

Abstract
Next-generation sequencing technologies have greatly expanded our understanding of cancer genetics. Antisense technology is an attractive platform with the potential to translate these advances into improved cancer therapeutics, because antisense oligonucleotide (ASO) inhibitors can be designed on the basis of gene sequence information alone. Recent human clinical data have demonstrated the potent activity of systemically administered ASOs targeted to genes expressed in the liver. Here, we describe the preclinical activity and initial clinical evaluation of a class of ASOs containing constrained ethyl modifications for targeting the gene encoding the transcription factor STAT3, a notoriously difficult protein to inhibit therapeutically. Systemic delivery of the unformulated ASO, AZD9150, decreased STAT3 expression in a broad range of preclinical cancer models and showed antitumor activity in lymphoma and lung cancer models. AZD9150 preclinical activity translated into single-agent antitumor activity in patients with highly treatment-refractory lymphoma and non-small cell lung cancer in a phase I dose escalation study.
Introduction
Large-scale cancer genome studies have dramatically increased our ability to classify cancers on the basis of specific mutations and gene expression signatures. This success has resulted in the identification of genetic events that occur with high frequency in various malignancies (1, 2). These data, in combination with pathway analysis (3) and functional genomics studies, made possible by tools such as small interfering RNA (siRNA) and antisense technology, indicate that tumor cells have specific sets of “driver” mutations that can render them vulnerable to the targeted inhibition of specific pathways (4, 5). These insights bolster hope that selective molecularly targeted therapies will ultimately translate into life-saving cancer treatments that do not have the toxicities associated with traditional chemotherapy. However, despite some notable successes (6-8), this promise has not yet been broadly realized, partly because available small molecule and protein therapeutic modalities have been unsuccessful in targeting many of the most attractive pathways driving cancer.
Therapeutic nucleic acid-based approaches like siRNA and antisense technology hold enormous potential to help bridge this pharmacogenomic divide because with these modalities, inhibitors can be rationally designed based solely on gene sequence information, thus expanding the range of targets that are druggable to include transcription factors, structural proteins, and even non-coding RNAs. Therapeutic antisense technology has greatly advanced during the 20 plus years since its inception. Recently, systemically delivered unformulated antisense oligonucleotides (ASOs) have proven effective as human therapeutics in several non-oncology disease settings, especially when directed to genes expressed in liver (9-13). Moreover, the second generation (Gen 2.0) 2′-O-methoxyethyl containing ASO, Kynamro, which targets the mRNA encoding the liver-derived factor apolipoprotein B-100 for the treatment of homozygous familial hypercholesterolemia, recently became the first systemically administered ASO to gain FDA approval. Gen 2.0 ASOs designed to target a nuclear-retained non-coding RNA involved in Myotonic Dystrophy type I (DM1) were recently reported to have unexpectedly potent activity in extra-hepatic tissues, such as skeletal muscle, that have been historically difficult to target with ASOs (11). The depletion of the non-coding RNA in this case, by systemically delivered ASOs, completely reversed the muscle pathology in all skeletal muscles in an animal model of DM1 (11). The sensitivity of this non-coding RNA to ASOs is believed to be due to prolonged nuclear co-localization of the target RNA and the enzyme ribonuclease H, which is responsible for the catalytic degradation of the target RNA molecule of the RNA:ASO duplex (11). Building on these data, we hypothesized that ASOs with greater intrinsic potency than the Gen 2.0 chemistry ASOs would also be capable of targeting traditional coding mRNAs and thus could potentially therapeutically target previously difficult to drug pathways in tumors. Towards this aim, we have developed a high-affinity next-generation class of ASO that contains 2′-4′ constrained ethyl (cEt)-modified residues. These ASOs typically consist of 8-10 base phosphorothioate (PS)-modified deoxynucleotide ‘gap’ flanked on either end with 2-3 cEt nuclelotides (14). The Gen 2.5 cEt-containing ASOs exhibit enhanced in vitro and in vivo potency over Gen 2.0 ASOs (14, 15). To determine the potential of next generation chemistry cEt ASOs in cancer, we chose to target a transcription factor, because this is a protein class that has historically been difficult to inhibit therapeutically. We selected the transcription factor STAT3, a member of the signal transduction and activator of transcription family, as a test-case transcription factor cancer target which, despite extensive effort, has proved difficult to inhibit specifically with conventional therapeutic approaches (16). Here, we report the superior pharmacologic activity of the cEt STAT3 ASO, AZD9150, compared to optimized earlier generation (Gen 2.0) STAT3 ASOs in multiple preclinical cancer models and the preclinical anti-tumor efficacy of ADZ9150 in models of human lymphoma and lung cancer. Finally, we describe the early clinical efficacy of AZD9150, where single-agent antitumor activity was observed at low doses in patients with highly treatment-refractory lymphomas and non-small cell lung cancer in a phase I dose-escalation study.
Results
Depletion of STAT3 RNA and protein with lipid-independent delivery of AZD9150 in vitro
ASO potency can be influenced by the target site on the RNA and by the chemistry of the ASO (14, 17). Thus, to identify the most potent Gen 2.0 and cEt STAT3 ASOs for comparison, we performed gene walk screens with ASOs containing either chemistry. The activity of the optimized Gen 2.0 and cEt ASOs identified was first evaluated in a panel of cancer cell lines in culture. The in vitro evaluation of nucleic acid-based agents (siRNA or antisense) typically relies on cationic lipid-mediated transfection to deliver them into cells (18, 19). Because the cEt chemistry was designed to increase the potency of ASOs, we sought to determine whether these inhibitors would be active in cancer cells growing in culture without any delivery agent (termed free-uptake conditions). The human-selective cEt STAT3 ASO, AZD9150, was 10 to 20 times more potent than the Gen 2.0 STAT3 ASOs as measured by analysis of cellular STAT3 mRNA 24 hours after treatment. The half-maximal inhibitory concentration (IC50) values for the most sensitive cells were in the low nanomolar range (Fig. 1A). The cEt STAT3 ASO was highly active by free uptake in both adherent solid tumor and non-adherent hematologic cancer cells in culture (fig. S1). However, the sensitivity to ASO-mediated target RNA reduction varied in the cell lines evaluated, potentially due to differences in productive and non-productive (for example, lysosome-mediated) uptake pathways in these cells, as has been described previously (20). Consistent with a loss of ASO productive uptake as a result of growth in cell culture, primary mouse hepatocytes lose ASO uptake capability in as little as 24 hours after being placed in cell culture (fig. S2). The potency advantage of cEt over Gen 2.0 chemistry across cell lines was also evident when STAT3 protein concentrations were measured at 72 h, by which point near-complete depletion of STAT3 protein was achieved in all cell lines with AZD9150 (Fig. 1B and fig. S3). The inhibition of STAT3 protein was dose-dependent and selective for STAT3. Amounts of related protein family members STAT1 and STAT5 were unchanged upon treatment of cells with STAT3 ASOs. The reduction in STAT3 was ASO sequence dependent, because the control ASOs had no effect even at the highest doses tested (Fig. 1B and fig. S3). The inhibition of STAT3 protein by AZD9150 was observed as early as 24 hours after treatment and was maintained until day 5 without additional ASO treatment (fig. S4).
Fig. 1. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in tumor cells in vitro.
