

Abstract
The soil-dwelling α-proteobacterium Sinorhizobium meliloti engages in a symbiosis with legumes: S. meliloti elicits the formation of plant root nodules where it converts dinitrogen to ammonia for use by the plant in exchange for plant photosynthate. To study the coordinate differentiation of S. meliloti and its legume partner during nodule development, we designed a custom Affymetrix GeneChip with the complete S. meliloti genome and ≈10,000 probe sets for the plant host, Medicago truncatula. Expression profiling of free-living S. meliloti grown with the plant signal molecule luteolin in defined minimal and rich media or of strains altered in the expression of key regulatory proteins (NodD1, NodD3, and RpoN) confirms previous data and identifies previously undescribed regulatory targets. Analyses of root nodules show that this Symbiosis Chip allows the study of gene expression in both partners simultaneously. Our studies detail nearly 5,000 transcriptome changes in symbiosis and document complex transcriptional profiles of S. meliloti in different environments.
Keywords: expression, Medicago, microarray, Sinorhizobium meliloti
Prokaryote–eukaryote interactions span a variety of outcomes from disease to simple commensalisms to beneficial symbiosis, a relationship in which one or both partners profit from the presence and activities of the other. Symbiosis between Rhizobium bacteria and legume plant hosts results in the reduction of inert dinitrogen to biologically usable ammonia. A host legume and free-living Sinorhizobium meliloti exchange signals during the earliest stages of the symbiosis (1–3). The roots secrete flavonoid signal compounds like luteolin, an inducer of bacterial nod genes. These nod genes encode enzymes that synthesize a second signal compound, Nod factor, which causes the plant to initiate the morphogenetic program to form the nitrogen-fixing root nodule.
There is an intimate interaction between the two partners throughout the course of the symbiosis. After initial signal exchange and bacterial attachment at the surface (4, 5), root inner cortical cells begin dividing to form the nodule meristem. Invasion involves delivery and release of the bacteria via an infection thread into the plant cell cytoplasm (6), where differentiated bacteria (bacteroids) reduce dinitrogen into ammonium that be assimilated by the plant. The host provides bacteroids with carbon compounds that fuel the energy-expensive process of nitrogen fixation (7, 8).
S. meliloti and other nitrogen-fixing symbionts belong to the α purple subgroup of proteobacteria, a diverse group containing the plant pathogen Agrobacterium, the reduced genome parasite Rickettsia, photosynthetic bacteria such as Rhodobacter, and the mammalian pathogens Brucella and Bartonella. It is common for members of the α-proteobacteria to have complex genome architectures (9). S. meliloti has three circular replicons: a chromosome (3.65 Mb) that encodes most of the housekeeping and essential genes (10) and two megaplasmids, pSymA (1.4 Mb) and pSymB (1.7 Mb) (11). pSymA is not essential for viability but contains many of the genes required for symbiosis [nod, nif, and most fix genes (12, 13)]. pSymB contains exo and dct genes required for symbiosis, as well as several genes essential for viability (14).
Determining the complete genome sequence of S. meliloti set the stage for exploring this wealth of information by transcriptional and proteomic studies (15–22). Here we describe the construction of a dual-genome Symbiosis Chip and how we have used this unique tool, which allows us to probe global gene expression in both partners simultaneously.
Materials and Methods
Design of the Symbiosis GeneChip. GeneChip manufacture combines photolithographic masking methods and combinatorial chemistry to generate hundreds of thousands of unique DNA probes (23). Affymetrix design rules and analysis algorithms called 13 perfect match and 13 1-bp mismatch oligos for each annotated ORF. Intergenic (IG) sequences were covered by evenly tiling both strands with 25-nt oligomers. To avoid the relatively short gaps frequently found between genes that are likely organized into operons, we tiled IG sequences only >150 bp. Spacing between tiled oligomers ranged from 1 to 7 nt.
Selection of probe sets corresponding to Medicago truncatula genes and tentative consensus sequences (TCs) are described in ref. 24.
Bacterial Strains and Culture Conditions. Rm1021 is the streptomycin (Sm)-resistant derivative of WT S. meliloti strain SU47 used for genome sequencing (11). Rm1680 contains a transposon Tn5 insertion in rpoN (25). A2102 is a triple nodD mutant (nodD1::Tn5, nodD2::Tn5-Tp, nodD3::Tn5-233) (26). VO2675 contains a Tn5 in fixJ (6). Broad host range plasmids overexpressing either nodD1 (pE43) or nodD3 (pE65) (27) from the trp promoter, or vector without insert (pTE3), were introduced into A2102. M9 minimal medium contained either 15 mM succinate or 15 mM glucose as the carbon source. Unless otherwise noted, all strains were grown overnight with constant agitation at 30°C in TY medium (6 g of tryptone/3 g of yeast extract/0.5 g of calcium chloride dihydrate per liter/Sm at 500 μg/ml).
At least three biological replicates, grown to midexponential phase (0.51–0.75 OD600), were used for each data set. For luteolin induction, late exponential cultures were diluted to 0.15–0.2 OD600 and induced for 4 h with 3 μM luteolin (Calbiochem). Negative controls were treated with an equivalent volume of dimethylformamide (DMF), the solvent for luteolin. A2102 pE43 was induced overnight (≈16 h) with 3 μM luteolin, whereas A2102 pE65 and A2102 pTE3 were grown with DMF.
Plant Growth. M. truncatula cv. AS17 was grown in vermiculite and watered with nitrogen-free 1/2× Buffered Nodulation Medium (28). Plants were inoculated 1 and 3 days after planting with either Rm1021 or the fixJ – strain, VO2675 (6). Nodules were removed from the roots 33–35 days after the first inoculation and frozen in liquid nitrogen. Uninoculated roots were harvested 2 weeks after planting.
