

Abstract
Objective:
To identify differences in the metabolomic profile in the serum of patients with multiple sclerosis (MS) compared to controls and to identify biomarkers of disease severity.
Methods:
We studied 2 cohorts of patients with MS: a retrospective longitudinal cohort of 238 patients and 74 controls and a prospective cohort of 61 patients and 41 controls with serial serum samples. Patients were stratified into active or stable disease based on 2 years of prospective assessment accounting for presence of clinical relapses or changes in disability measured with the Expanded Disability Status Scale (EDSS). Metabolomic profiling (lipids and amino acids) was performed by ultra-high-performance liquid chromatography coupled to mass spectrometry in serum samples. Data analysis was performed using parametric methods, principal component analysis, and partial least square discriminant analysis for assessing the differences between cases and controls and for subgroups based on disease severity.
Results:
We identified metabolomics signatures with high accuracy for classifying patients vs controls as well as for classifying patients with medium to high disability (EDSS >3.0). Among them, sphingomyelin and lysophosphatidylethanolamine were the metabolites that showed a more robust pattern in the time series analysis for discriminating between patients and controls. Moreover, levels of hydrocortisone, glutamic acid, tryptophan, eicosapentaenoic acid, 13S-hydroxyoctadecadienoic acid, lysophosphatidylcholines, and lysophosphatidylethanolamines were associated with more severe disease (non-relapse-free or increase in EDSS).
Conclusions:
We identified metabolomic signatures composed of hormones, lipids, and amino acids associated with MS and with a more severe course.
Metabolomics provides the opportunity to identify molecular patterns from serum or other tissues associated with multiple sclerosis (MS), such as the presence of metabolites involved in the control of the immune response as well as markers of brain damage that leak from tissue into the serum.1–6 Identification of biomarkers of disease severity (either relapse activity or disability worsening) or response to therapy is critical in order to improve patient management and the search for new therapies for patients with MS.7 Previous metabolomic studies in the serum or CSF of patients with MS have found evidence of differential levels of lipid or amino acid concentrations compared to controls,8–17 supporting the rationale behind this approach.
The aim of this study was to perform a metabolomic analysis in the serum of patients with MS in order to identify metabolomic signatures associated with the disease. Moreover, we aimed to identify biomarkers predicting disease severity, defined by either the presence of relapses (disease activity) or sustained increase in disability (disability worsening).18 To this aim, we make use of 2 well-characterized cohorts in which clinical phenotype was defined prospectively. We used a prospective cohort with longitudinal serum samples, allowing the assessment of metabolite stability over time, and another cross-sectional cohort with longitudinal clinical data and a larger sample size to account for interindividual variability. By using both groups as screening cohorts, we attempted to identify robust metabolites associated with the disease. Furthermore, their presence in both cohorts served to validate the metabolites. We made use of ultra-high-performance liquid chromatography coupled to mass spectrometry (UHPLC-MS), which allowed for the semiquantitative analysis of a wide array of lipids and amino acids, although this was not suitable for the detection of metabolites related to central carbon metabolism (e.g., glycolytic metabolites).19,20
METHODS
Patients.
We studied 2 independent cohorts of patients with MS: a retrospective longitudinal cohort from 5 centers (with longitudinally collected clinical information for 2 years) and a prospective cohort with serial serum samples for 2 years from a single center. Inclusion criteria included fulfilling McDonald criteria21 and agreeing to donate a blood sample for the study. Exclusion criteria included any significant comorbidity influencing the metabolome (e.g., diabetes, hypertriglyceridemia). Consecutive patients were recruited by their neurologists.
The retrospective longitudinal cohort was a multicenter cohort composed of 238 patients and 74 controls recruited between 2010 and 2014 from the Hospital Clinic of Barcelona; Hospital Vall d'Hebron, Barcelona; Hospital Ramon y Cajal, Madrid; Hospital Universitario Regional, Malaga; and Hospital Clinico San Carlos, Madrid, Spain. Patients were of any disease subtype and were prospectively followed for 2 years in their centers, with clinical variables, including age at disease onset, disease duration, presence of relapses, and disability status using the Expanded Disability Status Scale (EDSS) (table 1), collected every 6–12 months. To decrease variability in the retrospective cohort, the use of disease-modifying drugs was restricted to only interferon-β (as it was the most common therapy at this time). Serum samples were collected in the mornings (between 9 am and 12 pm) and stored at −80°C until metabolomics analysis. This cohort was compared to a group of 74 matched healthy controls (age 33.2 ± 4.2 years, 50 women and 24 men) with no diseases or medications.
Table 1.
Demographics, clinical variables, and samples of the multiple sclerosis cohorts
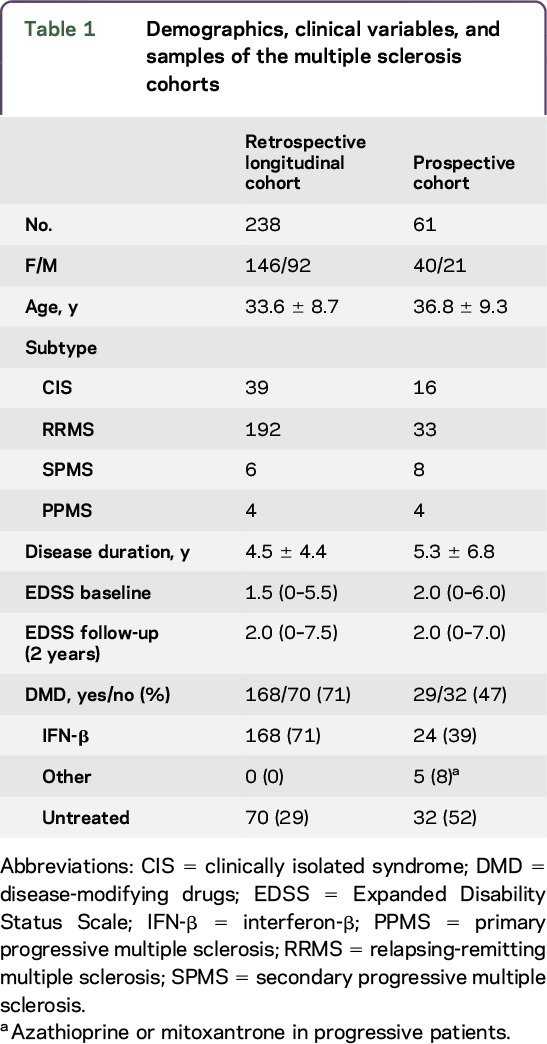
The prospective cohort included 61 patients and was recruited between 2008 and 2012 (table 1). Consecutive patients were prospectively followed for 2 years, collecting clinical variables such as age at disease onset, disease duration, disease subtype, presence of relapses, and disability using the EDSS at baseline and every 6 months. The use of disease-modifying drugs was allowed and patients were stable in their therapy for the last 6 months. In addition, we recruited 41 healthy controls (age 33.8 + 6.4 years; 29 women and 12 men). Serum samples were collected every 3 months in patients and healthy controls (9 samples per participant). As in the previous cohort, serum samples were collected in the mornings after overnight fasting and stored at −80°C until use.