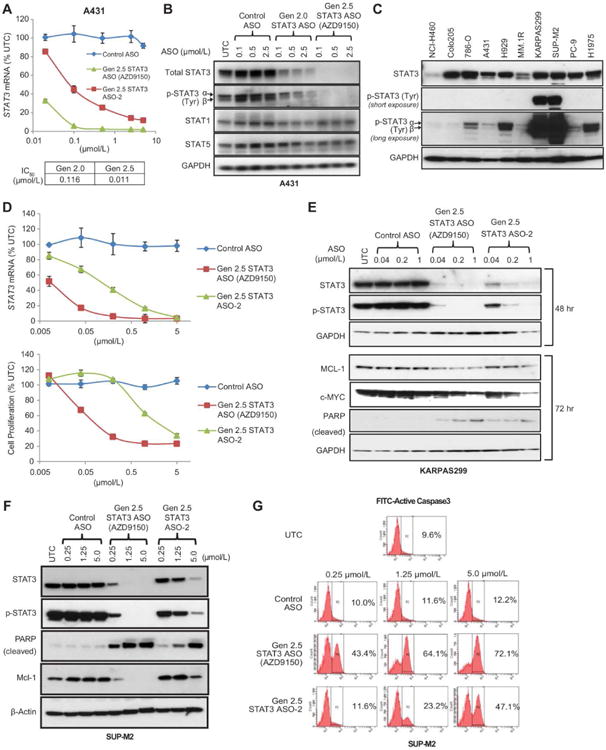
(A) Gen 2.0 MOE or cEt STAT3 ASO along with control ASO were delivered to human epidermoid cancer A431 cells via free uptake. STAT3 knockdown was determined by qRT-PCR. IC50 values for Gen 2.0 and Gen 2.5 cEt STAT3 ASOs calculated using GraphPad Prism software are shown. (B) Total cell lysates were collected for immunoblot analysis 3 days after ASO treatment. Amounts of total and phosphorylated forms of STAT3 protein as well as other STAT members, STAT1 and STAT5, were compared. (C) Amounts of total and phosphorylated STAT3 were measured in a variety of human tumor cell lines by immunoblot analysis. (D-G) Two cEt STAT3 ASOs along with a control ASO with the same chemistry were delivered to human anaplastic large T-cell lymphoma (ALCL) lines, SUP-M2 and KARPAS299, by free uptake. STAT3 mRNA concentration and cell proliferation were determined in KARPAS299 at 24 h and on day 5, respectively (D). The amounts of c-MYC and MCL-1 proteins, which are regulated by STAT3, also decreased at 72 h, after the decrease in STAT3 protein (E). Downregulation of STAT3 by the cEt STAT3 ASOs also resulted in strong apoptosis in SUP-M2 cells, as demonstrated by immunoblot analysis for cleaved PARP, a surrogate marker for apoptosis at 72 h (F), and FACS analysis for active caspase-3 at 72 h (G). UTC: untreated cells. These experiments have been replicated at least 3 times. Graphs in A and D show the means ± standard deviations (SD) of n = 3.
The potent free-uptake ASO activity described here differs from the activity referred to as “gymnotic” delivery previously described for ASOs with a different chemistry (21). In the previously reported study, target RNA reduction was only observed in the micromolar range (>100-fold less potent than the ASOs described here) after several days of treatment under highly culture-specific conditions. In contrast to the activity of AZD9150, which demonstrated low nanomolar IC50 values for target RNA inhibition in cell culture by free-uptake, double-stranded siRNA inhibitors were critically dependent on transfection reagents to induce RNA silencing and showed no cellular activity by lipid-independent delivery, although the siRNAs had good potency when transfected with cationic lipids (fig. S5). This is also consistent with previous reports demonstrating that in vivo siRNAs have limited efficacy without liposomal or nanoparticle formulation (22, 23).
Antiproliferative effect of AZD9150 in human lymphoma lines in vitro
To identify cancer cells that might rely on STAT3 for growth, we evaluated the degree of STAT3 pathway activation, as measured by the amount of phosphorylated STAT3 protein, in a broadened panel of cells that included the lymphoma cell lines KARPAS299 and SUP-M2. Although most cells had comparable amounts of total STAT3 protein, the two lymphoma lines had highly activated STAT3 pathways, suggesting that these cells may require STAT3 for survival (Fig. 1C). This was in fact the case, because ASO-mediated depletion of STAT3 in KARPAS299 and SUP-M2 cells rapidly induced apoptosis as measured by the production of cleaved PARP protein or the activation of Caspase-3 (Fig. 1, D and E for KARPAS299 and Fig. 1, F and G for SUP-M2). Moreover, in these lymphoma cells both AZD9150 and a second, less potent cEt STAT3 inhibitor (ASO-2) induced dose-dependent reductions in STAT3 mRNA (Fig. 1D) and STAT3 protein (Fig. 1, E and F) and reduced expression of STAT3 target genes MCL-1, c-MYC (Fig. 1E), BCL6, CYCLIN D1, BIRC5 (SURVIVIN), and IL-2Rα (fig. S6). These phenotypic effects correlated with the degree of STAT3 depletion induced by each of these independent STAT3 ASOs, in concordance with the potency of each ASO, demonstrating the specificity of the effect for STAT3 inhibition. To further demonstrate that the effects observed in SUP-M2 and KARPAS299 cells were specific to STAT3 inhibition, we modulated the STAT3 pathway with three additional approaches. First, we treated cells with the small molecule inhibitor AZD1480, an inhibitor of the kinases JAK1 and 2, which lie upstream of STAT3 and control its phosphorylation and activation (24). As expected, AZD1480 induced a dose-dependent reduction of p-STAT3 in KARPAS299 cells, and this correlated with an inhibition of cell proliferation (fig. S7A, left panel). Similarly, SUP-M2 cells were also sensitive to AZD1480 treatment, and both p-STAT3 and cell proliferation were markedly inhibited (fig. S7A, right panel). Finally, we evaluated the effect of STAT3 inhibition by a drug, crizotinib, an inhibitor of the upstream kinase ALK (25). As expected, treatment of KARPAS299 and SUP-M2 cells with crizotinib resulted in a marked decrease in cell proliferation (fig. S7B). Finally, we used multiple STAT3 siRNA inhibitors, delivered to the cells via electroporation to reduce STAT3 expression, and found that these reagents similarly inhibited the proliferation of lymphoma cells, whereas control siRNA molecules did not (fig. S7C). Furthermore, downregulation of STAT3 correlated tightly with effects on cell proliferation when several additional ASOs of varying length and chemistries were evaluated (Gen 2.0 vs Gen 2.5 or 16-mer vs 20-mer) (fig. S7D). Taken together, these results demonstrate that the effects observed with cEt ASOs are mediated by STAT3 inhibition and are consistent with previous reports suggesting that STAT3 is important for lymphoma cell proliferation and survival (26).
In contrast to the high sensitivity of KARPAS299 and SUP-M2 cells to STAT3 depletion, the near complete depletion of STAT3 had minimal effect on the proliferation of the majority of the other cancer cell lines evaluated in vitro (fig. S8A), even though the STAT3 pathway was active and phosphorylated STAT3 (p-STAT3) was detected in many of these cells (Fig. 1C and fig. S3). As expected, a positive control ASO targeting the mRNA encoding the mitotic kinesin Eg5 produced a strong antiproliferative effect in all cells tested (fig. S8A). These findings are also consistent with previous findings that no antiproliferative effects were observed in multiple solid cancer cell lines in vitro when the STAT3 pathway was inhibited by the JAK small molecule inhibitor AZD1480 (24). To confirm this, we also tested AZD1480 and STAT3 siRNA inhibitors in two of these cell lines, NCI-H460 and PC-9, and we did not observe any significant antiproliferative effects with either approach despite the strong reduction of p-STAT3 and STAT3 mRNA (fig. S8, B to D for NCI-H460 and fig. S8, E to G for PC9). These results further demonstrate that the antiproliferative effects of STAT3 ASOs are specific to the lymphoma cells and to the selective depletion of STAT3.