RNA Purification, cDNA Synthesis, Labeling, and Hybridization. Supporting materials and methods are available in Supporting Text, which is published as supporting information on the PNAS web site. To prepare samples for bacterial RNA purification, cultures were quickly transferred to chilled centrifuge tubes containing 2 ml of 5% buffer-equilibrated phenol in ethanol and chilled and centrifuged to pellet cells (29). Supernatants were discarded, and pellets were frozen in liquid nitrogen. To prepare samples for root and nodule RNA purification, tissue was ground in liquid nitrogen. To purify RNA, samples were processed by using Qiagen RNeasy bacterial RNA purification kits (Qiagen, Chatsworth, CA) with slight modifications. RNeasy columns retain RNA >200 nt; although we detected expression of genes <200 nt (e.g., tRNA), expression changes for these genes are unreliable.
First-strand cDNA was synthesized by using the GeneChip Pseudomonas aeruginosa Genome Array Expression Analysis protocol (Affymetrix, Santa Clara, CA) with volume adjustments. Fragmented labeled cDNA (4 μg for free-living S. meliloti, 8 μgfor roots, and 12 μg for nodules) was added to hybridization solution (final volume 200 μl), and GeneChips were hybridized at 48°C at 60 rpm for 16 h in a GeneChip Hybridization Oven 640. An internal standard (spike-in) hybridization control was used for symbiosis experiments. Arrays were washed and stained according to Affymetrix protocols.
Data Analysis. Data from experiments comparing culture-grown bacteria were processed by using microarray suite Ver. 5, microdb Ver. 3, and data mining tool Ver. 3 (Affymetrix). For analysis, GeneChips were scaled to a target signal intensity of 500 by using all probe sets (global scaling). For the symbiosis experiments, we analyzed data as described above and by normalizing each chip to the signal of 18 probe sets that hybridized to a “spike-in” internal standard.
Generally, we used comparison expression analysis where an experimental array was compared with a baseline array. Hence, an experiment with three control and three experimental arrays yielded nine pairwise comparisons, and a 4 × 4 experiment, 16 comparisons. We deemed an increase or decrease of average signal log ratio ≥ 0.96 to be significant if all, or all but one, of the pairwise comparisons were evaluated by the software as significantly changed (P ≤ 0.05). We used absolute expression analysis to compare signal values of a range of chips or to determine whether genes were expressed above background. For symbiosis experiments, only those changes found by both scaling and normalization methods were deemed significant.
Quantitative PCR (qPCR). We checked expression changes of selected probe sets using real-time qPCR with DyNAmo SYBR Green qPCR kits (Finnzymes, Helsinki) in conjunction with the DNA Engine Opticon 2 continuous fluorescence detection system (MJ Research, Cambridge, MA). Results are available as Tables 2–5, which are published as supporting information on the PNAS web site.
Results and Discussion
Transcription is a primary indicator of the state of differentiation for any organism. In a complex interaction between two organisms, such as the Rhizobium–legume symbiosis, the ability to assess global transcription patterns of both partners with a single RNA preparation provides internal consistency to longitudinal studies of differentiation. We used Affymetrix photolithographic technology to manufacture a virtually unlimited number of reproducible chips, thereby dramatically reducing issues of quality control that arise in use of in-house microarrays. A unique feature of our custom microarray is the inclusion of the complete prokaryotic genome together with 9,935 probe sets from root libraries of the eukaryotic host. This Symbiosis Chip thus gives us the potential to examine differentiation and response resulting from signal exchange between the two symbiotic partners simultaneously.
As a starting point, we conducted experiments that allowed a direct comparison of microarray results with those from earlier studies using reporter gene fusions or enzymatic assays of gene products. For a representative subset of genes, we also compared microarray results with those using a more sensitive technique, real-time qPCR, to get a sense of the advantages and limitations of these different techniques.
For six replicates of TY-grown cells, two-thirds of the putative coding regions were expressed above background (Data Set 1 and Table 6, which are published as supporting information on the PNAS web site). This is similar to the number of genes expressed above background for Escherichia coli grown in rich medium (30); the percentage of genes expressed is lower in S. meliloti, because its overall genome is >50% larger than that of E. coli.
Due to the unique nature of each probe set and the lack of a hybridization control containing known amounts of each mRNA, we cannot quantify transcript abundance from chip data. Transcriptome data, however, can approximate relative message abundance across a genome (31). We found that the probe sets with highest average signal values (within the top 100 for average signal) correlated well with computer-based predictions of highly expressed genes in S. meliloti (32). It must be kept in mind that the function of most S. meliloti predicted genes has not been confirmed: annotations are typically based on database similarity searches. Therefore, expression data should be useful in refining the genome's annotation. A data set containing all bacterial gene expression changes for this study is available as Data Set 2, which is published as supporting information on the PNAS web site.
Growth on Minimal and Rich Media. Table 1 summarizes the comparisons undertaken in this study. Much evidence suggests that the plant partner supplies C4-dicarboxylic acids, such as succinate, malate, or fumarate, to nitrogen-fixing bacteroids (33). We grew cells in M9 minimal medium with either succinate or glucose as carbon source to see how the expression profile changes under conditions that partially mimic bacteroids in a nodule (Table 7, which is published as supporting information on the PNAS web site). pSymB genes were overrepresented among those with changed expression, consistent with its predicted specialization in sugar and polysaccharide metabolism (14).