Standard protocol approvals, registrations, and patient consents.
The ethical committee of each institution approved the study and patients were included after signing the informed consent.
Clinical variables.
We collected a set of relevant clinical variables at baseline, including age at study inclusion, sex, disease subtype, disease duration, and EDSS score. Presence of relapses, EDSS, and changes in the use of disease-modifying drugs were assessed every 6 months for 2 years. After the 2-year follow-up, patients were classified as having stable or active disease using following definitions: (1) stable disease—no relapses and no changes in the EDSS (confirmed at 3 and 6 months) during the 2 years of follow-up; (2) active disease—2 or more relapses or a 1-point increase in the EDSS score (0.5 points for EDSS between 5.0 and 6.5) confirmed at 3 months during the 2 years of follow-up. Disability severity was tested by comparing patients with EDSS <3.0 (a cutoff being used in the definition of benign MS18) against those with EDSS >4.5 (a cutoff identified in natural studies associated with the onset of progressive MS22) and patients between both cutoffs were defined as uncategorized and excluded from the analysis.
Metabolomics analysis.
Two different approaches were followed for the analysis of both cohorts (figure e-1 at Neurology.org/nn). Serum samples included in the prospective cohort were analyzed following the untargeted procedure described by Barr et al.23 Briefly, the samples were defrosted, vortex mixed, and centrifuged. An aliquot of the sample (50 μL) was diluted with methanol (200 μL), vortex mixed, and stored at −20°C overnight. Then, samples were centrifuged at ×16,000 g for 10 minutes; the supernatant was analyzed by UHPLC-MS. In order to avoid systematic bias during the procedure, all samples were randomized prior to the metabolite extraction and analyzed blinded to the diagnosis or disease phenotype.23
The analysis of the retrospective longitudinal cohort involved a targeted approach, applying 3 separate UHPLC-MS platforms as previously described19 (figure e-1). Briefly, UHPLC–single quadrupole–MS was used for amino acid analysis and combined with 2 separate UHPLC–time-of-flight (TOF)–MS-based platforms analyzing methanol and chloroform/methanol serum extracts. The first UHPLC-TOF-MS platform analyzed methanol serum extracts, including the following identified ion features: nonesterified fatty acids, oxidized fatty acids, acylcarnitines, N-acyl ethanolamines, bile acids, steroids, and lysoglycerophospholipids.19 The second UHPLC-TOF-MS platform was optimized for the analysis of chloroform/methanol extracts, covering glycerolipids, glycerophospholipids, cholesteryl esters, sphingolipids, and primary fatty acid amides. In this case, all the metabolic features were identified prior to the analysis.19
Because the first approach did not allow for identification of metabolites, but rather peaks from the spectra, both studies were considered screening studies. However, we were able to retrospectively identify several of the peaks from the first analysis using the second UPLC platform and were thereby able to validate several of the metabolites (although we were unable to completely identify all the metabolites defining the signatures associated with disease activity from the prospective cohort).
Data preprocessing.
The untargeted data analysis of the prospective cohort was performed as follows. A list of intensities of the chromatographic peak areas detected was generated for each sample, identifying each peak as retention time (Rt) and mass-to-charge (m/z) data pairs. This process was initiated with the first sample and repeated for each UHPLC-MS run in the batch, sorting the data such that the correct peak intensity for each Rt–m/z pair was aligned in the final table. Data were then normalized to correct for instrument sensitivity drift over time and sample-to-sample preparation concentration variations. Normalization between batches was carried out by relating the 10 extractions of reference serum samples injected per batch. The sample-to-sample normalization constant was the median of the ratios of intensities for all Rt–m/z pairs between the considered injection and a reference injection (which was the first reference serum sample injected; choice of the reference injection is arbitrary).
In the case of the targeted approach used in the retrospective longitudinal study, data preprocessing was performed following the procedure described perviously.20 Briefly, data obtained were preprocessed using the TargetLynx application manager for MassLynx (Waters Corp., Milford, MA).20 The UHPLC-MS features were identified prior to the analysis, either by comparison of their chromatographic Rt and accurate mass spectra with those of reference standards or by accurate mass MS/MS fragment ion analysis if the reference standard was not available, as described previously.19 Intrabatch and interbatch normalization was performed based on the criteria of multiple internal standards and pool calibration samples approach.20
Statistical analysis.