Reduction of STAT3 in tumor xenografts and primary human tumor explants in vivo after systemic administration of AZD9150
The potency advantage of the cEt ASO relative to the Gen 2.0 ASO suggested that efficient target RNA inhibition might be achieved in vivo in extra-hepatic tissues and in tumors. We chose to first test AZD9150 in the A431 subcutaneous human epidermoid tumor xenograft model because A431 cells were among the cells most sensitive to ASO under free uptake conditions in vitro (IC50 11 nmol/L) (Fig. 1A) and because the depletion of STAT3 did not result in inhibition of A431 cell proliferation (fig. S8A). Thus, effects on STAT3 in this tumor model would not be a result of indirect effects on tumor growth. To clearly demonstrate the in vivo potency advantage of the Gen 2.5 over Gen 2.0 ASO, we evaluated AZD9150 at half the dose of the Gen 2.0 ASO. Systemic delivery of 25 mg/kg per day of AZD9150 to mice bearing established A431 tumors produced an approximately 90% reduction in STAT3 mRNA in tumors (P = 0.0001; Fig. 2A). However, the Gen 2.0 ASO administered at 50 mg/kg per day reduced STAT3 mRNA in tumors by only 30% (Fig. 2A). Western blot analysis confirmed both the strong inhibition of STAT3 protein by AZD9150 (93% inhibition) and the potency advantage over Gen 2.0 ASO, where the 50 mg/kg dose produced only approximately 40% reduction of STAT3 protein (Fig. 2B). AZD9150 treatment had little effect on STAT1 or STAT5 protein, confirming the drug's specificity for STAT3 in vivo. Treatments with either Gen 2.0 or cEt control ASOs did not have an effect on STAT3 RNA or protein (Fig. 2, A and B). The presence of ASOs in tumors was demonstrated by both immunohistochemistry (IHC) analysis with an anti-ASO antibody and by quantitative HPLC (fig. S9, upper panel and table S1, respectively). Consistent with the broad distribution in tumors, AZD9150 treatment produced dose-dependent, near-complete depletion of human STAT3 protein throughout the tumor, as determined by IHC analysis of STAT3 protein (Fig. 2C); as expected, the Gen 2.0 ASO treatment was much less effective (fig. S9, lower panel). Despite the strong knock down of STAT3 in the A431 tumor cells, there was no effect on the growth of A431 tumors even at higher doses of AZD9150 (37.5 and 50 mg/kg) in a separate study (fig. S10), demonstrating that this activity is not an indirect result of effects on tumor growth, and that this model is not sensitive to STAT3 depletion. In addition, because AZD9150 is a human-specific STAT3 ASO with limited activity against the mouse STAT3, much of the residual STAT3 protein in the tumor may be due to the presence of mouse stromal cells in the tumor microenvironment. To confirm this, tumor-bearing mice were treated with a mouse active cEt STAT3 ASO (Fig. 3A). Treatment with this ASO resulted in STAT3 depletion in mouse tumor-associated stromal cells but not the human tumor cells, as shown by IHC and qRT-PCR with species-specific primers/probe (Fig. 3, B and C). Although muscle is a tissue typically insensitive to ASOs, the mouse active cEt STAT3 ASO also produced near complete depletion of STAT3 protein in skeletal muscle of treated mice (Fig. 3D).
Fig. 2. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in A431 xenograft tumors.
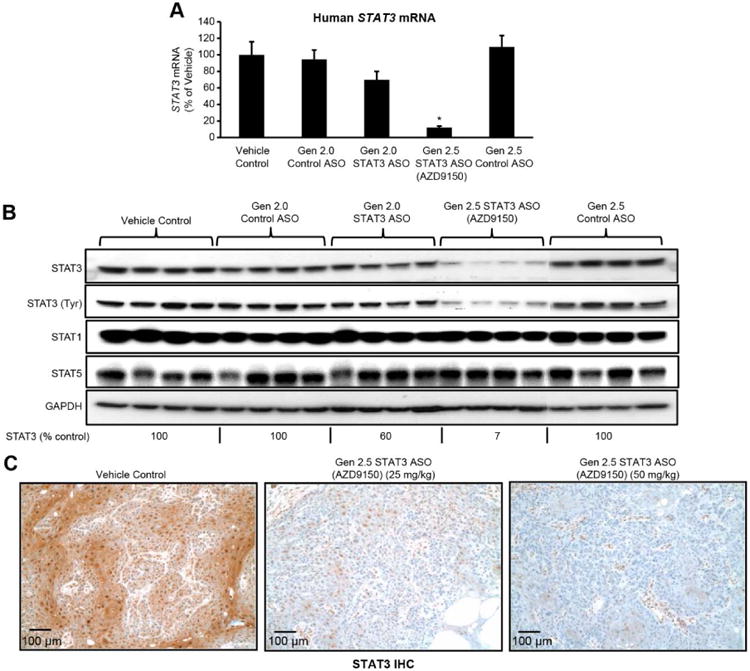
(A-B) Animals bearing A431 human epidermoid tumors were treated with Gen 2.0 STAT3 ASO, cEt STAT3 ASO, or control Gen 2.0 or cEt ASO at either 50 mg/kg (for Gen 2.0 chemistry) or 25 mg/kg (for Gen 2.5 chemistry), 5 times per week for 3 weeks. Target reduction at RNA or protein levels was determined by q-RT-PCR (A) (*P = 0.0001; n = 6 mice per group) and immunoblot analysis (B), respectively. (C) Dose-dependent decrease in human STAT3 by AZD9150 was confirmed by immunohistochemistry (IHC). Graph A shows the means ± SD. Scale bars, 100 μm.
Fig. 3. Downregulation of mouse or human STAT3 expression with species-specific cEt STAT3 ASOs in A431 xenograft model.
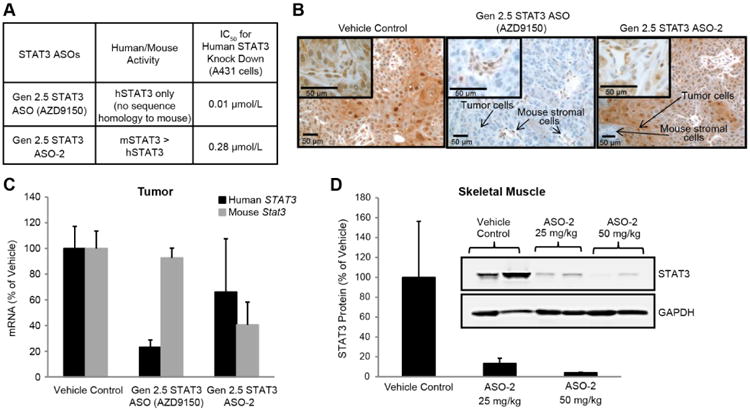
(A) Mice transplanted with A431 human epidermoid tumor cells were treated with either a cEt STAT3 ASO that is specific for human STAT3 (AZD9150) or with an ASO that has greater activity for mouse STAT3 (ASO-2), at 25 mg/kg, daily, 5 days a week for 3 weeks (n = 4 mice per group). (B) Immunohistochemistry demonstrated selective inhibition of either human STAT3 in tumor cells or mouse stromal STAT3 with AZD9150 and ASO-2, respectively, which was consistent with q-RT-PCR results for STAT3 mRNA determined by using species-specific probe/primer sets, as shown in (C) (n = 4 mice per group). (D) Mouse-active STAT3 ASO (ASO-2) also reduced the endogenous STAT3 protein in the quadriceps muscle of treated animals in a dose-dependent manner (n = 2 mice per group). Graphs in C and D show the means ± SD. Scale bars, 50 μm.
As mentioned above, if the diminished ASO uptake capacity of some cancer cell lines in vitro were a reflection of the loss of free uptake when establishing cell lines in culture, similar to that seen in hepatocytes in culture (fig. S2), we might expect ASO uptake pathways to be intact in primary tumors and primary tumor explants to be sensitive to ASO-mediated target knock down in vivo. To test this, we evaluated the pharmacologic activity of AZD9150 in a series of human primary patient-derived tumor explant (PDX) models, where tumors have never been exposed to tissue culture. These included non-small cell lung cancer (NSCLC), colorectal cancer, and lymphoma PDX models. Treatment of mice bearing established primary human tumors with AZD9150 resulted in strong inhibition of STAT3 protein in the tumors of all three primary cancer models, as determined by western blot analysis (80%, 76%, and 54% inhibition of STAT3 for the NSCLC, CRC, and lymphoma tumors, respectively) (Fig. 4, left panel). Because all three PDX models contained a large amount of mouse stromal cells within tumors themselves, and because the antibody for the western blots does not distinguish between mouse and human STAT3, the inhibition calculated for the depletion of human STAT3, by the human-specific STAT3 ASO AZD9150, is likely an underestimation. IHC on tumor sections for STAT3 protein demonstrated strong inhibition of STAT3 protein in the tumor cells in all 3 PDX models (Fig. 4, right panel), although quantification of STAT3 on IHC showed less reduction compared to the western blots, most likely due to the limitation of this method as reported (27, 28). AZD9150 had no effect on STAT1 and STAT5 protein (fig. S11).
Fig. 4. Pharmacological activity of AZD9150 in patient-derived primary explants (PDX).
