

Abstract
Erdheim–Chester disease (ECD) is a rare histiocytic disorder, characterized by the xanthomatous infiltration of tissues by CD68-positive and CD1a-/CD100-negative foamy histiocytes. In childhood, ECD is exceptionally rare, and only a dozen cases have been published so far. The cooccurence of Langerhans cell histiocytosis (LCH) and ECD is even rarer. Here, we report a 2-year-old boy, the youngest patient in the literature so far, who was diagnosed with concomitant BRAF mutation-positive LCH and ECD. In his case, conventional LCH treatment proved to be ineffective, but he is the youngest patient who was successfully treated with the BRAF inhibitor vemurafenib.
Keywords: vemurafenib, treatment, Erdheim–Chester disease, Langerhans cell histiocytosis, pediatric histiocytic disorders
Introduction
ECD is a rare non-Langerhans histiocytic disorder, which typically causes bone lesions, but virtually any organ can be involved, and the presentation symptoms may be also varied, ranging from asymptomatic to multisystem, life-threatening forms. Although, according to the WHO classification, ECD is a neoplasm deriving from CD68-positive and CD1a- and S100-negative histiocytes, the exact pathogenesis has not been fully clarified yet, whether it has a malignant nature or an inflammatory nature.1 In recent years, a growing number of studies that have investigated the underlying mechanisms of ECD have identified an activating point mutation of BRAF V600E in >50% of ECD patients. This has given a basis for encouraging targeted therapies. The BRAF inhibitor vemurafenib has demonstrated activity in small cohorts of adults treated with multisystem and refractory ECD.2,3 Data about the usage and efficacy of vemurafenib in pediatric ECD are lacking. We report an excellent therapy response to vemurafenib in a 2-year-old ECD patient, who is the youngest patient in the literature so far.
Case report
A 20-month-old boy was presented with visible soft masses on the left temporal and right parietal bone after a ~1-year-long history of “seborrheic dermatitis” of the scalp, “diaper rash”, and recurrent episodes of upper airway infections and otitis media. MRI revealed a 5×5×2.5 cm size frontal destructive mass on the right, a 6×3×2 cm size frontotemporoparietal mass, and a 1×1×1 cm size frontal mass on the left side, and all of them caused bone lesions (Figure 1A and B). Based on the patient history, physical findings, and imaging studies, LCH came up as a possible diagnosis with skin and multifocal bone involvement, which was confirmed histologically: the biopsy specimen from one of the skull masses was histologically composed of CD1a- and S100-positive mononuclear cells with coffee bean-shaped nuclei, eosinophil granulocytes, and multinucleated giant cells (Figure 2A and B). Morphology and immunophenotype were typical for LCH. Furthermore, skeletal manifestations and solid organ involvement were excluded by bone scintigraphy and MRI. We started his chemotherapy based on LCH-III protocol’s appropriate arm, according to the risk of the disease. Thanks to the effectivity of the therapy, rapid regression of the skull masses was detectable in the beginning (Figure 1C and D), but in the fourth month of chemotherapy, hepatosplenomegaly, hypalbuminemia, serious pancytopenia occurred, the activity of the left frontal bone lesion came back (Figure 1E and F), and the skin symptoms flared up. Bone marrow biopsy was negative at that time. Thereafter, he required dose reduction of cytostatic drugs, his transfusion need was extreme, and he required platelet support almost daily and blood transfusion regularly as well. He required albumin products for severe hypalbuminemia, desferoxamine treatment for secondary hemosiderosis, hospital care almost continuously for frequent septic episodes, and durable heparin therapy for deep vein thrombosis. Due to the serious symptoms, we were forced to stop cytotoxic therapy and started corticosteroid maintenance therapy with indomethacin supplementation for a short period, which was stopped because of its platelet aggregation inhibitory effect and simultaneous gravis thrombocytopenia. Eight months after the initial diagnosis, splenectomy was performed due to symptoms of hypersplenia, and histology confirmed splenic fibrosis represented