The advantages of using both univariate and multivariate approaches in data processing are complementary and their results do not necessarily coincide.24 Considering that untargeted metabolomic analysis provided peaks of spectra but not individual metabolites, we used machine-learning techniques for the analysis, whereas for targeted metabolomics analysis providing individual metabolite levels, we used statistical analysis. Unsupervised principal components analysis (PCA) reduces the dimensionality of the complex dataset to enable easy visualization of any metabolic clustering of the different groups of samples. The supervised orthogonal projection to latent structures (OPLS) method is a supervised approach for discriminating between groups. As it is a supervised analysis, larger differences between groups are obtained. This approach is usually used in the data analysis process for the identification of metabolites contributing to the clustering observed in the PCA plots. PCA and OPLS multivariate data analysis were applied (SIMCA-P+ software package, version 12.0.1; Umetrics, Umea, Sweden). Analyses of variance (ANOVAs) were performed for each metabolite as a statistical test for evaluating possible differences between the means of each peak of the disease subgroups (e.g., EDSS by month 24: 0–3.0, 3.5–4.5, and >4.5). Unpaired Student t test p values (or Welch T test for unequal variances) were also calculated for the following comparisons (R v2.13.0 [R Development Core Team, 2010; cran.r-project.org] with MASS, xlxs, robustbase, and pwr packages): (1) active vs nonactive disease (clinical relapses); (2) disability worsening by EDSS: decrease or stable EDSS vs increased EDSS (≥1 point); and (3) EDSS change from baseline to month 24. A p value < 0.05 was considered significant.
An alternative approach to time series biomarkers discovery was applied.13 First, we used the smoothing splines mixed effects (SME) model that treats each longitudinal measurement as a smooth function of time, and second we utilized an associated functional test statistic (ANOVA for repeated measurements).13 Statistical significance was assessed by a nonparametric bootstrap procedure. Results are described following Strengthening the Reporting of Observational Studies in Epidemiology criteria and the guidelines for uniform reporting of body fluid biomarker studies in neurologic disorders.25 Sample sizes were defined by the availability of a convenience dataset due to the lack of similar studies for performing power calculations and the exploratory nature of this study.
RESULTS
Identification of metabolomic signatures in patients with MS.
We analyzed serum samples from a prospective cohort of 61 patients with MS and 41 matched controls, collected every 3 months over 2 years (8 samples per participant) by UPLC-MS. We searched for different signatures of the metabolomic profile between patients and controls. PCA analysis grouped cases and controls based on the metabolomic profile with moderate accuracy (R2 = 0.247, Q2 = 0.128, figure 1A) and the PLS-DA classifier classified participants as healthy participants or patients with MS with high accuracy (R2X = 0.414, R2Y = 0.481, Q2Y = 0.348, figure 1E).
Figure 1. Metabolic signatures in the longitudinal cohort.

(A–D) Principal components analysis (PCA) models. Each model is a linear combination of the original retention time (Rt)–mass-to-charge (m/z) pair peak areas. Hotelling T2 statistic is shown on the plot, defining a 95% confidence ellipse for the samples included in the analysis. (A) Patients vs healthy controls (R2 = 0.247; Q2 = 0.128). (B) Patients with relapses during follow-up vs relapse-free patients (R2 = 0.380; Q2 = 0.128). (C) Patients reaching Expanded Disability Status Scale (EDSS) >4 from baseline to end of follow-up by month 24 (R2 = 0.380; Q2 = 0.126). (D) Patients with increase in the EDSS (ΔEDSS) from baseline to end of follow-up by month 24 (R2 = 0.381; Q2 = 0.129). R2 = Degree of fit (goodness of prediction); Q2 = predictive ability (goodness of prediction). (E–H) Orthogonal projection to latent structures (OPLS) models. (E) Patients vs healthy controls (R2X = 0.414; R2Y = 0.481; Q2Y = 0.348). (F) Patients with relapses during follow-up vs relapse-free patients (R2X = 0389; R2Y = 0.425; Q2Y = 0.231). (G) Patients reaching EDSS >4.0 from baseline to end of follow-up by month 24 (R2X = 0.512; R2Y = 0.756; Q2Y = 0.555). (H) Patients with ΔEDSS from baseline to end of follow-up by month 24 (R2X = 0.148; R2Y = 0.222; Q2Y = 0.036). R2X = degree of fit for X axis; R2Y = degree of fit for y-axis; Q2Y = predictive ability for y-axis. MS = multiple sclerosis.
Due to the fact that serum samples were obtained prospectively every 3 months over 2 years, we performed a time-series analysis in order to assess the stability of the metabolomic signature. We found 29 peaks of the spectra associated with MS compared to controls along the 2-year follow-up, using ANOVA and SME approaches (p < 0.01). Metabolite identification was achieved for a subset of these peaks and revealed sphingomyelin and a lysophosphatidylethanolamine as the main metabolites of the MS signature (table e-1). We observed that changes in lysophosphatidylethanolamine over time were associated with the use of disease-modifying drugs (interferon-β) (p = 0.01) (figure e-2).
Second, we were interested in identifying differences in the metabolomic profile associated with disease severity. Patients were classified after 2 years of follow-up in active or stable disease based on presence of relapses or changes in disability measured with EDSS (see Methods for definitions). Therefore, patients were compared based on relapse-free status or increase in the EDSS by the end of follow-up. We found a metabolomic signature that was able to identify patients with MS with an increase in their EDSS 2 years later with high accuracy as well a metabolomic signature that identified relapse-free patients with moderate accuracy. Specifically, both PCA and PLS analysis differentiate between relapse-free patients during 2-year follow-up compared with patients with relapses (PCA: R2 = 0.380, Q2 = 0.128, figure 1B; PLS: R2X = 0.389, R2Y = 0.425, Q2Y = 0.231, figure 1F). Regarding disability, the classifiers were able to distinguish between patients remaining with EDSS <3.0 compared with patients reaching an EDSS >4.5 after 2 years of follow-up based on the metabolic profile (PCA: R2 = 0.380, Q2 = 0.126, figure 1C; PLS: R2X = 0.512, R2Y = 0.756, Q2Y = 0.555, figure 1G) or the change in the EDSS after 2 years of follow-up (PCA: R2 = 0.381, Q2 = 0.129, figure 1D; PLS: R2X = 0.148, R2Y = 0.222, Q2Y = 0.036, figure 1H) with medium to high accuracy.
Metabolites associated with MS severity point to an imbalance in phospholipid metabolism.
In order to validate the ability of the metabolomics signature to discriminate between patients and controls, we analyzed another independent and multicenter cohort of 238 patients with MS and 74 matched controls. We identified a set of metabolites significantly different between patients and controls (figure 2). Metabolites included in this signature comprised amino acids, saturated, monounsaturated, and polyunsaturated fatty acids, diglycerols, triglycerides, cholesteryl esters, bile acids, steroids, lysophosphatidylethanolamine, lysophosphatidylcholines, phosphatidyl-inositol, ceramide, sphingomyelin, and monohexosylceramides (figure 3).