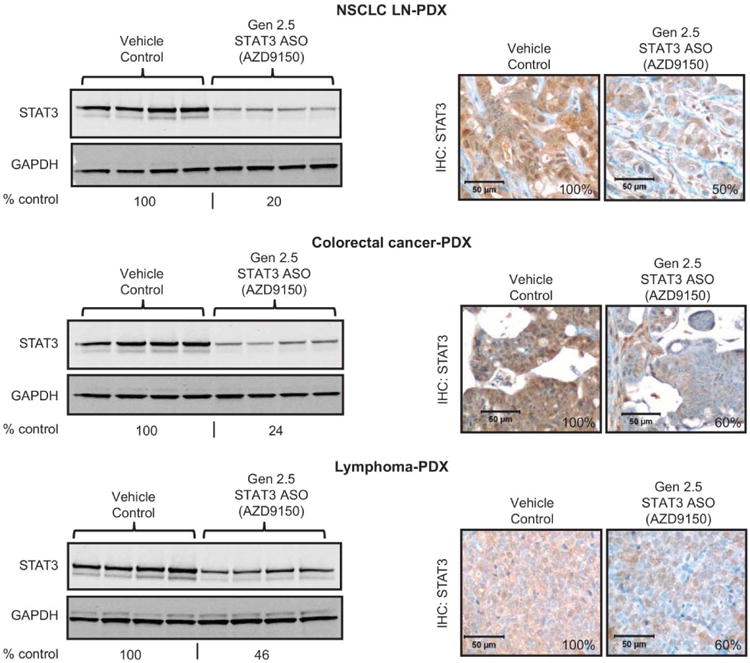
Mice transplanted with human patient-derived primary explants (PDX) from NSCLC metastasized to lymph node (NSCLC-LN), colorectal cancer, or lymphoma were treated with cEt (Gen 2.5) STAT3 ASO at 50 mg/kg, daily, 5 days a week for 3 weeks, then tumors were harvested and analyzed for STAT3 protein by immunoblot (left panel) and IHC (right panel) (n = 4 mice per group). Amounts of STAT3 protein on the immunoblots and the IHC were quantified and expressed as % control. Scale bars, 50 μm.
Quantitative measures of drug concentrations for Gen 2.0 STAT3 ASO and cEt ASO in tumor were 98.9 μg/g vs. 59.8 μg/g of tumor tissue, respectively (table S1) and were consistent with the respective doses evaluated, indicating that the distribution of ASOs in tumor is similar for both ASO chemistries and that the intrinsically higher cellular potency of cEt ASOs rather than pharmacokinetic differences drives the efficacy gain observed in vivo. Tissue concentrations of ASO in preclinical models have been used to predict efficacious dose levels in man (29). The tumor concentrations of AZD9150 achieved at doses associated with strong STAT3 depletion in tumors (25 mg/kg and 50 mg/kg) suggest that the corresponding doses in man would be expected to be in the range of 5-10 mg/kg per week, doses that are well within the range of what has been achieved previously with ASOs in man (10).
Together these results demonstrate that systemically delivered cEt ASOs formulated in simple saline solutions, and administered at well-tolerated and clinically meaningful doses, exhibit good in vivo pharmacological activity against a difficult to drug target in tumors and extra-hepatic tissues such as skeletal muscle (Fig. 3D).
Antitumor activity of unformulated, systemically administered AZD9150 in tumor models in vivo
To further explore the therapeutic potential of AZD9150 itself, we evaluated the in vivo antitumor efficacy of AZD9150 in preclinical cancer models. Because of the high sensitivity of lymphoma cells to STAT3 depletion in vitro, we first evaluated the in vivo anti-tumor activity of AZD9150 in several lymphoma models, including subcutaneous cell line-derived xenografts, patient-derived primary explants, and a systemically disseminated model of lymphoma. SUP-M2 cells showed high sensitivity to STAT3 depletion for survival in vitro and therefore, we chose to evaluate the activity of AZD9150 in SUP-M2 tumors in vivo. Consistent with the in vitro findings, SUP-M2 xenograft tumors in vivo were sensitive to AZD9150, and tumor growth was significantly inhibited (P = 0.0018; 62% Tumor Growth Inhibition (TGI) vs control ASO treated group, Fig. 5A, left panel) without any associated change in body weight (fig. S12). The extent of STAT3 knock down by AZD9150 was lower in SUP-M2 tumors than that seen in A431 tumors, ∼40% vs ∼ 90% (Fig. 5A, right panel and Fig 2A). This was anticipated because of the lower ASO free uptake ability of SUP-M2 cells (IC50 = 0.09 μmol/L; fig. S7D) vs A431 (IC50 = 0.011 μmol/L; Fig. 1A) seen in vitro. However, the in vivo dependence of SUP-M2 tumors on STAT3 for growth and survival was similar to SUP-M2 cells in vitro, where ∼40% knockdown of STAT3 mRNA also resulted in growth inhibition (fig. S7D). To extend the evaluation of AZD9150 in lymphoma to a more disease-relevant setting in vivo, we next established a model of systemically disseminated lymphoma, where SUP-M2 cells injected into immune-compromised mice through the tail vein formed tumors in axillary lymph nodes and in the peritoneal cavity. AZD9150 treatment reduced STAT3 mRNA in tumors isolated from both lymph nodes and the peritoneum (∼50% reduction at each site compared to vehicle or control ASO treated groups, Fig. 5B, left panel). In this model, overall systemic disease burden could also be monitored indirectly by sIL-2Rα or sCD30 concentrations in the plasma of tumor bearing animals (30). As expected, both sIL-2Rα and sCD30 concentrations were significantly reduced by AZD9150 treatment, indicating the systemic anti-lymphoma activity of AZD9150 treatment (P = 0.0023 for sIL-2Rα and P = 0.0043 for sCD30, respectively; Fig. 5B, middle panel). The anti-lymphoma activity of AZD9150 was further confirmed by the direct evaluation of tumor weights (P = 0.001; Fig. 5B, right panel) and reduced ascites at the end of the experiment (fig. S13). We further extended our lymphoma studies of cell line-derived models to a primary PDX model of human lymphoma. As demonstrated histologically (Fig. 4), the lymphoma PDX model contained a marked mouse stromal component. To better reflect the cumulative activity of STAT3 inhibition in lymphoma tumor cells and in tumor-associated stromal cells, we tested the combination of the human-selective AZD9150 and the mouse active cEt STAT3 ASO characterized in Fig. 3. The combined inhibition of STAT3 with AZD9150 and the mouse active STAT3 ASO produced significant antitumor activity in this lymphoma PDX model (TGI = 67%, P = 0.001 compared to the control ASO treatment; Fig. 5C) with no associated changes in body weight (fig. S14). Species-specific PCR analysis confirmed that the combined treatment inhibited both human STAT3 in the tumor cells and murine STAT3 in tumor-associated mouse cells (Fig. 5D, left panel), and IHC analysis confirmed the inhibition of pSTAT3 protein in both human tumor and tumor-associated mouse stromal cells (Fig. 5D, right panel).
Fig. 5. Antitumor activity of AZD9150 in tumor models.