Table 1. Number of expression changes in coding and intergenic (IG) probe sets.
| Experimental comparison | INC coding | INC IG | DEC coding | DEC IG |
|---|---|---|---|---|
| M9 succinate vs. TY medium | 255 | 46 | 271 | 47 |
| M9 glucose vs. TY medium | 418 | 79 | 385 | 65 |
| M9* vs. TY medium | 200 | ND | 227 | ND |
| M9 succinate vs. M9 glucose medium | 25 | 2 | 113 | 14 |
| Rm1021 + luteolin vs. Rm1021 | 27 | 7 | 4 | 1 |
| Rm1021 + streptomycin vs. Rm1021 | 4 | 2 | 6 | 0 |
| Rm1021 + dimethylformamide vs. Rm1021 | 29 | 10 | 23 | 3 |
| nodD1 overexpression vs. nodD- | 14 | 2 | 2 | 0 |
| nodD3 overexpression vs. nodD- | 106 | 35 | 95 | 10 |
| Rm1021 vs. Rm1680 (rpoN-) | 8 | ND | 12 | ND |
| WT vs. Tn5-containing strains† | 20 | ND | 62 | ND |
| Rm1021 nodule bacteria vs. free-living | 345 | 248 | 943 | 141 |
| fixJ- nodule bacteria vs. free-living | 18 | 8 | 1,325 | 227 |
| Rm1021 vs. fixJ- nodule bacteria | 750 | 425 | 6 | 0 |
| Rm1021 vs. fixJ- (free-living) | 3 | 2 | 0 | 0 |
| M. truncatula Rm1021 nodule vs. root | 259 | — | 3,507 | — |
| M. truncatula fixJ- nodule vs. root | 202 | — | 1,467 | — |
| Rm1021 nodule vs. fixJ- nodule | 28 | — | 186 | — |
Data were analyzed by using Affymetrix mas and dmt. The cutoff for increase (INC) or decrease (DEC) is a signal log ratio ≥0.96, changing across all or all but one pairwise comparisons. ND, not determined.
Succinate and glucose.
This set includes those genes whose expression changed in the Rm1021 vs. Rm1680 comparison and in at least one other comparison of a Tn5-carrying strain to its corresponding non-Tn5 control.
The largest expression increases in succinate-grown cells were for genes encoding phosphoenolpyruvate carboxykinase (pckA), the DctA dicarboxylate transporter, and fructose bisphosphate aldolase (fbaA). Both PckA and FbaA are important in the synthesis of hexose sugars when citric acid cycle intermediates such as succinate are the sole carbon source. Our results agree with data showing that dctA and pckA mutants fail to grow on succinate, and that succinate-grown cells show large increases in PckA activity (34). In glucose-grown cells, the greatest expression increases included genes encoding enzymes in the Entner–Douderoff pathway; a set of genes adjacent to each other on pSymA encoding an ABC transporter, a histidine kinase/response regulator pair, and a gluconolactonase; and two additional ABC transport clusters. Our results are consistent with data showing that the Entner–Douderoff pathway is used for glucose metabolism (34). We hypothesize that at least one of the ABC transporters brings glucose into the cell; future experiments can test this hypothesis.
In E. coli, many of the expression decreases seen during growth in minimal medium correlate with decreased growth rate, and many expression increases result from increased biosynthesis of essential compounds needed in the minimal defined medium (30, 31). Strikingly, there was no similar global change in S. meliloti. The expression differences between S. meliloti and E. coli suggest a different regulatory linkage of nutritional state to overall transcription.
Of the genes whose expression increased during growth in minimal medium compared to rich medium, at least five encode iron and hemin transport functions. Regulation of iron uptake is important because iron availability is limited due to insolubility in the soil, and because a number of proteins essential to symbiosis, such as nitrogenase, contain iron. exo gene expression increased up to 4-fold in minimal medium, and colonies are more mucoid than when grown on TY. Previous work shows that these genes are metabolically regulated: exopolysaccharide (EPS) production increases when nitrogen, sulfur, or phosphorus is limiting (35).
Expression of chemotaxis and flagellar biosynthetic genes decreased sharply in minimal medium; these made up one-quarter of the 250 genes whose expression decreased. Loss of motility and partial loss of flagella were previously found in starved S. meliloti cultures (36). However, because our exponentially growing cultures were obviously not starving, this suggests that motility in S. meliloti is additionally linked to metabolic state. Alternatively, down-regulation of motility and chemotaxis may be linked to increased exo expression in minimal medium. Faster-swarming S. meliloti mutants show an increased proportion of motile and flagellated cells, more and longer flagella per cell, and decreased EPS synthesis (37). Consistent with this idea, we saw similar decreases in flagellar and chemotaxis gene expression when nodD3 is overexpressed during growth in TY, another condition that increases EPS synthesis (see below). Moreover, strains overproducing succinoglycan, either by deletion of exoR or by constitutive expression of exoS, show greatly reduced expression of flagellar and chemotaxis genes (data not shown); motility assays and microscopy show that these two mucoid strains are nonmotile and lack flagella (D. H. Wells, E. J. Chen, and S.R.L., unpublished data). This pattern of coordinate synthesis suggests that genes involved in EPS production and motility may be part of a complex multitrait adaptation, perhaps related to biofilm formation, that responds to diverse environmental stimuli.
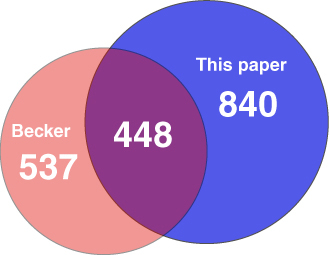
Using a PCR-based microarray, Becker et al. (20) compared S. meliloti grown in TY with Vincent's minimal medium (20). Of the changes they reported, our technique detected 81%; additionally, the Affymetrix chip tests detected five times more gene expression changes. Our larger data set may reflect a greater sensitivity of Affymetrix GeneChips or may arise from differences in growth and other experimental conditions.