by multiple fibrohistiocytic nodules composed of foamy macrophages loaded with hemosiderin accompanied by maturing extramedullary hematopoiesis. Bone marrow biopsy at the same time revealed highly similar pathological findings: excessive (~75%) engagement of marrow by CD68-positive and CD1a- and S100-negative macrophages inducing mild fibrosis. These macrophages had water clear wide cytoplasm and rounded nuclei (Figure 3A and B). A significant number of Langerhans cells were not detected in these bone marrow and spleen samples. On plain radiograph of the painful knees, symmetric osteosclerosis could be seen (Figure 4A). Interestingly, low signal intensities could be seen in all visualized bones (not only in the long bones) on every MRI sequence (Figure 5). These findings raised the diagnosis of non-Langerhans cell ECD with extensive bone marrow and spleen involvement. BRAF V600E mutation positivity was confirmed by pyrosequencing in the initial LCH skull lesion and in the recent ECD bone marrow biopsy specimen as well. According to the diagnosis and the literature, we started IFN-alpha therapy, and then, in default of clinical improvement and after obtaining the result of BRAF V600E mutation positivity of his disease, we stopped IFN-alpha administration and started BRAF inhibitor vemurafenib therapy (according to the license by National Institute of Pharmacy and Nutrition; license number: OGYI/40060-1/2014). As there are no data about the pediatric dosage of vemurafenib, we calculated it by correlating the child’s body surface with the adults’ 2 m2 body surface and the associated adult dose (240 mg BID for 0.73 m2 body surface). Within a few days, we observed a rapid and impressive improvement in the general state; after 4 days of vemurafenib treatment, his thrombocyte count started to increase, and he did not require transfusions any more. The control bone marrow histological examination after 3 months of BRAF inhibitor therapy showed 60% residual disease, which decreased to 15% in the 11th month. In the 16th month of vemurafenib therapy we could detect complete remission in the bone marrow, which was also verified by BRAF mutation analysis. Our findings reveal that the histological disappearance of the disease from the bone marrow can last for several months; however, the radiological improvement takes a couple of weeks/month (Figure 4B), and the positive clinical response is the first indicator of the effective treatment. This improvement could be seen in a couple of days after starting the vemurafenib therapy.
Figure 1.

The role of head MRI in the evaluation of therapy response.
Notes: (A) T2W image shows left temporal bone involvement of LCH (white arrow). (B) This lesion shows inhomogenous diffusion restriction (black arrow) on the diffusion-weighted image. (C and D) Normal appearance of the left temporal area after the initial chemotherapy on T2W and diffusion-weighted images. (E and F) Signal abnormality in the left temporal area on the T2W (white arrow) and diffusion-weighted (black arrow) images.
Abbreviations: LCH, Langerhans cell histiocytosis; MRI, magnetic resonance imaging.
Figure 2.

Skull lesion in LCH.
Notes: (A) Hematoxylin–eosin stain. Majority of the cells have coffee bean-shaped nuclei with open chromatin pattern and nuclear membrane grooves. (B) Strongly positive CD1a immune reaction, which is specific for LCH.
Abbreviation: LCH, Langerhans cell histiocytosis.
Figure 3.

Massive bone marrow infiltration with ECD (original magnification).
Notes: (A) Hematoxylin–eosin stain. The marrow spaces are extensively engaged by foamy macrophages. (B) S100 immune reaction. The immune phenotype of macrophages is consistent with the phenotype of the conventional tissue histiocytes (CD68 positive, S100 negative). S100 is a highly sensitive but a nonspecific marker of Langerhans cells, therefore a good marker for screening Langerhans cells, or exclude Langerhans cell origin of a tumorous proliferation.
Abbreviation: ECD, Erdheim–Chester disease.
Figure 4.

Plain radiograph of the knees (A) before and (B) after vemurafenib treatment.
Notes: (A) Bilateral, symmetric, irregular sclerotic changes in the metaphysis of the bones. (B) Normal bone structure with a radiolucent band and small cystic lesions next to the epiphyseal plates.