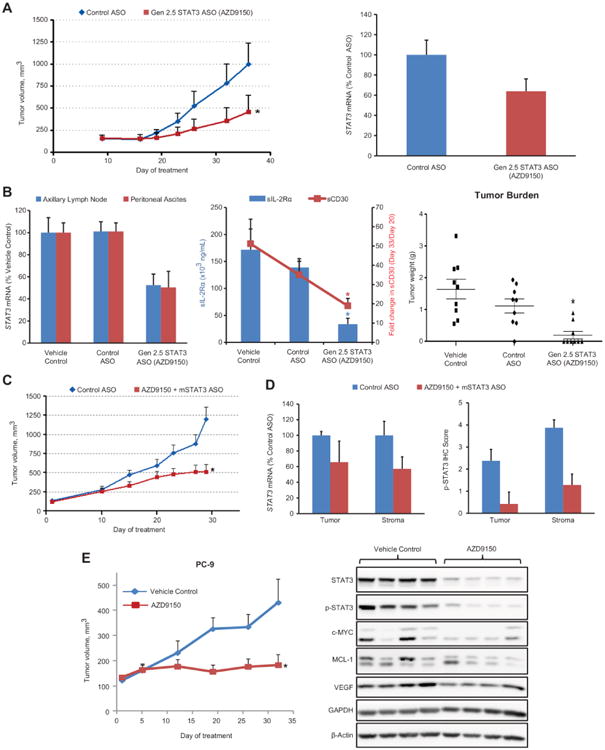
(A) Animals bearing SUP-M2 cells in the left flank region were treated with cEt (Gen 2.5) STAT3 ASO or control ASO at 50 mg/kg, 5 times per week for 5 weeks. cEt STAT3 ASO (AZD9150) significantly inhibited tumor growth compared to the control ASO (TGI = 62%) (*P = 0.0018) (n=16 mice per group) (left panel) even with modest STAT3 downregulation in the tumor (right panel). (B) SUP-M2 cells were intravenously injected into NSG mice. Twenty days later, animals were randomized on the basis of soluble CD30 (sCD30) concentration and treated with either control ASO or cEt STAT3 ASO at 50 mg/kg, 5 times per week for 2 weeks. Reductions in STAT3 mRNA in axillary lymph nodes and peritoneal ascites of the STAT3 ASO-treated group (left panel), in plasma sIL-2Rα (*P = 0.0023) and sCD30 (*P = 0.0043) concentrations (middle panel), and in tumor burden (*P = 0.001) (n = 9 mice per group) (right panel) were assessed. (C-D) Mice transplanted with human patient-derived primary DLBCL tumors in the flank region were treated with either control ASO or AZD9150 plus mouse STAT3-targeting ASO at 50 mg/kg, 5 times per week for 4 weeks. Tumor growth was significantly inhibited in the STAT3 ASO-treated group compared to the control ASO group (TGI = 67%) (*P = 0.001) (n = 7-8 mice per group) (C). STAT3 mRNA knockdown in tumor and stroma cells was demonstrated in STAT3 ASO-treated group by qRT-PCR using species-specific probe/primer sets (D, left panel). A decrease in phospho-STAT3 protein in both tumor and stromal cells with STAT3 ASO treatment was also quantified from IHC (D, right panel). (E) Animals bearing human NSCLC PC-9 cells in the flank region were treated with AZD9150 alone at 25 mg/kg, 5 times per week for about 5 weeks. Tumor growth was significantly delayed with AZD9150 treatment compared to control (left panel) (TGI = 90%) (*P = 0.027) (n = 7 mice per group), which also correlated with marked reductions in both total STAT3 and phospho-STAT3 protein as well as STAT3 targets (right panel). Graphs in A to E show the means ± SEM.
In addition to the potential of STAT3 as a target in lymphoma, it has also been proposed as a therapeutic target in various solid tumors, including lung cancer (31, 32). However, as mentioned above, neither cEt STAT3 ASOs nor the JAK/STAT inhibitor AZD1480 inhibit the proliferation of NSCLC cell lines growing in culture (fig. S8). In contrast, AZD1480 has shown marked antitumor activity in vivo when these same cancer lines were grown as tumor xenografts (24, 33). This suggests that in vivo, these tumor cells acquire dependence on STAT3 for growth. To determine whether AZD9150 behaves similarly to the small molecule STAT3 pathway inhibitor, AZD1480, in vivo, we tested it in the PC-9 NSCLC subcutaneous tumor xenograft model that has previously been shown to be sensitive to JAK/STAT inhibition in vivo but not in vitro (33). Consistent with previous findings with JAK inhibitor AZD1480, systemic treatment of mice bearing PC-9 tumors with AZD9150 produced near complete inhibition of tumor growth in vivo (TGI = 90%, P = 0.027; Fig. 5E, left panel). AZD9150 treatment of PC-9 tumors in vivo resulted in the inhibition of expression of the STAT3 downstream target genes c-MYC, MCL-1, and VEGF (Fig. 5E, right panel). Nevertheless, strong depletion of STAT3 in PC-9 cells growing in vitro did not alter either the proliferation of these cells or the amount of the survival protein MCL-1 (fig. S8E), suggesting that when grown in vivo, PC-9 tumor cells become dependent on STAT3 for growth and that selective inhibition of STAT3 has activity in NSCLC tumors in vivo.
Taken together, the data from mouse models demonstrated that systemically delivered cEt ASOs can be used to inhibit expression of difficult-to-drug cancer driver pathways, and also supported the further development of AZD9150. AZD9150 was evaluated in Investigational New Drug (IND) enabling toxicology studies in both mice and non-human primates and was found to be well tolerated (34). In both mice and non-human primates, up to 90% inhibition of STAT3 protein was observed after systemic treatment with ASO formulated in saline (34).
Toxicological findings were consistent with effects typically observed with ASOs (10), and the class effects of cEt chemistry were observed at similar doses to Gen 2.0 chemistry, suggesting that cEt ASOs may similarly be well-tolerated in man up to doses of 10 mg/kg.
Single-agent antitumor activity of AZD9150 in treatment-refractory cancer patients
Because of the antitumor activity seen in lymphoma models and the strong linkage of STAT3 pathway activation with this disease and other tumor types (35), both advanced lymphoma and solid tumor patients were included in the phase I dose-escalation study of AZD9150. Of the 25 patients enrolled, 12 were advanced lymphoma patients; of these, seven had been diagnosed with diffuse large B-cell lymphoma (DLBCL), two each with Hodgkin's lymphoma and follicular non-Hodgkin's lymphoma, and one with mantle cell lymphoma. For patient eligibility selection and inclusion criteria see Materials and Methods; for patient demographics see table S2. The initial two patients were treated with AZD9150 starting at 2 mg/kg of ideal body weight per week by intravenous infusion, with a loading dose of three infusions the first week followed by once weekly infusions until unacceptable toxicity or disease progression occurred. In successive cohorts, 12 patients were treated with 3 mg/kg and nine with 4 mg/kg doses of AZD9150.
Pharmacokinetic studies demonstrated that AZD9150 behaved similarly to previously described ASO molecules (36) (see full pharmacokinetic data in fig. S15). The maximum tolerated dose (MTD) of AZD9150 was determined to be 3 mg/kg. A rapidly evolving thrombocytopenia (in the first month of dosing) was observed in two of nine patients at 4 mg/kg and was considered the dose-limiting toxicity. A more chronic slowly progressing thrombocytopenia occurs after 4 to 6 months of dosing at 2 and 3 mg/kg (and for most patients at 4 mg/kg) and was effectively managed with pauses and dose frequency adjustments. Figure S16 demonstrates the chronic slowly progressing thrombocytopenia observed at all doses. The drug-related adverse events that occurred in >5% of patients are listed in table S3. The drug-related adverse events that occurred most commonly in patients dosed with AZD9150 were AST elevation (44%), ALT elevation (44%), and thrombocytopenia (40%). One dose-limiting toxicity (DLT) event occurred in a patient (DLT#1) who received the highest dose tested (4 mg/kg). After four doses of AZD9150, this patient was hospitalized for generalized weakness in the setting of a rapid fall in platelet counts, which was unlike the slow platelet reductions seen at 2 and 3 mg/kg. The kinetics of this thrombocytopenia is consistent with thrombotic microangiopathy that is known to occur with increased frequency in patients with advanced cancers, however, due to the temporal relationship to the start of dosing, the case was considered possibly related to study drug. This patient's condition deteriorated rapidly and she expired after 9 days of hospitalization. A second DLT event (DLT#2), also possibly treatment-related thrombocytopenia, occurred after two 4 mg/kg doses of AZD9150. In this patient, platelet counts also dropped quickly, however, they recovered to normal after discontinuation of study drug. The slowly progressing thrombocytopenia seen in patients at or below the MTD is consistent with the reported role of STAT3 in megakaryopoeisis (37, 38), whereas the rapidly progressing thrombocytopenia seen above the MTD in the two patients with DLTs described above (DLT#1 and DLT #2) had an unknown etiology. Forty-four percent of all patients (n=11/25) achieved stable disease (SD) or a partial response (PR) and encouragingly, three of six patients (50%) with treatment-refractory DLBCL had evidence of tumor shrinkage and two patients (33%) achieved a confirmed durable PR (Fig. 6A). The one follicular non-Hodgkin's lymphoma patient achieved SD as defined by International Working Group Criteria (IWGC). One of the DLBCL patients (patient 1001) with PR was treated with AZD9150 at 2 mg/kg. Patient 1001 was a 63-year-old female originally diagnosed with follicular lymphoma in 2007, which transformed to a large B-cell lymphoma in 2010. The patient had relapsed disease or was refractory to 9 prior chemotherapy-containing regimens (table S4). After 7 weeks of treatment with AZD9150, patient 1001 had a PR with an overall decrease in tumor burden of 50% when assessed by fluorodeoxyglucose positron emission tomography (FDG-PET) scan (Fig. 6B). This patient remained on treatment with a very durable partial response for a year and 3 months after treatment initiation.