Comparison of Rm1021 and an rpoN (ntrA) Mutant and Tn5-Influenced Gene Expression. Genome-wide transcription assays provide a means to identify the targets for regulation by a particular RNA polymerase σ factor. In mature nodules, after bacterial release into host cells and bacteroid differentiation, genetic circuits that control nitrogen fixation and dicarboxylate uptake are activated. These circuits are regulated by the σ factor RpoN, which recognizes -24/-12-type promoters (38) and requires enhancer-binding proteins (EBP) to activate the specific transcriptional units. Because each EBP is controlled by its own signal transduction pathway, different sets of RpoN-dependent genes can be transcribed under diverse conditions. For example, RpoN in S. meliloti activates expression of nitrogen fixation genes during symbiosis using the NifA EBP, C4-dicarboxylate metabolism genes using DctD, and ammonium transport and assimilation genes using NtrC (25). Sequence analysis predicts that S. meliloti encodes four additional EBPs, for which roles are so far not known.
By comparing expression profiles of WT Rm1021 and a rpoN mutant generated using transposon Tn5, we attempted to define these genetic circuits (Table 7). We expected to confirm a known set of differences and perhaps discover new RpoN targets. The results confirmed the previously reported dependence of glutamine synthetase II (glnII) and C4-dicarboxylate transport (dctD) on RpoN. As expected, RpoN-regulated genes that are not expressed in free-living bacteria (nif and fix) did not show altered expression in the rpoN mutant (Table 1). Only one of these, encoding a putative monooxygenase (SMb20103), contains a potential -24/-12-type promoter (38). It cannot be determined from these data whether the genes lacking a -24/-12 sequence depend directly or indirectly on RpoN.
During the course of our experiments, we compared expression profiles of WT cells to those of several different mutants that were generated by using transposon Tn5 insertions. In most of these Tn5-containing strains, the same set of ≈80 genes changed expression (Table 7). Many have predicted functions in the respiratory chain, including genes encoding cytochrome oxidases and their biogenesis proteins. Another group of changes involve those predicted to be involved in 1C metabolism.
Although surprising, these results are not inexplicable. The Tn5-encoded bleomycin resistance gene confers improved survival and growth advantage to E. coli, independent of bleomycin (39). The exact mechanism is unknown, but cytochrome c biogenesis is involved in both bleomycin resistance and the fitness effect, both of which might be explained by a reduction in toxic oxygen-activated or reduced iron-containing molecules (40). Moreover, it is clear that bacterial respiration, with multiple branched pathways and several terminal electron acceptors, provides for flexibility in adapting to diverse environments (41, 42). Additional research can determine whether Tn5 provides a growth or survival advantage to S. meliloti, and if so, whether it is mediated through changes in respiratory pathways and 1C metabolism as predicted by transcriptional analysis.
Gene Induction with Luteolin and the Role of nodD1 and nodD3. The bacterium–plant symbiosis is a complex developmental process with many steps of signal and response. The earliest known signals are plant flavonoids that induce expression of the bacterial nod genes. In S. meliloti, this induction is mediated by the three members of the nodD family of transcriptional activators: NodD1 and NodD2 require plant compounds such as flavonoids and betaines, but NodD3 is inducer-independent. NodD3 also appears to control at least one more gene, syrM, that is not regulated by NodD1 (43).
We examined this initial symbiotic interaction through study of the NodD regulators and inducers so that we could compare chip results with earlier studies using gene fusions and search for new NodD1-dependent genes that are induced with luteolin. As expected, luteolin induced large changes in nod gene expression. In addition, we saw smaller and more variable increases in hybridization to probe sets corresponding to 12 putative genes located in three different clusters (Table 7). Although qPCR confirmed the increases seen in the GeneChip analyses (Table 2), for completeness we also created transcriptional β-glucuronidase fusions to test luteolin induction by an mRNA-independent technology. Only SMc03168, whose induction was inconsistent (data not shown), followed the hybridization data. Because expression of these 12 genes was variable and because their expression was not increased when either nodD1 or nodD3 was overexpressed, we conclude that these genes do not show true luteolin induction but instead vary in expression from culture to culture. These results underscore the need for further analysis of those genes that show the most variability (Table 8, which is published as supporting information on the PNAS web site) and independent confirmation of results.
Overexpressing nodD allowed us to test for NodD dependence of the changes seen upon luteolin induction, as well as to test the behavior of plasmid-containing strains. We used strains that were mutated for all three nodD genes and that contained either nodD1 or nodD3 expressed constitutively on a plasmid. As expected, strains overexpressing nodD1 and grown in the presence of luteolin showed increased expression of the inducible nod genes. Moreover, nodD1's own expression increased 36-fold, confirming that elevated nodD1 expression from the constitutive trp promoter in the overexpression plasmid could be measured on the chips.
GeneChip measurement of transcripts in cells carrying overexpressed nodD3 on a plasmid correlated well with genetic studies (43). nodD3 expression itself was elevated 258-fold, and inducible nod gene expression showed comparable increases. We also saw very high expression of syrM and syrA, consistent with our discovery that syrM and nodD3 participate in an amplifying positive feedback loop, and that SyrM elevates syrA expression (43, 44).
In agreement with the previous observation that strains overexpressing nodD3 form mucoid colonies (44), we found 2.3- to 2.7-fold increases in EPS biosynthetic (exo) gene expression. Beyond confirmation of previous results, we discovered that overexpression of nodD3 results in elevated expression of >70 genes, half of which lack a predicted function. There is a strong intriguing replicon bias in the pattern of the NodD3 effect: genes whose expression increased were more likely to be carried on the symbiotic plasmids, whereas most repressed genes were chromosomal (Fig. 1).