Figure 2. Differential metabolomics profile in patients with multiple sclerosis and healthy controls.
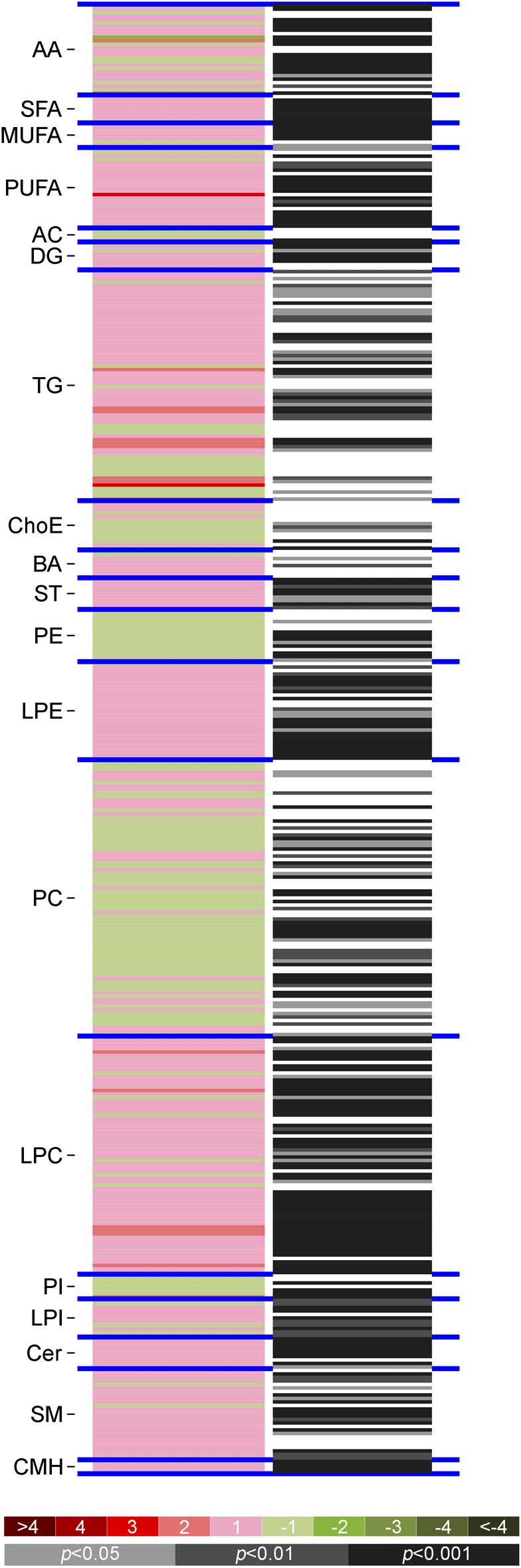
Heatmaps of the differences between patients (retrospective longitudinal cohort) and controls (vertical axis; ordered by metabolite chemical group and according to their carbon number and unsaturation degree of their esterified acyl chains). The log2 transformed ion abundance ratios (colors from green to red show drops or elevations of the metabolite levels in patients) and unpaired Student t test (or Welch t test where unequal variances were found) p values (gray lines correspond to significant fold changes of individual metabolites) per metabolite are displayed. AA = amino acids; AC = acylcarnitines; BA = bile acids; Cer = ceramides; ChoE = cholesteryl esters; CMH = monohexosylceramides; DG = diglycerols; LPC = lysophosphatidylcholines; LPE = lysophosphatidylethanolamines; LPI = lysophosphatidylinositols; MUFA = monounsaturated fatty acids; PC = phosphatidylcholines; PE = phosphatidylethanolamines; PI = phosphatidylinositols; PUFA = polyunsaturated fatty acids; SFA = saturated fatty acids; SM = sphingomyelins; ST = steroids; TG = triglycerides.
Figure 3. Metabolic pathways of the metabolites identified.

Lipid biosynthesis. Fold-change (patients with multiple sclerosis/controls) trends are indicated in red, green, and gray arrows for significant upregulated, deregulated, and nonsignificant chemical classes in patients with MS, respectively. Areas in orange represent processes carried out in the mitochondria. AC = acylcarnitines; CE = cholesteryl esters; Cer = ceramides; CL = cardiolipins; CS = cholesterol sulfate; DAG = diacylglycerols; FAA = primary fatty amides; G3P = glycerol 3-phosphate; LPC = lysophosphatidylcholines; LPE = lysophosphatidylethanolamines; LPG = lysophosphatidylglycerols; LPI = lysophosphatidylinositols; MAG = monoacylglycerols; NAE = N-acylethanolamines; NEFA = nonesterified fatty acids; oxFA = oxidized fatty acids; PA = phosphatidic acids; PC = phosphatidylcholines; PE = phosphatidylethanolamines; PG = phosphatidylglycerols; PI = phosphatidylinositols; PS = phosphatidylserines; SAMe = S-adenosylmethionine; SM = sphingomyelins; ST = steroids; TAG = triacylglycerols; UC = unesterified cholesterol.