Fig. 6. Clinical antitumor activity of AZD9150.
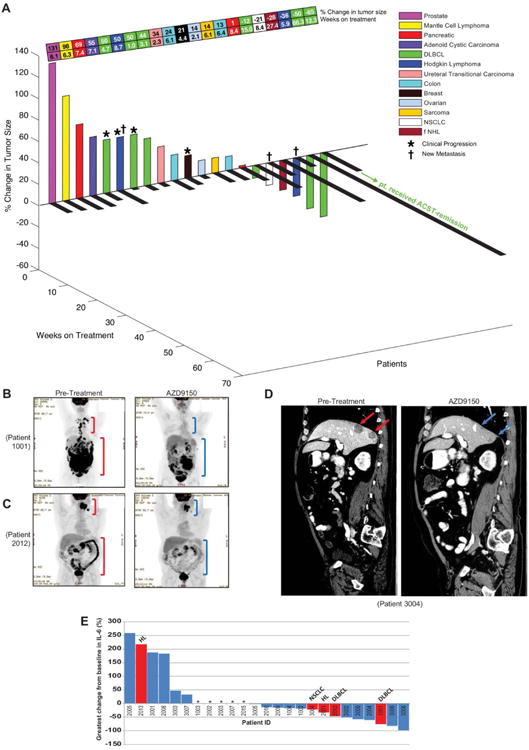
(A) 3D waterfall plot of change from baseline of tumor size with time on treatment and tumor type for all patients with pre- and post-dose tumor measurements. ASCT; autologous stem-cell transplantation. (B) Positron emission tomography (PET) scan of DLBCL patient 1001 at baseline (before AZD9150 treatment, left panel) and after 7 weeks of treatment with AZD9150 (2 mg/kg per week, right panel). (C) PET scan for DLBCL patient 2012 at baseline and 7 weeks after AZD9150 (4 mg/kg per week). (D) PET scans for NSCLC patient 3004 demonstrating reduction in 2 liver lesions approximately 8 weeks after AZD9150 (2 mg/kg on study day 1, 3, and 5 and then weekly from study day 8) treatment. (E) Waterfall plot of greatest IL-6 change from baseline for all 25 patients. Plasma IL-6 concentrations in patients 1001 and 2012 (DLBCL), 2001 and 2013 (HL), and 3004 (NSCLC) are marked in red. In B to D, red brackets or arrows indicate regions of highly metabolically active tumors, and blue brackets or arrows indicate regions where tumors have regressed. Asterisks in E indicate the patients who came off the treatment prior to the first restaging FDG-PET scan at 2 months after treatment initiation.
Patient 2012, a 53-year-old male diagnosed with advanced DLBCL in 2012, received 4 mg/kg of AZD9150. This patient was refractory to the front-line therapies R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone) and R-ICE (rituximab, ifosfamide, carboplatin, and etoposide) (table S4). As with patient 1001, patient 2012 had a strong partial response (65% reduction in target lesion) after 7 weeks of AZD9150 treatment. There were marked reductions in both mesenteric and retroperitoneal lesions (Fig. 6C) and in an oropharyngeal mass that was causing dysphagia in this patient (fig. S17A). He maintained a PR for approximately 4 months, and with his improved and stabilized disease he became a candidate for an autologous stem cell transplantation, which he received. He currently remains in complete metabolic remission, with no lesions having metabolic activity above background on follow up FDG-PET scans.
Two patients with refractory Hodgkin's lymphoma also had evidence of antitumor responses after treatment with AZD9150 where both patients experienced marked reductions in target lesions at the end of 7 weeks of treatment (fig. S17B) but were reported as mixed responders due to the appearance of new lesions and came off study.
Finally, our preclinical data suggested that NSCLC tumors may respond to AZD9150. Intriguingly, the only NSCLC patient evaluated showed evidence of near complete resolution of highly treatmentrefractory NSCLC liver metastasis upon first restaging, with additional stabilization of mediastinal lymph nodes in response to 3 mg/kg of AZD9150 treatment (Fig. 6D). Given the occurrence of new lesions on second restaging by RECIST 1.1, the patient was taken off protocol. A summary of all tumor responses and effects on disease stabilization for the 21 patients for whom baseline and restaging scans were performed is shown schematically in Fig. 6A.
Elevated circulating concentrations of IL-6 have been associated with poor prognosis in several cancers including DLBCL (39, 40). IL-6 receptor signals through STAT3 increase the transcription and production of IL-6, resulting in a feed forward autocrine loop (41). Thus, circulating IL-6 could potential serve as an early biomarker of STAT3 pathway inhibition. Eight patients had post-treatment reductions in serum concentrations of IL-6 greater than 30% (Fig. 6E). Three of the four responding lymphoma patients were among those with >30% decreases in IL-6 (Fig. 6E). Moreover, we did not observe increases in circulating IL-6 in patients with confirmed PRs, and no patients reported constitutional symptoms such as fever or chills that would suggest a proinflammatory activity for AZD9150, as has been reported previously with certain ASOs at higher doses (36, 42).
Discussion
The findings reported here highlight antisense technology as it applies to cancer drug discovery and treatment. We have identified and characterized the in vitro and in vivo activity of optimized next-generation (cEt) ASO AZD9150 targeted to the transcription factor STAT3.
Additional recent clinical approaches to target STAT3 directly include the evaluation of STAT3 DNA binding site decoys (43, 44) and small molecule agents that disrupt STAT3 SH2 dimerization domains (45, 46), however, these used employed intra-tumoral injection of drug or were associated with notable toxicities, respectively. Targeting the upstream JAK receptor tyrosine kinase has proven a more feasible means of indirectly targeting the STAT pathway (26, 33), however, multiple JAK-independent mechanisms causing STAT3 activation have been identified (47-53), and thus direct STAT3 inhibition remains an attractive cancer strategy.
In cancer cell lines in culture, AZD9150, the cEt STAT3 ASO, had low-nanomolar activity in the absence of lipid-mediated permeability enhancers. Our preclinical characterization of AZD9150 supports the findings with an earlier generation of STAT3 ASOs that lymphoma tumor may be particularly sensitive to STAT3 inhibition (54) and extends this to demonstrate that NSCLC tumors in vivo can be sensitive to STAT3 inhibition. Of principal therapeutic importance, we demonstrated that systemically administered, cEt STAT3 ASOs formulated in simple saline solutions were more potent than previous generation ASOs and potently reduced STAT3 mRNA and protein in tumors, tumor-associated stromal cells, and multiple normal tissues in mice. These preclinical findings translated into the clinical setting, where we observed encouraging clinical efficacy in patients with advanced highly treatment-refractory lymphomas and NSCLC in the phase I dose escalation study. Consistent with the increased potency of cEt ASOs demonstrated in preclinical models, the clinical efficacy of AZD9150 was achieved at doses that are 3-7 fold lower than those of Gen 2.0 ASOs evaluated in cancer patients previously (55-57).
Although our initial clinical data suggest that AZD9150 has potential as a cancer therapeutic, it will be important to reassess this early activity in phase II and III clinical trials and, if possible, understand why certain lymphoma patients (or patients with other tumor types) respond to AZD9150 but others do not. In the current study, baseline and on-treatment tumor biopsies were not available for most patients. In future studies, it will be important to include pre- and post-treatment biopsies to identify the cell type specificity and the degree of STAT3 inhibition associated with clinical efficacy and to determine whether a particular genetic or gene expression signature predicts which patients will respond or be resistant to AZD9150. The biopsies will also help clarify the contribution of STAT3 knockdown in the tumor cell vs. tumor stromal compartment. Preclinical modeling predicted that doses between 5 and 10 mg/kg/week in man would be required for robust STAT3 depletion in tumor cells, however, AZD9150 was clinically active at doses as low as 2 mg/kg. It is possible that limited inhibition of STAT3 produced the observed antitumor effects, as was the case in lymphoma cells in culture.