Fig. 1.

Replicon distribution of coding probe sets increased in bacteroids or with nodD3 overexpression.
Similar to the gene set and extent of decrease as elaborated above, when nodD3 is overexpressed, half of the ≈100 repressed genes encode motility and chemotaxis functions. In agreement with this idea, the nodD3 overexpressing strain showed reduced motility on swarm plates (data not shown). Because the coordinate EPS/motility changes can be effected by more than one regulatory condition, and because it is not observed when nodD1 is overexpressed, we suggest that the decreases in motility-related genes are indirect effects of nodD3 overexpression rather than direct action of nodD3 on transcription.
Symbiotic Gene Expression. Because symbiosis is a developmental process, signal and response between the two partners probably occur at multiple stages. There must be an ongoing communication by which the bacteria and host monitor the progress of the infection as it proceeds through the invasion and bacterial release phases, and as the bacteria differentiate into nitrogen-fixing bacteroids.
The dual-genome chip provides a unique opportunity to assess changes occurring in both symbiotic partners simultaneously. Plant RNA copurifies with bacterial RNA from harvested root nodules, so both RNAs were present and able to hybridize to their respective targets. We compared expression between normal and mutant nodules and uninfected root tissue (Fig. 3, which is published as supporting information on the PNAS web site). Over 200 M. truncatula TCs increased expression in WT functioning nodules compared to roots, most showing large fold changes (Table 9, which is published as supporting information on the PNAS web site). These TCs corresponded to leghemoglobin, nodulins, transporters, proteases, calmodulin-like proteins, and proteins of unknown function. The majority of these had been previously shown to increase in legume nodules, confirming the validity of this approach. As a control, we examined expression in nodules infected with fixJ– mutant bacteria. Most of the TCs up-regulated in WT nodules were also induced in these Fix– nodules, an indication that many of the differences result from nodule morphogenesis and bacterial occupancy rather than from the nitrogen fixing status of the bacteria within (Fig. 2). Hundreds of TCs showed decreased expression in nodules; we are now conducting nodulation time courses to determine whether these are gene expression patterns that correlate with different stages of nodule development (C. G. Starker, A. Parra-Colmenares, and S.R.L., unpublished data).
Fig. 2.

Relative proportion of plant and bacterial probe sets increased in WT vs. mutant nodules. The groups of plant (Left) and bacterial (Right) probe sets that increased expression only in WT nodules are shown in orange. Those that increased only in fixJ– mutant nodules are shown in blue. Increases that occurred in both WT and mutant nodules are represented by the overlap.
Comparison of bacterial gene expression in nodules vs. free-living cultures showed that about one-fifth of the bacterial genome exhibited an expression change (Table 10, which is published as supporting information on the PNAS web site). In WT S. meliloti, 345 putative genes showed increased expression, and 943 showed a decrease. The sets of genes showing a decrease in WT nodules and mutant nonfixing nodules were almost the same. In contrast, very few of the normal increases in bacterial gene expression in nodules were found in the fixJ mutant, which indicates that most of the bacterial gene induction in nodules correlates with the ability to fix nitrogen (Fig. 2). As expected, there was virtually no difference between free-living aerobically grown WT and fixJ mutant strains (Table 1).
Consistent with what is known about this symbiotic developmental stage from earlier studies, bacteria in WT nodules showed induction of syrM, syrA, and the nif and fix genes (8, 43–45). Most genes involved in EPS biosynthesis were shut down. An exception was exoX, a posttranscriptional inhibitor of EPS synthesis that increased in bacteroids. Most nod genes were not expressed. However, nodL and adjacent genes were increased up to 50-fold in bacteroids, a surprising result that implies an additional role for NodL besides O-acetylation of Nod factors. Genes for phenylacetic acid catabolism (paaABCD) increased in nodule bacteria, in agreement with the finding that S. meliloti degrades plant flavonoids to phenylacetic acid (46).
Consistent with our earlier proposal regarding replicon specialization (11), we found a striking correlation of gene expression change with replicon. Over half of the genes induced in bacteroids are located on pSymA and pSymB; those on pSymA constitute one-third of the total (Fig. 1) and include many with unknown function. In contrast, genes whose expression decreased in bacteroids mainly fill “housekeeping” roles and are mostly chromosomal. Expression of cytochrome oxidases, NADH dehydrogenase (nuo), and ATP synthase decreased, reflecting the change in respiratory pathways toward those specialized for nitrogen fixation and perhaps altered energy requirements. Not all housekeeping genes showed reduced expression. In bacteroids, we saw strong induction of the ribosomal protein gene, rpsU1, and pSymB-encoded DNA topoisomerase and DNA ligase (SMb2144 and SMb21044).
In the nodule, marked decrease of expression was found for many bacterial genes involved in cell surface functions (capsular polysaccharides, lipopolysaccharides, sulfolipids, lipoproteins, and outer membrane proteins). Two genes whose expression increased in nitrogen-fixing bacteroids, but not in fixJ nodule bacteria, were ropB2 (outer membrane protein) and algI2 (putative cell surface saccharide acetylase). Because bacteroids exist in a novel compartment, these transcriptional changes may be important indicators of cell surface differentiation required for endocellular stability or function.
Dozens of regulatory proteins showed differential expression in bacteroids. Although most genes in the flagellar/chemotaxis regulon are repressed in bacteroids, two chemotaxis regulators, VisR and VisN, showed increased expression. SMc00887, which has GGDEF and EAL domains, and SMc00888, a putative kinase with a PAS oxygen-sensing domain, showed reduced expression in minimal medium in the nodD3 overexpressing strain and in bacteroids, suggesting some feature in common in all three situations resulted in their lowered expression.