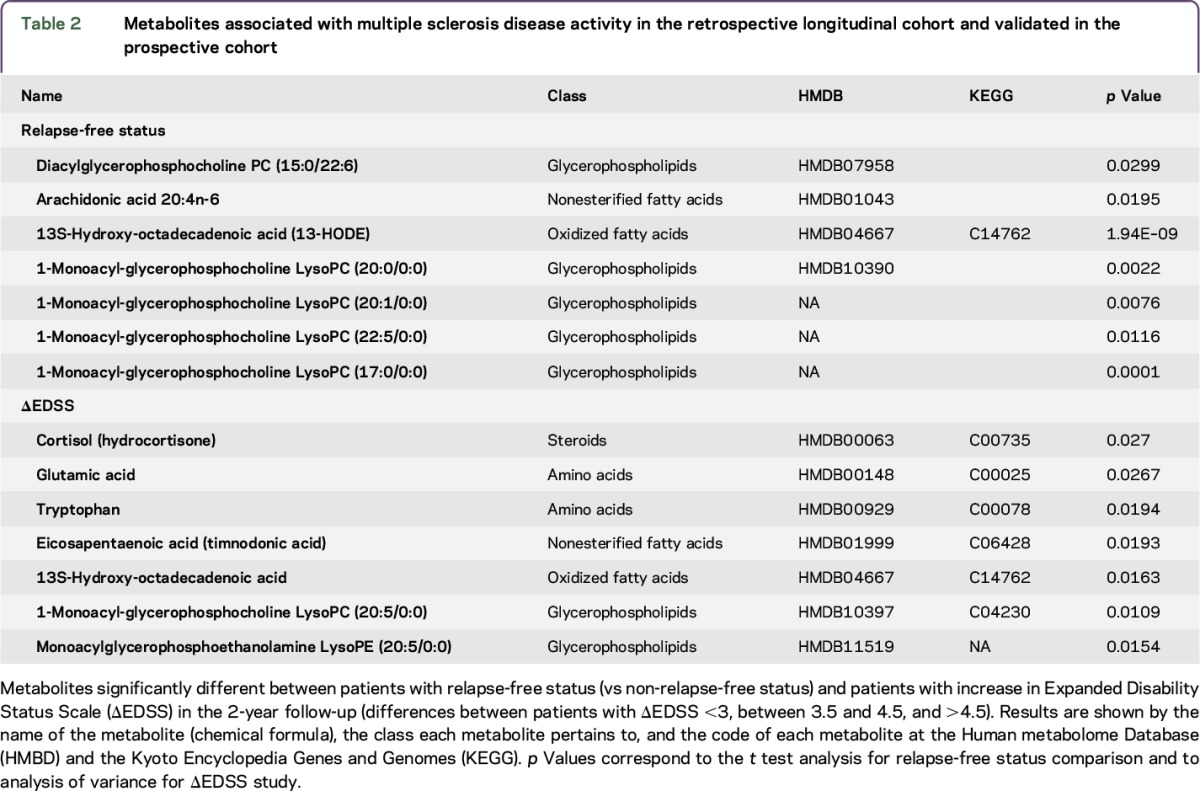
Considering that one of the clinical uses of a biomarker for MS would be to classify individuals with different levels of disease severity, we analyzed the metabolomic profile in this second cohort of 238 patients with MS stratified by disease severity (presence of relapses or increase in EDSS) (table e-2). Regarding disease activity (relapse-free vs having relapses), we found a total of 116 different metabolites (uncorrected p < 0.05) in the unpaired Student t test for stable (n = 77) vs active (n = 134) disease (unclassified n = 27). Table 2 shows the metabolites that remained significant after correction for multiple testing for each subgroup in the retrospective longitudinal cohort, as well as those that were also significant in the prospective cohort and were therefore considered validated. Specifically, we found that levels of PC (15:0/22:6), arachidonic acid, 13- hydroxyoctadecadienoic acid, and the lysophosphatidylcholines PC (20:0/0:0), PC (201/0:0), PC (22:5/0:0), and PC (17:0/0:0) were associated with the relapse-free status. Moreover, we found that levels of cortisol (hydrocortisone) showed a trend for association with disability measured by EDSS after 2 years of follow-up (p = 0.051, ANOVA test). The differences showed a moderate accuracy and high specificity (area under the curve 0.6, sensitivity 0.38, specificity 1.0) for predicting increase in the EDSS 2 years later. Moreover, the metabolites associated with disability status (EDSS <3.0 compared with patients reaching EDSS >4.5 after 2 years follow-up) included glutamic acid, tryptophan, eicosapentaenoic acid (timnodonic acid), 13S-hydroxyoctadecadienoic acid, lysophosphatidylcholine (20:5/0:0), and lysophosphatidylethanolamine (20:5/0:0). Finally, considering the use of disease-modifying drugs, among all metabolites identified in both cohorts, LysoPC (20:0/0:0) and LysoPC (22:5/0:0) were associated with relapse-free status and glutamic acid was associated with increase in EDSS in patients treated with interferon-β (table e-3).
Table 2.
Metabolites associated with multiple sclerosis disease activity in the retrospective longitudinal cohort and validated in the prospective cohort
DISCUSSION
In this study, we identified a set of metabolites that were differentially present in patients with MS compared to controls. In addition, we identified a set of metabolites that were differentially present in patients with more severe disease based on the presence of relapses or changes in the disability scale EDSS. Moreover, we were able to validate sphingomyelin and phosphatidylethanolamine in the 2 cohorts as discriminant between patients with MS and controls. Overall, our results point to an imbalance in the phospholipids and sphingolipids composition of the serum, as well as changes in several amino acids such as glutamic acid or tryptophan (figure 3). It is important to remember that the current metabolomic assays in the serum or CSF have been highly optimized for lipid and amino acid characterization; therefore other metabolites such as intermediate metabolism are not adequately analyzed.
The source of the differences in lipid and amino acid levels in patients with MS compared to healthy controls and along disease severity may suggest several hypotheses. First, such changes may be related to increased activation of the immune system in which phospholipids and amino acids play a role either as signaling molecules or as regulators of membrane synthesis, cytokinesis, or lipid rafts.26 Second, these metabolomic signatures may reflect changes in the CNS lipid composition due to myelin destruction (mainly composed by sphingolipids) and astroglia proliferation (mainly requiring phospholipids). Another alternative hypothesis is that genetic susceptibility associated with MS may drive changes in lipid and amino acid composition of the serum, although at present none of the polymorphisms associated with MS have been involved directly in lipid metabolism.27
Regarding the main metabolites identified, most of them play a key role in the signaling and regulation of the immune system.26 Sphingomyelins are one of the main lipid classes in myelin, but also influence the immune response through S1P receptor signaling.28,29 Phosphatidylethanolamine modulates the immune response by activating the CD300 receptors.30,31 Phosphatidylcholine is the main phospholipid class in cell membranes and plays a critical role in proliferative growth and programmed cell death.32 Arachidonic acid is present in phospholipids and also works as a second messenger for phospholipase activation and as a key inflammatory mediator.33 Regarding amino acids, in addition to being part of protein sequences and the main excitatory neurotransmitter, glutamic acid and glutamate activate purinergic receptors P2X7 in macrophages and also contribute to oligodendrocyte death in the white matter.34,35 Tryptophan's key role is serving as a checkpoint in the activation of the immune system by the enzyme indoleamine 2,3-dioxygenase.36 For all of these reasons, and in line with many genetic and immunologic studies, our results support the first hypothesis that changes observed in the serum metabolome are a reflection of chronic activation of the immune system in patients with MS and that the severity of the disease also corresponds with greater activation of the immune system, rather than differential involvement of pathways.