Figure 5.
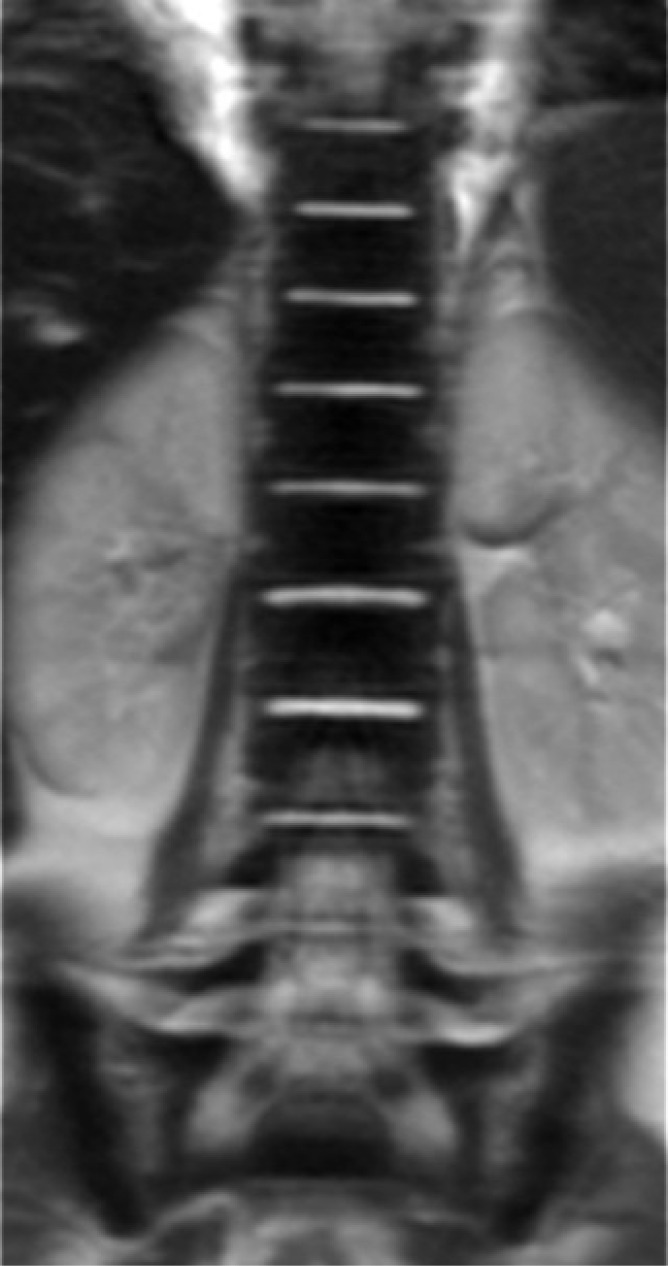
Coronal T2W image of the spine shows very low signal intensity of the bone marrow that is specific for ECD.
Abbreviation: ECD, Erdheim–Chester disease.
Now the patient is 4 years old, and he is in the 18th month of vemurafenib therapy given in the same dose from the beginning. His general status is excellent, no hepatosplenomegaly is found, blood count and routine laboratory parameters are normal, and his ECD is in complete remission. The detected side effects of vemurafenib are the lack of eyebrows, very dry, rough skin all over the body, and hypertension.
As there is no experience with the long-term use of vemurafenib in the literature and it can cause serious side effects, regarding the young age and the high-risk disease of our patient, the National Pediatric Stem Cell Transplantation Committee decided to treat him with allogenic stem cell transplantation; hence, the search for an appropriate donor is in progress. Parents’ consent has been obtained for the publication of this case report and its accompanying images.