We used qPCR to independently test expression of several genes, using the same samples of cDNA that were hybridized to chips, and thus directly compared the two techniques (Tables 4 and 5). We found consistent agreement between the chip and qPCR data and investigated one exception more closely. Although chips showed nodD3 expression to be unchanged between nodule and free-living bacteria, the more sensitive qPCR technique showed that nodD3 was expressed 40-fold higher in nodules than in free-living cells. To substantiate this result, we used three different pairs of primers for qPCR and determined the DNA sequence of these nodD3 products. Analysis of all three qPCR products showed the same 40-fold difference in expression between nodule and free-living bacteria. Thus some genes expressed at low levels may yield misleading information from chip studies. This can be important when considering a gene encoding a regulatory protein, because of the downstream consequences of its action.
In nodules, dicarboxylic acids like succinate and malate are preferred metabolic fuel substrates for nitrogen-fixing bacteroids (33). We found that the genes expressed at high levels in free-living succinate-grown cells are not similarly expressed in nodules. In fact, pckA, which encodes phosphoenolpyruvate carboxykinase, is 6.5-fold lower in nodules. Recent studies using knockout mutations show that a complete bacterial citric acid cycle is not required for nitrogen fixation in the Bradyrhizobium japonicum–soybean symbiosis (47, 48). This finding points to a need for more detailed studies on basic nodule physiology, so that we can learn how dicarboxylic acids are catabolized to subsidize nitrogen fixation.
Comparison of GeneChips with Spotted Microarrays. Affymetrix GeneChips represent one of several platforms for genome-wide hybridization analysis. Direct comparisons among platforms can be problematic. On PCR- or oligonucleotide-based nylon filter macro- and glass slide microarrays, the weight of detection depends on a single probe for each gene. Any inappropriate crosshybridization to that spotted PCR product probe will result in an inaccurate measurement of mRNA level. On oligo-based Affymetrix GeneChip microarrays, each gene is represented by multiple oligonucleotides making up a probe set. If one member of the probe set hybridizes aberrantly to the appropriate target, there remain many other probes in that probe set to help determine the expression level for that gene. Studies generally show that crossplatform differences are a consequence of intrinsic properties of the arrays themselves; these can include probe sequence differences, differences in labeling and hybridization conditions, and quality control issues such as PCR product quantification and spotting reproducibility, the last two of which are eliminated in use of Affymetrix GeneChips (49, 50).
Comparison of our studies with those of Becker et al. (20), who used spotted arrays, showed both overlap and difference (Data Set 3 and Fig. 4, which are published as supporting information on the PNAS web site). We observed 30% more expression changes than found in their study; one-third of our changes overlap with their work. Likewise, only 45% of their changes are found in ours. Some of these differences may be related to a difference between arrays and chips, as discussed above, and/or to differences in RNA preparation methods. In addition, the comparison of results may point to biologically interesting information. Because S. meliloti forms continually growing or indeterminate nodules, preparations of intact nodules are heterogeneous, containing bacteria in all stages of invasion and differentiation. Becker et al. (20) isolated RNA from much younger nodules of a different host, Medicago sativa, for their studies; their nodule preparations probably contained more invading bacteria than ours, whereas our samples were probably more enriched for bacteroids and contained senescing bacteroids. Indeed, we found induction of more nif and fix genes than they did but failed to see increased expression of nod and exo genes, a hallmark of invading bacteria. Also, we did not see induction of the oxidative stress genes katA and oxyR (20), perhaps indicative that the oxidative burst associated with early infection had mostly passed.
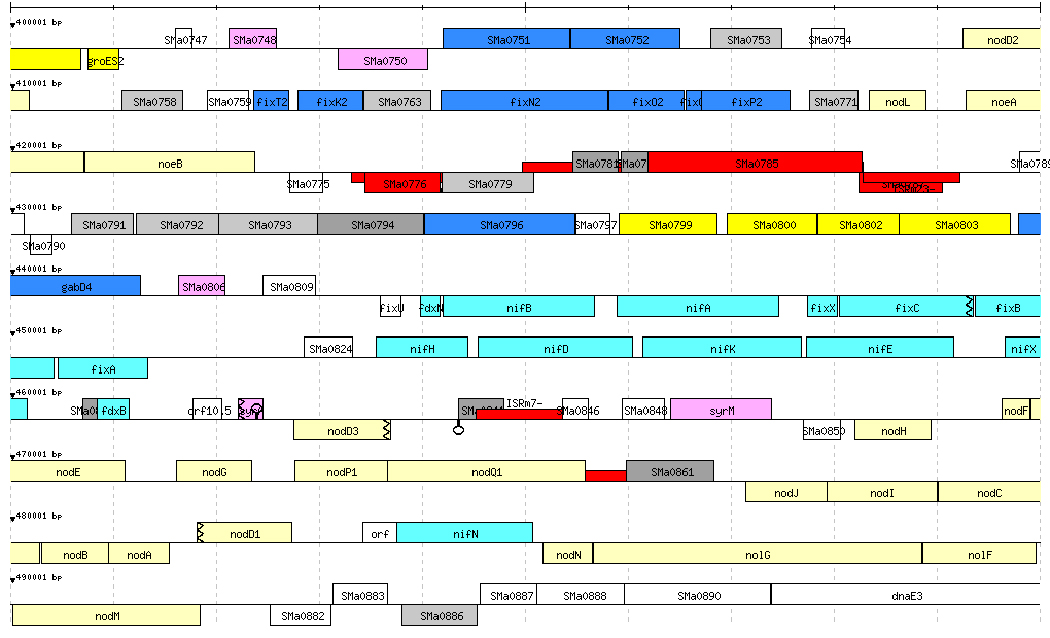
Expression of Orphan Genes and Intergenic Regions. S. meliloti contains many ORFs with no homology to protein databases (11). Although some of these, like noeB, have been assigned a role, other hypothetical or “orphan” genes may not be genes at all. Examining orphan gene expression during growth in rich and minimal medium and in the nodule, we found that over half were expressed above (Data Set 4, Fig. 5, and Table 11, which are published as supporting information on the PNAS web site). Although the average signal was usually low, many were expressed highly under one or several of our different experimental conditions, suggesting that they are specialized and needed only under certain environmental conditions. Of the nearly 100 orphan genes expressed above background in bacteroids, nearly half changed expression compared with free-living bacteria, with most being higher in bacteroids.