Our results are supported by previous small metabolomics studies in the serum of patients with MS, which have identified differences in levels of phosphatidyl-choline, phosphatidyl-inositol, glutamate, and other amino acids between patients and controls.10,12,37 Furthermore, a magnetic resonance spectroscopy (MRS) analysis identified differences in serum metabolites between MS and neuromyelitis optica, namely scyllo-inositol (the second largest isomer of inositol, after myo-inositol) increased in MS and acetate (a metabolite of the neurotransmitter synthesis of glutamate to glutamine in astrocytes) increased in neuromyelitis optica.11 Moreover, a recent well-powered MRS study of the serum was able to identify a metabolomic signature differentiating relapsing from progressive disease in 3 independent cohorts.9 Metabolites involved in such patterns included reduced levels of fatty acids, phosphatidyl-choline, N-acetyl species, lactate, and glucose and increased levels of other fatty acids and β-hydroxybutyrate in patients with progressive disease.
Regarding metabolomic differences in the CSF, MRS analyses have revealed differences in the levels of small metabolites (increase of choline, myo-inositol, and threonate and decrease of 3-hydroxybutyrate, citrate, phenylalanine, 2-hydroxyisovalerate, and mannose) associated with energy and lipid metabolism in patients with MS,14 as well as increased levels of lipid (8-iso-prostaglandin F2α) or protein peroxidation products (carboxymethylated, neuroketal, and malondialdehyde-mediated protein modifications).8 Similarly, another small metabolomic study of the CSF found differences in metabolite concentration based on disease activity, with increased levels of hydroxyeicosatetraenoic acid, prostaglandin E2, and resolvin D1 in patients with highly active MS.13
In addition, MRS allows for assessment of the metabolomic profile of the CNS, having found significant differences in metabolite concentration (e.g., glutamate, N-acetylaspartate, γ-aminobutyric acid, and aspartate) between patients with MS and controls.15 Indeed, brain tissue analysis by electrospray identified higher phospholipid and lower sphingolipid content in normal-appearing gray matter of patients with MS associated with a metabolic defect that causes sphingolipids to be shuttled to phospholipid production.38 It seems that in MS there is an increase of phospholipids and a decrease of sphingolipids, perhaps reflecting a reduction of myelin-specific lipid content or production in the CNS associated with increased cellularity due to glial scars.
Our study has several limitations. In the first prospective cohort, identification of metabolites was limited in the platform used; therefore the analysis was done based on peaks of the spectra. As such, we were unable to perform the analysis in a more informative way (e.g., using the retrospective cohort to identify metabolites with high classification power [by univariate analysis], then selecting the top performing features and training a classifier [with internal cross-validation], and finally testing this classifier in the other cohort, using area under the receiver operating characteristic curve as the metric to evaluate performance). Second, use of disease-modifying drugs was allowed in order to represent the type of patients currently monitored in MS centers; therefore, it is not possible to ascertain whether differences were truly due to disease activity or related to the use of such drugs. However, the fact that most of the treated patients received interferon-β decreased heterogeneity and noise by this factor. Moreover, disease activity was based on clinical activity since MRI monitoring was not available. Consequently, active patients may have been misclassified as stable patients if new lesions happened in noneloquent areas. Although this limitation may have decreased the power of our study and introduced false-negatives, it is unlikely to have introduced false-positive metabolites associated with disease activity. Moreover, lack of MRI studies prevented relating metabolites with specific imaging markers, as described by Baranzini et al.39 Other contemporary clinical parameters such as oligoclonal banding presence were not analyzed here and would require further studies in order to clarify their relationship. Although the sample size of the study is significant, it does not allow assessment of differences at various steps of the EDSS or for specific subgroups. Finally, although the identified classifiers of worsening disability were significant, their diagnostic accuracy was not very high and future studies should refine them to improve upon this.
ACKNOWLEDGMENT
The authors thank Mark Sefton and Erika Lampert for English review of the manuscript.
GLOSSARY
- ANOVA
analysis of variance
- EDSS
Expanded Disability Status Scale
- m/z
mass-to-charge
- MRS
magnetic resonance spectroscopy
- MS
multiple sclerosis
- OPLS
orthogonal projection to latent structures
- PCA
principal components analysis
- Rt
retention time
- SME
splines mixed effects
- TOF
time of flight
- UHPLC-MS
ultra-high-performance liquid chromatography coupled to mass spectrometry
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
P.V. designed the study, recruited patients and clinical information, performed the analysis, and wrote the article. A.C. designed the metabolomic study, supervised the analysis, and reviewed the manuscript. C.A. performed bioinformatic analysis and reviewed the manuscript. I.A. performed bioinformatic analysis and reviewed the manuscript. I.Z. performed the analysis of clinical phenotypes and reviewed the article. I.P.-V. performed the analysis of clinical phenotypes and reviewed the article. A.S. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. M.C. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. X.M. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. L.V. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. J.C.A.-C. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. O.F. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. R.A. recruited patients and clinical information, performed the analysis of clinical phenotypes, and reviewed the article. R.A.-L. collected clinical information, performed the analysis of clinical phenotypes, and reviewed the article. E.K. performed bioinformatic analysis and reviewed the manuscript.