The preclinical antitumor activity reported here for the human selective ASO AZD9150, in immunocompromised mice, clearly demonstrates that inhibition of STAT3 in tumor cells themselves can result in antitumor activity. However, STAT3 is reported to have both tumor cell-autonomous and non-autonomous functions, including well-described roles in the cross-talk between tumor cells and immune cells of the tumor microenvironment (31-33), and thus it is possible that these activities may contribute to the clinical efficacy seen with AZD9150. Thus, dissecting the roles STAT3 plays in all cell types of the tumor microenvironment in both immunocompromised and immunocompetent mouse models of cancer will be important questions for future studies. The human- and mouse-selective STAT3 inhibitors described here should serve as valuable tools for these efforts.
AZD9150 treatment was both well-tolerated and efficacious at the 2 and 3 mg/kg doses. However, dose escalation with AZD9150 above 4 mg/kg was not possible because of effects on platelets. This toxicity is likely an on-target effect mediated by STAT3 depletion, because it is consistent with effects seen in mice with genetic STAT3 deficiency in megakaryocyte progenitor cells (37). Future cEt ASOs directed to additional cancer pathways, which might not be similarly limited by on-target toxicities, should be able to achieve much higher doses in man and consequently produce even stronger effects. This is supported by the pre-clinical toxicology indicating that non-specific toxic effects of cEt ASOs are similar to those of Gen 2.0 ASOs that have achieved 10-12 mg/kg dose levels in patients (14).
In addition, the toxicities associated with AZD9150 treatment must be characterized in larger numbers of patients and must also include the careful evaluation of susceptibility to pathogenic infections, because inactivating mutations in STAT3 have recently been identified as the underlying genetic event in Hyperimmunoglobulin E syndrome (HIES), characterized by high plasma IgE concentrations and recurrent staphylococcal infections of the lung and skin as well as chronic mucocutaneous candidiasis (58).
In conclusion, the cEt ASO AZD9150, containing a constrained ethyl modification, decreased STAT3 expression in a broad range of preclinical cancer models and had antitumor activity in lymphoma and lung cancer models. Antitumor activity of AZD9150 was also demonstrated in patients with highly treatment-refractory lymphoma and non-small cell lung cancer in a phase I dose escalation study. These findings indicate that this antisense chemistry can provide a rational means to translate cancer genomic information into therapeutic molecules capable of targeting previously intractable cancer drivers.
Materials and Methods
These are provided in the supplementary materials.
Supplementary Material
Fig. S1. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in tumor cells in vitro determined by qRT-PCR.
Fig. S2. Loss of sensitivity of primary cells to ASO over time when plated in culture.
Fig. S3. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in tumor cells in vitro determined by immunoblot.
Fig. S4. Superior activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) over Gen 2.0 STAT3 MOE ASO.
Fig. S5. Lack of activity of STAT3 siRNAs when delivered to tumor cells by free uptake.
Fig. S6. A decrease in the expression of STAT3-target genes after downregulation of STAT3 by STAT3 ASOs in ALCL, KARPAS299 cells.
Fig. S7. Sensitivity of ALCL cell lines, KARPAS299 and SUP-M2, to STAT3 inhibition.
Fig. S8. Effect of STAT3 inhibition on cell proliferation in a variety of cell types in vitro.
Fig. S9. IHCs of ASOs and STAT3 in A431 xenograft tumors.
Fig. S10. Effect of AZD9150 on the growth of A431 xenograft tumors.
Fig. S11. Selectivity of AZD9150 for STAT3 over STAT1 or STAT5 in lymphoma patient-derived xenografts (PDX).
Fig. S12. Toxicity testing of AZD9150 in animals bearing SUP-M2 tumors.
Fig. S13. Antitumor activity of cEt STAT3 ASO (AZD9150) in SUP-M2 dissemination model.
Fig. S14. Toxicity testing of AZD9150 in animals bearing patient-derived lymphoma xenografts.
Fig. S15. Pharmacokinetic data for AZD9150.
Fig. S16. Changes in platelet count.
Fig. S17. Clinical antitumor activity of AZD9150.
Table S1. Quantification of ASO accumulation in A431 tumor.
Table S2. AZD9150 phase I patient demographics.
Table S3. AEs greater than 5% for all dosed.
Table S4. Prior treatment regimens of DLBCL patients, 1001 and 2012.
Table S5. List of primer and probe sequences used for qRT-PCR.
Table S6. Changes in tumor volume during the course of ASO treatment.
Summary.
This work describes the activity in multiple preclinical cancer models of the unformulated, systemically administered novel chemistry (cEt) antisense oligonucleotide inhibitor AZD9150 an inhibitor of the previously undruggable transcription factor STAT3 and the translation of these preclinical findings to phase 1 clinical studies where AZD9150 has demonstrated single agent antitumor activity in several highly treatment refractory cancer patients.
Acknowledgments
We thank Nancy Zhang, Chris May, and Kirsten Bell for help with tumor studies; Minwei Ye for conducting western blot analysis; Gene Hung and Yuhong Zhang for performing IHC studies; Sarah Greenlee for PK analysis; Maheen David, Tracy Reigle, Lauren Elder, and Betsy Stephen for preparing graphics and formatting of the final manuscript; Ron Baldwin for coordination of clinical operations.
Funding: The funding for this study was provided by Isis Pharmaceuticals Inc.
Footnotes
Author contributions: Y.K., R.W., D.C.B., and A.R.M. designed preclinical experiments; Y.K., R.W., J.S., M.J., S.J., M.VP N., D.L., C.R., X.X., and J.H., and A.R. performed preclinical experiments and analyzed data; D.H., R.K., J.M., N.F., M.Y., S.H., L.F., and S.P-P. performed clinical studies; the manuscript was written by D.H., R.K.,Y.K., B.P.M., and A.R.M and edited by all authors.
Competing interests: Y.K., T.Z., J.S., M.J., S.J., M.Y., S.H., X.X., J.H., A.R., B.P.M., and A.R.M are employees of Isis Pharmaceuticals Inc. and R.W., M.VP N., D.L., C.R., and D.C.B. are employees of AstraZeneca Pharmaceuticals. The other authors declare that they have no competing interests.