Hybridization to intergenic probe sets may occur due to: expression of genes missed during the original annotation, detection of the untranslated 5′ and 3′ ends of genes, and noncoding RNAs. Examination of intergenic region expression upstream of nod boxes and known translation start sites indicates that leader transcripts can be detected by hybridization to specific tiled oligonucleotides (Fig. 6 and Table 12, which are published as suporting information on the PNAS web site; D. Almassian, S.R.L., and S. L. Shaw, unpublished data). We are developing software tools to aid in analysis of the intergenic regions.
Conclusion
The dual-genome microarray is a powerful tool not only for examination of bacterial strains in diverse growth conditions but also for simultaneous assay of both bacterial and plant gene expression within the context of nodule development. The global changes in gene expression we have documented show that S. meliloti adapts to abiotic and symbiotic environments in unique ways, and that the three bacterial replicons serve specialized roles in this adaptation. Among the findings of this study is a striking disparity of plant and bacterial transcription changes in a mutant nodule–WT nodule comparison. This suggests that analyses of nodules resulting from several mutants with distinct points of developmental arrest will point to the differing stages at which the two partners control each other.
Supplementary Material
Acknowledgments
We are grateful to D. Almassian for his role in chip design, to G. Barnett for help with software, to R. Kurtz and D. Batey of MJ Research for advice on qPCR and use of equipment, to I. Russell and C. Rosenow of Affymetrix for helpful advice, and to E. Roberts for assistance in growing plants and harvesting nodules. We thank M. Kahn and V. Oke for critical reading of the manuscript and fellow lab members for useful discussions. This work was supported by Department of Energy Grant DE-FG03-90ER20010, National Science Foundation Grant U/G001311, and Stanford University.
Author contributions: M.J.B., C.J.T., R.F.F., and S.R.L. designed research; M.J.B., C.J.T., and R.F.F. performed research; M.J.B., C.J.T., and R.F.F. analyzed data; M.J.B., R.F.F., and S.R.L. wrote the paper; and S.R.L. was the Principal Investigator.
Abbreviations: TC, tentative consensus sequence; qPCR, quantitative PCR; EPS, exopolysaccharide.
References
- 1.Long, S. R. (2001) Plant Physiol. 125, 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultze, M. & Kondorosi, A. (1998) Annu. Rev. Genet. 32, 33–57. [DOI] [PubMed] [Google Scholar]
- 3.Stougaard, J. (2000) Plant Physiol. 124, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brewin, N. J. (1998) in The Rhizobiaceae, eds. Spaink, H. P., Kondorosi, A. & Hooykaas, P. J. J. (Kluwer, Dordrecht, The Netherlands), pp. 418–429.
- 5.Hirsch, A. M. (1992) New Phytol. 122, 211–237. [DOI] [PubMed] [Google Scholar]
- 6.Oke, V. & Long, S. R. (1999) Mol. Microbiol. 32, 837–849. [DOI] [PubMed] [Google Scholar]
- 7.Poole, P. & Allaway, D. (2000) Adv. Microb. Physiol. 43, 117–163. [DOI] [PubMed] [Google Scholar]
- 8.Kaminski, P. A., Batut, J. & Boistard, P. (1998) in The Rhizobiaceae, eds. Spaink, H. P., Kondorosi, A. & Hooykaas, P. J. J. (Kluwer, Dordrecht, The Netherlands), pp. 431–460.