STUDY FUNDING
This study was supported by grants from the Instituto de Salud Carlos III to P.V. (PI12/01823) and to P.V., A.S., M.C., L.V., O.F., and R.A. (RED07-0060) as part of the Spanish MS Network.
DISCLOSURE
P. Villoslada served on scientific advisory boards for Roche, Novartis, Bionure, and Genzyme; received travel funding and/or speaker honoraria from Novartis, Roche, and Genzyme; is an academic editor for PloS One; is on the editorial board for Neurology and Therapy, Curr Treat Options Neurol, and Multiple Sclerosis and Demyelinating Disorders; holds patents for Methylthioadenosine for the treatment of MS, and neurotrophic compounds for the treatment of brain diseases, Gene signature pattern as a biomarker for MS, and Algorithm for reconstructing white matter tracts from diffusion images; received research support from Novartis, Roche, Biogen, Genzyme, Insituto de Salud Carlos III, European Commission, National MS Society, Fundaction Maraton TV3, Bionure Inc., Spire Bioventures, and Mint-Labs Health Engineering; and provided expert testimony for Synthon. C. Alonso is an employee of OWL. I. Agirrezabal is employed by Amgen (Europe) GmbH and received research support from Ayudas para Formacion de Profesorado Universitario, Spanish Government. E. Kotelnikova received research support from H2020 Programme, Marie Curie. I. Zubizarreta reports no disclosures. I. Pulido-Valdeolivas received travel funding from Roche Spain, European Academy of Neurology, and European Committee for Treatment and Research in Multiple Sclerosis; holds a patent for an affordable eye tracking system to measure eye movement in neurologic diseases; and holds stock or stock options in Aura Innovative Robotics. A. Saiz served on scientific advisory boards for Biogen Idec and Merck-Serono; received travel funding and/or speaker honoraria from Bayer-Schering, Merck-Serono, Biogen Idec, Sanofi-Aventis, Teva Pharmaceutical Industries, and Novartis; and consulted for Bayer-Schering, Merck-Serono, Biogen-Idec, Sanofi-Aventis, Teva Pharmaceutical Industries, and Novartis. M. Comabella received speaker honoraria from Bayer Schering Pharma, Merck Serono, Biogen-Idec, Teva Pharmaceuticals, Sanofi-Aventis, Genzyme, and Novartis; is on the editorial advisory boards for Journal of Neuroimmunology, Multiple Sclerosis Journal, and PLoS One; and received research support from Red Española de Esclerosis Múltiple, Fondo de Investigación Sanitaria. X. Montalban served on scientific advisory boards for Actelion, Bayer, Biogen, Celgene, Genzyme, Merck, Novartis, Oryzon Genomics, Roche, Sanofi-Genzyme, and Teva Pharmaceutical; received travel funding from Biogen, Genzyme, Merck, Novartis, Celgene, Roche, Sanofi-Genzyme, and Teva Pharmaceutical; is on the editorial board for Multiple Sclerosis Journal, Journal of Neurology, The International MS Journal, Revista de Neurologica, and Therapeutic Advances in Neurologic Disorders; has consulted for Actelion, Bayer, Biogen, Celgene, Genzyme, Merck, Novartis, Oryzon Genomics, Roche, Sanofi-Genzyme, and Teva Pharmaceutical; and is vice-president of Multiple Sclerosis Foundation of Barcelona. L.M. Villar received travel funding and/or speaker honoraria from Biogen, Novartis, Genzyme, Teva, Bayer, and Roche; holds a patent for a biomarker for selecting good responders to IFN-β in multiple sclerosis; has consulted for Bayer and Biogen Idec; and received research support from Genzyme, Merck-Serono, Biogen, and Spanish Ministry of Economy and Competitiveness. J.C. Alvarez-Cermeño served on scientific advisory boards for Biogen Idec, Novartis, Genzyme, Roche, and Merck-Serono; received speaker honoraria from Biogen Idec, Novartis, Genzyme, Merck-Serono, and Roche; and received research support from Biogen Idec, Novartis, Merck-Serono, Carlos III Institute, and Spanish Ministry of Economy. O. Fernandez has received honoraria as a consultant in advisory boards and as a chairman or lecturer in meetings and has also participated in or participates in clinical trials and other research projects promoted by Biogen-Idec, Bayer-Schering, Merck-Serono, Teva, Novartis, Actelion, Roche Almirall, and Allergan; has travel and accommodation covered by the ECF (Europen Charcot Foundation); is on the editorial board for Revista de Multiple Sclerosis; and holds a patent for IFNR2 molecule and a bioassay to measure it. R. Alvarez-Lafuente received travel funding and/or speaker honoraria from Novartis, Merck, Biogen, Bayer, Teva, Sanofi, and Roche; is on the editorial board for ISRN Infectious Diseases, Austin Virology and Retrovirology, and Austin Journal of Multiple Sclerosis and Neuroimmunology; is on the speakers' bureau for Novartis, Merck, and Biogen; and received research support from Novartis, Merck, Bayer, Teva, ISCIII-FEDER, Fundación Mutua Madrileñam Fundación Lair, Fundación Salud, and Fundación Genzyme. R. Arroyo served on the scientific advisory board for and received travel funding and/or speaker honoraria from Teva, Biogen, Novartis, Merck-Serono, Roche, Bayer, Genzyme, and Almirall. A. Castro is an employee of Owl Metabolomics SA. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Ibrahim SM, Gold R. Genomics, proteomics, metabolomics: what is in a word for multiple sclerosis? Curr Opin Neurol 2005;18:231–235. [DOI] [PubMed] [Google Scholar]
- 2.Villoslada P. Biomarkers for multiple sclerosis. Drug News Perspect 2010;23:585–595. [DOI] [PubMed] [Google Scholar]
- 3.Villoslada P, Baranzini S. Data integration and systems biology approaches for biomarker discovery: challenges and opportunities for multiple sclerosis. J Neuroimmunol 2012;248:58–65. [DOI] [PubMed] [Google Scholar]
- 4.Comabella M, Montalban X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol 2014;13:113–126. [DOI] [PubMed] [Google Scholar]
- 5.Tumani H, Hartung HP, Hemmer B, et al. . Cerebrospinal fluid biomarkers in multiple sclerosis. Neurobiol Dis 2009;35:117–127. [DOI] [PubMed] [Google Scholar]