References and Notes
- 1.Chin L, Gray JW. Translating insights from the cancer genome into clinical practice. Nature. 2008 Apr 3;452:553. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim H, et al. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proceedings of the National Academy of Sciences of the United States of America. 2010 Feb 2;107:2183. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Adelstein SJ, Kassis AI. Integrated bioinformatics analysis for cancer target identification. Methods Mol Biol. 2011;719:527. doi: 10.1007/978-1-61779-027-0_25. [DOI] [PubMed] [Google Scholar]
- 4.Weinstein IB, Joe A. Oncogene addiction. Cancer research. 2008 May 1;68:3077. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
- 5.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO molecular medicine. 2011 Nov;3:623. doi: 10.1002/emmm.201100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004 Mar 1;22:785. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 7.Quintas-Cardama A, Kantarjian H, Cortes J. Imatinib and beyond--exploring the full potential of targeted therapy for CML. Nature reviews Clinical oncology. 2009 Sep;6:535. doi: 10.1038/nrclinonc.2009.112. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AT, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. The lancet oncology. 2011 Oct;12:1004. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raal FJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010 Mar 20;375:998. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 10.Saad F, et al. Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011 Sep 1;17:5765. doi: 10.1158/1078-0432.CCR-11-0859. [DOI] [PubMed] [Google Scholar]
- 11.Wheeler TM, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012 Aug 2;488:111. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buller HR, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. The New England journal of medicine. 2015 Jan 15;372:232. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaudet D, et al. Targeting APOC3 in the familial chylomicronemia syndrome. The New England journal of medicine. 2014 Dec 4;371:2200. doi: 10.1056/NEJMoa1400284. [DOI] [PubMed] [Google Scholar]
- 14.Seth PP, et al. Short antisense oligonucleotides with novel 2′-4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. Journal of medicinal chemistry. 2009 Jan 8;52:10. doi: 10.1021/jm801294h. [DOI] [PubMed] [Google Scholar]
- 15.Murray S, et al. TricycloDNA-modified oligo-2′-deoxyribonucleotides reduce scavenger receptor B1 mRNA in hepatic and extra-hepatic tissues--a comparative study of oligonucleotide length, design and chemistry. Nucleic acids research. 2012 Jul;40:6135. doi: 10.1093/nar/gks273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furqan M, et al. STAT inhibitors for cancer therapy. Journal of hematology & oncology. 2013;6:90. doi: 10.1186/1756-8722-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostergaard ME, et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic acids research. 2013 Nov;41:9634. doi: 10.1093/nar/gkt725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monia BP, Johnston JF, Geiger T, Muller M, Fabbro D. Antitumor activity of a phosphorothioate antisense oligodeoxynucleotide targeted against C-raf kinase. Nature medicine. 1996 Jun;2:668. doi: 10.1038/nm0696-668. [DOI] [PubMed] [Google Scholar]
- 19.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. The Journal of clinical investigation. 2007 Dec;117:3623. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koller E, et al. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic acids research. 2011 Jun;39:4795. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein CA, et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic acids research. 2010 Jan;38:e3. doi: 10.1093/nar/gkp841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozema DB, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 2007 Aug 7;104:12982. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuckerman JE, Choi CH, Han H, Davis ME. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proceedings of the National Academy of Sciences of the United States of America. 2012 Feb 21;109:3137. doi: 10.1073/pnas.1200718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedvat M, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer cell. 2009 Dec 8;16:487. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zamo A, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002 Feb 7;21:1038. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 26.Scuto A, et al. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer research. 2011 May 1;71:3182. doi: 10.1158/0008-5472.CAN-10-2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor CR, Levenson RM. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006 Oct;49:411. doi: 10.1111/j.1365-2559.2006.02513.x. [DOI] [PubMed] [Google Scholar]
- 28.Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PloS one. 2014;9:e96801. doi: 10.1371/journal.pone.0096801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu RZ, et al. Predictive dose-based estimation of systemic exposure multiples in mouse and monkey relative to human for antisense oligonucleotides with 2′-o-(2-methoxyethyl) modifications. Molecular therapy Nucleic acids. 2015;4:e218. doi: 10.1038/mtna.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang M, et al. Effective therapy for a murine model of human anaplastic large-cell lymphoma with the anti-CD30 monoclonal antibody, HeFi-1, does not require activating Fc receptors. Blood. 2006 Jul 15;108:705. doi: 10.1182/blood-2005-11-4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song L, Rawal B, Nemeth JA, Haura EB. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Molecular cancer therapeutics. 2011 Mar;10:481. doi: 10.1158/1535-7163.MCT-10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Looyenga BD, et al. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PloS one. 2012;7:e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami T, et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer. 2014 Jan;83:30. doi: 10.1016/j.lungcan.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 34.Burel SA, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic acid therapeutics. 2013 Jun;23:213. doi: 10.1089/nat.2013.0422. [DOI] [PubMed] [Google Scholar]
- 35.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011 Feb 3;470:115. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talbot DC, et al. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010 Dec 15;16:6150. doi: 10.1158/1078-0432.CCR-10-1932. [DOI] [PubMed] [Google Scholar]
- 37.Kirito K, et al. A functional role of Stat3 in in vivo megakaryopoiesis. Blood. 2002 May 1;99:3220. doi: 10.1182/blood.v99.9.3220. [DOI] [PubMed] [Google Scholar]
- 38.Mantel C, et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012 Sep 27;120:2589. doi: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visconti L, et al. Prognostic value of circulating cytokines on overall survival and disease-free survival in cancer patients. Biomarkers in medicine. 2014 Feb;8:297. doi: 10.2217/bmm.13.122. [DOI] [PubMed] [Google Scholar]
- 40.Seymour JF, Talpaz M, Cabanillas F, Wetzler M, Kurzrock R. Serum interleukin-6 levels correlate with prognosis in diffuse large-cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1995 Mar;13:575. doi: 10.1200/JCO.1995.13.3.575. [DOI] [PubMed] [Google Scholar]
- 41.Faruqi TR, Gomez D, Bustelo XR, Bar-Sagi D, Reich NC. Rac1 mediates STAT3 activation by autocrine IL-6. Proceedings of the National Academy of Sciences of the United States of America. 2001 Jul 31;98:9014. doi: 10.1073/pnas.161281298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chi KN, et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010 Sep 20;28:4247. doi: 10.1200/JCO.2009.26.8771. [DOI] [PubMed] [Google Scholar]
- 43.Sen M, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer discovery. 2012 Aug;2:694. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sen M, et al. Systemic administration of a cyclic signal transducer and activator of transcription 3 (STAT3) decoy oligonucleotide inhibits tumor growth without inducing toxicological effects. Molecular medicine. 2014;20:46. doi: 10.2119/molmed.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bendell JC, et al. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer chemotherapy and pharmacology. 2014 Jul;74:125. doi: 10.1007/s00280-014-2480-2. [DOI] [PubMed] [Google Scholar]
- 46.Wong AL, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2015 May;26:998. doi: 10.1093/annonc/mdv026. [DOI] [PubMed] [Google Scholar]
- 47.Yu CL, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995 Jul 7;269:81. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 48.DeArmond D, et al. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene. 2003 Oct 30;22:7781. doi: 10.1038/sj.onc.1206966. [DOI] [PubMed] [Google Scholar]
- 49.Yoshikawa H, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nature genetics. 2001 May;28:29. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 50.Brantley EC, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008 Aug 1;14:4694. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koppikar P, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012 Sep 6;489:155. doi: 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gough DJ, et al. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009 Jun 26;324:1713. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee HJ, et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer cell. 2014 Aug 11;26:207. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Chiarle R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nature medicine. 2005 Jun;11:623. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 55.Hong DS, et al. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011 Oct 15;17:6582. doi: 10.1158/1078-0432.CCR-11-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villalona-Calero MA, et al. A phase I/II study of LY900003, an antisense inhibitor of protein kinase C-alpha, in combination with cisplatin and gemcitabine in patients with advanced non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004 Sep 15;10:6086. doi: 10.1158/1078-0432.CCR-04-0779. [DOI] [PubMed] [Google Scholar]
- 57.Saad F, et al. Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011 Sep 1;17:5765. doi: 10.1158/1078-0432.CCR-11-0859. [DOI] [PubMed] [Google Scholar]
- 58.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007 Aug 30;448:1058. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in tumor cells in vitro determined by qRT-PCR.
Fig. S2. Loss of sensitivity of primary cells to ASO over time when plated in culture.
Fig. S3. Activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) and Gen 2.0 STAT3 ASO in tumor cells in vitro determined by immunoblot.
Fig. S4. Superior activity of cEt (Gen 2.5) STAT3 ASO (AZD9150) over Gen 2.0 STAT3 MOE ASO.
Fig. S5. Lack of activity of STAT3 siRNAs when delivered to tumor cells by free uptake.
Fig. S6. A decrease in the expression of STAT3-target genes after downregulation of STAT3 by STAT3 ASOs in ALCL, KARPAS299 cells.
Fig. S7. Sensitivity of ALCL cell lines, KARPAS299 and SUP-M2, to STAT3 inhibition.
Fig. S8. Effect of STAT3 inhibition on cell proliferation in a variety of cell types in vitro.
Fig. S9. IHCs of ASOs and STAT3 in A431 xenograft tumors.
Fig. S10. Effect of AZD9150 on the growth of A431 xenograft tumors.
Fig. S11. Selectivity of AZD9150 for STAT3 over STAT1 or STAT5 in lymphoma patient-derived xenografts (PDX).
Fig. S12. Toxicity testing of AZD9150 in animals bearing SUP-M2 tumors.
Fig. S13. Antitumor activity of cEt STAT3 ASO (AZD9150) in SUP-M2 dissemination model.
Fig. S14. Toxicity testing of AZD9150 in animals bearing patient-derived lymphoma xenografts.
Fig. S15. Pharmacokinetic data for AZD9150.
Fig. S16. Changes in platelet count.
Fig. S17. Clinical antitumor activity of AZD9150.
Table S1. Quantification of ASO accumulation in A431 tumor.
Table S2. AZD9150 phase I patient demographics.
Table S3. AEs greater than 5% for all dosed.
Table S4. Prior treatment regimens of DLBCL patients, 1001 and 2012.
Table S5. List of primer and probe sequences used for qRT-PCR.
Table S6. Changes in tumor volume during the course of ASO treatment.