- 9.Jumas-Bilak, E., Michaux-Charachon, S., Bourg, G., Ramuz, M. & Allardet-Servent, A. (1998) J. Bacteriol. 180, 2749–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capela, D., Barloy-Hubler, F., Gouzy, J., Bothe, G., Ampe, F., Batut, J., Boistard, P., Becker, A., Boutry, M., Cadieu, E., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9877–9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galibert, F., Finan, T. M., Long, S. R., Pühler, A., Abola, A. P., Ampe, F., Barloy-Hubler, F., Barnett, M. J., Becker, A., Boistard, P., et al. (2001) Science 293, 668–672. [DOI] [PubMed] [Google Scholar]
- 12.Oresnik, I. J., Liu, L.-L., Yost, C. K. & Hynes, M. F. (2000) J. Bacteriol. 182, 3582–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnett, M. J., Fisher, R. F., Jones, T., Komp, C., Abola, P. A., Barloy-Hubler, F., Bowser, L., Capela, D., Galibert, F., Gouzy, J., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9883–9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finan, T. M., Weidner, S., Chain, P., Buhrmester, J., Wong, K., Vorhölter, F.-J., Hernandez-Lucas, I., Becker, A., Cowie, A., Gouzy, J., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9889–9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ampe, F., Kiss, E., Sabourdy, F. & Batut, J. (2003) Genome Biol. 4, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergès, H., Lauber, E., Liebe, C., Batut, J., Kahn, D., de Bruijn, F. & Ampe, F. (2003) Appl. Environ. Microbiol. 69, 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, H., Higgins, J., Oresnik, I., Hynes, M. F., Natera, S., Djordjevic, M. A., Weinman, J. J. & Rolfe, B. G. (2000) Electrophoresis 21, 3833–3842. [DOI] [PubMed] [Google Scholar]
- 18.Djordjevic, M. A., Chen, H. C., Natera, S., Noorden, G. V., Menzel, C., Taylor, S., Renard, C., Geiger, O., the Sinorhizobium DNA Sequencing Consortium & Weiller, G. F. (2003) Mol. Plant–Microbe Interact. 16, 508–524. [DOI] [PubMed] [Google Scholar]
- 19.Rüberg, S., Tian, Z.-X., Krol, E., Linke, B., Meyer, F., Wang, Y., Pühler, A., Weidner, S. & Becker, A. (2003) J. Biotechnol. 106, 255–268. [DOI] [PubMed] [Google Scholar]
- 20.Becker, A., Bergès, H., Krol, E., Bruand, C., Rüberg, S., Capela, D., Lauber, E., Meilhoc, E., Ampe, F., de Bruijn, F., et al. (2004) Mol. Plant–Microbe Interact. 17, 292–303. [DOI] [PubMed] [Google Scholar]
- 21.House, B. L., Mortimer, M. W. & Kahn, M. L. (2004) Appl. Environ. Microbiol. 70, 2806–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Djordjevic, M. A. (2004) Proteomics 4, 1859–1872. [DOI] [PubMed] [Google Scholar]
- 23.Lockhart, D. J., Dong, H., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S., Mittmann, M., Wang, C., Kobayashi, M., Horton, H., et al. (1996) Nat. Biotechnol. 14, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 24.Mitra, R. M., Shaw, S. L. & Long, S. R. (2004) Proc. Natl. Acad. Sci. USA 101, 10217–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronson, C. W., Nixon, B. T., Albright, L. M. & Ausubel, F. M. (1987) J. Bacteriol. 169, 2424–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honma, M. & Ausubel, F. M. (1987) Proc. Natl. Acad. Sci. USA 84, 8558–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher, R. F., Egelhoff, T. T., Mulligan, J. T. & Long, S. R. (1988) Genes Dev. 2, 282–293. [DOI] [PubMed] [Google Scholar]
- 28.Ehrhardt, D. W., Atkinson, E. M. & Long, S. R. (1992) Science 256, 998–1000. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein, J. A., Khodursky, A. B., Lin, P.-H., Lin-Chao, S. & Cohen, S. N. (2002) Proc. Natl. Acad. Sci. USA 99, 9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao, H., Bausch, C., Richmond, C., Blattner, F. R. & Conway, T. (1999) J. Bacteriol. 181, 6425–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei, Y., Lee, J. M., Richmond, C., Blattner, F. R., Rafalski, J. A. & LaRossa, R. A. (2001) J. Bacteriol. 183, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlin, S., Barnett, M. J., Campbell, A. M., Fisher, R. F. & Mrázek, J. (2003) Proc. Natl. Acad. Sci. USA 100, 7313–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDermott, T. R., Griffith, S. M., Vance, C. P. & Graham, P. H. (1989) FEMS Microbiol. Rev. 63, 327–340. [Google Scholar]
- 34.Finan, T. M., Oresnik, I. & Bottacin, A. (1988) J. Bacteriol. 170, 3396–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leigh, J. A., Signer, E. R. & Walker, G. C. (1985) Proc. Natl. Acad. Sci. USA 82, 6231–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei, X. & Bauer, W. D. (1998) Appl. Environ. Microbiol. 64, 1708–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei, X. & Bauer, W. D. (1999) Appl. Environ. Microbiol. 65, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dombrecht, B., Marchal, K., Vanderleyden, J. & Michiels, J. (2002) Genome Biol. 3, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blot, M., Hauer, B. & Monnet, G. (1994) Mol. Gen. Genet. 242, 595–601. [DOI] [PubMed] [Google Scholar]
- 40.Adam, E., Volkert, M. R. & Blot, M. (1998) Mol. Microbiol. 28, 15–24. [DOI] [PubMed] [Google Scholar]
- 41.Poole, R. K. & Cook, G. M. (2000) Adv. Microbiol. Physiol. 43, 165–224. [DOI] [PubMed] [Google Scholar]
- 42.Delgado, M. J., Bedmar, E. J. & Downie, J. A. (1998) Adv. Microbiol. Physiol. 40, 191–231. [DOI] [PubMed] [Google Scholar]
- 43.Swanson, J. A., Mulligan, J. T. & Long, S. R. (1993) Genetics 134, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnett, M. J., Swanson, J. A. & Long, S. R. (1998) Genetics 148, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma, S. B. & Signer, E. R. (1990) Genes Dev. 4, 344–356. [DOI] [PubMed] [Google Scholar]
- 46.Raghavendra Rao, J. & Cooper, J. E. (1994) J. Bacteriol. 176, 5409–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green, L. S. & Emerich, D. W. (1997) Plant Physiol. 114, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thöny-Meyer, L. & Künzler, P. (1996) J. Bacteriol. 178, 6166–6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogojina, A. T., Orr, W. E., Song, B. K. & Geisert, E. E. (2003) Mol. Vision 9, 482–496. [PMC free article] [PubMed] [Google Scholar]
- 50.Tan, P. K., Downey, T. J., Spitznagel, E. L., Xu, P., Fu, D., Dimitrov, D. S., Lempicki, R. A., Raaka, B. M. & Cam, M. C. (2003) Nucleic Acids Res. 31, 5676–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}