E462. doi: 10.1111/j.1399-0012.2011.01459.x. [DOI] [PubMed] [Google Scholar]
- 17.Saylor C, Dadachova E, Casadevall A. 2009. Monoclonal antibody-based therapies for microbial diseases. Vaccine 27(Suppl 6):G38–G46. doi: 10.1016/j.vaccine.2009.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan DC. 2001. Cytomegalovirus in renal transplantation. J Am Soc Nephrol 12:848–855. [DOI] [PubMed] [Google Scholar]
- 19.Varga M, Remport A, Hídvégi M, Péter A, Kóbori L, Telkes G, Fazakas J, Gerlei Z, Sárváry E, Sulyok B, Járay J. 2005. Comparing cytomegalovirus prophylaxis in renal transplantation: single center experience. Transpl Infect Dis 7:63–67. doi: 10.1111/j.1399-3062.2005.00094.x. [DOI] [PubMed] [Google Scholar]
- 20.Both L, Banyard AC, van Dolleweerd C, Wright E, Ma JK, Fooks AR. 2013. Monoclonal antibodies for prophylactic and therapeutic use against viral infections. Vaccine 31:1553–1559. doi: 10.1016/j.vaccine.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishida JH, Burgess T, Derby MA, Brown PA, Maia M, Deng R, Emu B, Feierbach B, Fouts AE, Liao XC, Tavel JA. 2015. Phase 1 randomized, double-blind, placebo-controlled study of RG7667, an anticytomegalovirus combination monoclonal antibody therapy, in healthy adults. Antimicrob Agents Chemother 59:4919–4929. doi: 10.1128/AAC.00523-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishida JH, Patel A, McBride J, Burgess T, Derby MA, Emu B, Feierbach B, Liao XC, Maia M, Deng R, Rosenberger CM, Gennaro LA, Striano N, Tavel JA. 2015. Phase 2 randomized, double-blind, placebo-controlled trial of RG7667 for prevention of cytomegalovirus infection in high-risk kidney transplant recipients. Abstr Joint 55th Intersci Conf Antimicrob Agents Chemother/28th Int Congr Chemother Infect, abstr 2015-A-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramanan P, Razonable RR. 2013. Cytomegalovirus infections in solid organ transplantation: a review. Infect Chemother 45:260–271. doi: 10.3947/ic.2013.45.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, Parish T, Scott G, Shankar G, Shores E, Swanson SJ, Taniguchi G, Wierda D, Zuckerman LA. 2004. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods 289:1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Schellekens H, Casadevall N. 2004. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol 251(Suppl 2):II4–II9. [DOI] [PubMed] [Google Scholar]
- 26.Büttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, Kramer D, Kropshofer H, Lloyd P, Lubiniecki A, Krause R, Mire-Sluis A, Platts-Mills T, Ragheb JA, Reipert BM, Schellekens H, Seitz R, Stas P, Subramanyam M, Thorpe R, Trouvin JH, Weise M, Windisch J, Schneider CK. 2011. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals 39:100–109. doi: 10.1016/j.biologicals.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Tovey MG, Lallemand C. 2011. Immunogenicity and other problems associated with the use of biopharmaceuticals. Ther Adv Drug Saf 2:113–128. doi: 10.1177/2042098611406318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torres-Madriz G, Boucher HW. 2008. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 47:702–711. doi: 10.1086/590934. [DOI] [PubMed] [Google Scholar]
- 29.Hodson EM, Jones CA, Webster AC, Strippoli GF, Barclay PG, Kable K, Vimalachandra D, Craig JC. 2005. Antiviral medications to prevent cytomegalovirus disease and early death in recipients of solid-organ transplants: a systematic review of randomised controlled trials. Lancet 365:2105–2115. doi: 10.1016/S0140-6736(05)66553-1. [DOI] [PubMed] [Google Scholar]
- 30.Abate D, Saldan A, Fiscon M, Cofano S, Paciolla A, Furian L, Ekser B, Biasolo MA, Cusinato R, Mengoli C, Bonfante L, Rossi B, Rigotti P, Sgarabotto D, Barzon L, Palù G. 2010. Evaluation of cytomegalovirus (CMV)-specific T cell immune reconstitution revealed that baseline antiviral immunity, prophylaxis, or preemptive therapy but not antithymocyte globulin treatment contribute to CMV-specific T cell reconstitution in kidney transplant recipients. J Infect Dis 202:585–594. doi: 10.1086/654931. [DOI] [PubMed] [Google Scholar]