- 6.Wishart DS. Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov 2016;15:473–484. [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis: a quiet revolution. Nat Rev Neurol 2015;11:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalo H, Brieva L, Tatzber F, et al. . Lipidome analysis in multiple sclerosis reveals protein lipoxidative damage as a potential pathogenic mechanism. J Neurochem 2012;123:622–634. [DOI] [PubMed] [Google Scholar]
- 9.Dickens AM, Larkin JR, Griffin JL, et al. . A type 2 biomarker separates relapsing-remitting from secondary progressive multiple sclerosis. Neurology 2014;83:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehrpour M, Kyani A, Tafazzoli M, Fathi F, Joghataie MT. A metabonomics investigation of multiple sclerosis by nuclear magnetic resonance. Magn Reson Chem 2013;51:102–109. [DOI] [PubMed] [Google Scholar]
- 11.Moussallieh FM, Elbayed K, Chanson JB, et al. . Serum analysis by 1H nuclear magnetic resonance spectroscopy: a new tool for distinguishing neuromyelitis optica from multiple sclerosis. Mult Scler 2014;20:558–565. [DOI] [PubMed] [Google Scholar]
- 12.Pieragostino D, D'Alessandro M, di Ioia M, et al. . An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol Biosyst 2015;11:1563–1572. [DOI] [PubMed] [Google Scholar]
- 13.Pruss H, Rosche B, Sullivan AB, et al. . Proresolution lipid mediators in multiple sclerosis: differential, disease severity-dependent synthesis: a clinical pilot trial. PLoS One 2013;8:e55859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinke SN, Broadhurst DL, Sykes BD, et al. . Metabolomic profiling in multiple sclerosis: insights into biomarkers and pathogenesis. Mult Scler 2014;20:1396–1400. [DOI] [PubMed] [Google Scholar]
- 15.Vingara LK, Yu HJ, Wagshul ME, et al. . Metabolomic approach to human brain spectroscopy identifies associations between clinical features and the frontal lobe metabolome in multiple sclerosis. Neuroimage 2013;82:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cocco E, Murgia F, Lorefice L, et al. . (1)H-NMR analysis provides a metabolomic profile of patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2016;3:e185. doi: 10.1212/NXI.0000000000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhargava P, Calabresi PA. Metabolomics in multiple sclerosis. Mult Scler 2016;22:451–460. [DOI] [PubMed] [Google Scholar]
- 18.Lublin FD, Reingold SC, Cohen JA, et al. . Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barr J, Caballeria J, Martinez-Arranz I, et al. . Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res 2012;11:2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Arranz I, Mayo R, Perez-Cormenzana M, et al. . Enhancing metabolomics research through data mining. J Proteomics 2015;127:275–288. [DOI] [PubMed] [Google Scholar]
- 21.Polman CH, Reingold SC, Edan G, et al. . Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.” Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 22.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain 2006;129:606–616. [DOI] [PubMed] [Google Scholar]
- 23.Barr J, Vazquez-Chantada M, Alonso C, et al. . Liquid chromatography-mass spectrometry-based parallel metabolic profiling of human and mouse model serum reveals putative biomarkers associated with the progression of nonalcoholic fatty liver disease. J Proteome Res 2010;9:4501–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saccenti E, Hoefsloot HCJ, Smilde AK, Westerhuis JA. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014;10:361–374. [Google Scholar]
- 25.Gnanapavan S, Hegen H, Khalil M, et al. . Guidelines for uniform reporting of body fluid biomarker studies in neurologic disorders. Neurology 2014;83:1210–1216. [DOI] [PubMed] [Google Scholar]
- 26.O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016;16:553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soltow QA, Jones DP, Promislow DE. A network perspective on metabolism and aging. Integr Comp Biol 2010;50:844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beyersdorf N, Muller N. Sphingomyelin breakdown in T cells: role in activation, effector functions and immunoregulation. Biol Chem 2015;396:749–758. [DOI] [PubMed] [Google Scholar]
- 29.Chakraborty M, Jiang XC. Sphingomyelin and its role in cellular signaling. Adv Exp Med Biol 2013;991:1–14. [DOI] [PubMed] [Google Scholar]
- 30.Zenarruzabeitia O, Vitalle J, Eguizabal C, Simhadri VR, Borrego F. The biology and disease relevance of CD300a, an inhibitory receptor for phosphatidylserine and phosphatidylethanolamine. J Immunol 2015;194:5053–5060. [DOI] [PubMed] [Google Scholar]
- 31.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 2013;1831:543–554. [DOI] [PubMed] [Google Scholar]
- 32.Ridgway ND. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit Rev Biochem Mol Biol 2013;48:20–38. [DOI] [PubMed] [Google Scholar]
- 33.Tripathi T, Alizadeh H. Significance of arachidonic acid in ocular infections and inflammation. Inflamm Cell Signal 2014;1:e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butt AM, Fern RF, Matute C. Neurotransmitter signaling in white matter. Glia 2014;62:1762–1779. [DOI] [PubMed] [Google Scholar]
- 35.Oyanguren-Desez O, Rodriguez-Antiguedad A, Villoslada P, Domercq M, Alberdi E, Matute C. Gain-of-function of P2X7 receptor gene variants in multiple sclerosis. Cell Calcium 2011;50:468–472. [DOI] [PubMed] [Google Scholar]
- 36.Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan-catabolizing enzymes: party of three. Front Immunol 2014;5:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Boccio P, Pieragostino D, Di Ioia M, et al. . Lipidomic investigations for the characterization of circulating serum lipids in multiple sclerosis. J Proteomics 2011;74:2826–2836. [DOI] [PubMed] [Google Scholar]
- 38.Wheeler D, Bandaru VV, Calabresi PA, Nath A, Haughey NJ. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 2008;131:3092–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baranzini SE, Srinivasan R, Khankhanian P, et al. . Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 2010;133:2603–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]