- 31.Å-sberg A, Jardine AG, Bignamini AA, Rollag H, Pescovitz MD, Gahlemann CC, Humar A, Hartmann A, VICTOR Study Group. 2010. Effects of the intensity of immunosuppressive therapy on outcome of treatment for CMV disease in organ transplant recipients. Am J Transplant 10:1881–1888. doi: 10.1111/j.1600-6143.2010.03114.x. [DOI] [PubMed] [Google Scholar]
- 32.Avery RK, Kaplan B. 2010. Immunosuppressive agents and CMV risk in the VICTOR study. Am J Transplant 10:1727–1728. doi: 10.1111/j.1600-6143.2010.03194.x. [DOI] [PubMed] [Google Scholar]
- 33.Nigro G, Adler SP, La Torre R, Best AM, Congenital Cytomegalovirus Collaborating Group. 2005. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med 353:1350–1362. doi: 10.1056/NEJMoa043337. [DOI] [PubMed] [Google Scholar]
- 34.Bonaros N, Mayer B, Schachner T, Laufer G, Kocher A. 2008. CMV-hyperimmune globulin for preventing cytomegalovirus infection and disease in solid organ transplant recipients: a meta-analysis. Clin Transplant 22:89–97. [DOI] [PubMed] [Google Scholar]
- 35.Manischewitz JE, Quinnan GV Jr. 1980. Antivirus antibody-dependent cell-mediated cytotoxicity during murine cytomegalovirus infection. Infect Immun 29:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watkins RR, Lemonovich TL, Razonable RR. 2012. Immune response to CMV in solid organ transplant recipients: current concepts and future directions. Expert Rev Clin Immunol 8:383–393. doi: 10.1586/eci.12.25. [DOI] [PubMed] [Google Scholar]
- 37.Gerna G, Percivalle E, Lilleri D, Lozza L, Fornara C, Hahn G, Baldanti F, Revello MG. 2005. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J Gen Virol 86:275–284. doi: 10.1099/vir.0.80474-0. [DOI] [PubMed] [Google Scholar]
- 38.Jacob CL, Lamorte L, Sepulveda E, Lorenz IC, Gauthier A, Franti M. 2013. Neutralizing antibodies are unable to inhibit direct viral cell-to-cell spread of human cytomegalovirus. Virology 444:140–147. doi: 10.1016/j.virol.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Atabani SF, Smith C, Atkinson C, Aldridge RW, Rodriguez-Perálvarez M, Rolando N, Harber M, Jones G, O'Riordan A, Burroughs AK, Thorburn D, O'Beirne J, Milne RS, Emery VC, Griffiths PD. 2012. Cytomegalovirus replication kinetics in solid organ transplant recipients managed by preemptive therapy. Am J Transplant 12:2457–2464. doi: 10.1111/j.1600-6143.2012.04087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Ruiz M, Arias M, Campistol JM, Navarro D, Gómez-Huertas E, Gómez-Márquez G, Díaz JM, Hernández D, Bernal-Blanco G, Cofan F, Jimeno L, Franco-Esteve A, González E, Moreso FJ, Gómez-Alamillo C, Mendiluce A, Luna-Huerta E, Aguado JM, OPERA Study Group. 2015. Cytomegalovirus prevention strategies in seropositive kidney transplant recipients: an insight into current clinical practice. Transpl Int 28:1042–1054. doi: 10.1111/tri.12586. [DOI] [PubMed] [Google Scholar]
- 41.Hayden RT, Preiksaitis J, Tong Y, Pang X, Sun Y, Tang L, Cook L, Pounds S, Fryer J, Caliendo AM. 2015. Commutability of the first World Health Organization International standard for human cytomegalovirus. J Clin Microbiol 53:3325–3333. doi: 10.1128/JCM.01495-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.National Cancer Institute. 2014. National Cancer Institute common toxicity criteria for adverse events (NCI CTCAE; v4.0). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40 Accessed 17 October 2014.
- 43.Koch GG, Carr GJ, Amara IA, Stokes ME, Uryniak TJ. 1989. Categorical data analysis. In Berry DA. (ed), Statistical methodology in the pharmaceutical sciences. Marcel Dekker, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.