Discussion
ECD is a rare histiocytic disorder, and since its first description in 1930, ~500 cases have been reported in the literature,4 and there are only a few publications about pediatric ECD patients.5–10 The pathogenesis is still incompletely understood, and there are an increasing number of contradictory results supporting a theory of either a reactive origin or a malignant origin. Recent discovery of the high prevalence of BRAF V600E oncogenic mutation in ECD and LCH but not in other histiocytic disorders suggests that both proliferations could derive from a common origin.11–13 In addition to the recognized oncogenic activity of BRAF V600E mutation, it has also been associated with OIS, a recently identified important mechanism against tumor proliferation. As a crucial part of this antitumor activity, BRAFV600E-mutated cells produce a wide range of cytokines and chemokines, and this local inflammatory reaction may prevent the transformation of the mutated cells to cancer. This hypothesis raises the possible rule of OIS in ECD as well.14,15 Although ECD can be distinguished from LCH by morphological and immunohistochemical evaluation, based on the same clonal mutations and similar clinical manifestations, the latest updated WHO classification includes them in the same “L” (Langerhans) group. Furthermore, studies suggest that the occurrence of both diseases in the same patient is not rare, and in 12%–20% of patients, they really present as LCH-ECD overlap syndrome.12,16,17 Hervier et al17 identified 23 patients with mixed histiocytosis, and both diseases (LCH and LCD) were diagnosed simultaneously in 11 patients (48%). Diagnosis of LCH preceded ECD – similarly to our case – in all remaining patients (52%). To our best knowledge, only one pediatric double diagnosis has been published so far: in 2015, Kim et al published a case of a 3-year-old boy with intracranial simultaneous LCH and ECD.18 In these mixed histiocytosis cases, both LCH and ECD bear the same BRAF V600E mutation. These data suggest the theory of the common clonal origin of these disorders.
As ECD is extremely rare in children, evidence regarding treatment is limited to case reports. Successful treatments with corticosteroid monotherapy, IFN-alpha, or anakinra have been published on a case-to-case basis.5,7–10 Since the prognosis of ECD has been reported as poor, search for new therapeutic approaches is essential. More than 50% prevalence of BRAF mutation in ECD patients gives a possible therapeutic target. Clinical experiences with the BRAF inhibitor vemurafenib in ECD are rapidly increasing, both in prospective clinical trials and in “off-label” administration.12 Initial case reports have demonstrated significant activity of vemurafenib in adult ECD patients: three different publications reported at least partial response following vemurafenib therapy, which involved ~30 ECD patients in all with multisystemic, refractory diseases.3,19,20 In the case of our patient, corticosteroid and IFN-alpha therapies proved to be ineffective, and hence we were forced to change to another therapy option. BRAF mutation positivity of his ECD gave us a potential therapeutic option, which proved to be very effective. Nevertheless, indefinite treatment with BRAF inhibition is potentially unsafe, given the risk of accelerating premalignant RAS-mediated neoplastic lesions, although to date no treatment course has been defined for vemurafenib in ECD.12 Attempting vemurafenib treatment cession and watch-and-wait attitude were not a real possibility in his case facing with his very high-risk disease. Since 2001, some case reports about autologous stem cell transplantation as a therapy in ECD have been published in the literature;21,22 in our case, due to extensive bone marrow infiltration, allogenic transplantation had to be preferred instead of autologous.
Extensive bone marrow involvement in ECD is also not common, but the exact frequency is unknown because of the rarity of the disease, although case reports have been published in 2016 about bone marrow involvement in two adult ECD patients.23,24
Recent publication reports the youngest ECD patient in the literature, and this is the first one about the successful application of the BRAF inhibitor vemurafenib in pediatric ECD. The case is also a rarity because of the consecutive presence of BRAF-positive LCH and ECD. Furthermore, according to our encouraging results with this boy, we suggest to think of vemurafenib as a possible, off-label therapy in pediatric ECD.
Acknowledgments
This article was not supported by any financial or other relationships.
Abbreviations
- ECD
Erdheim Chester disease
- IFN-alpha
interferon-alpha
- LCH
Langerhans cell histiocytosis
- MRI
magnetic resonance imaging
- OIS
oncogene-induced senescence
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Cives M, Simone V, Rizzo FM, et al. Erdheim–Chester disease: a systematic review. Crit Rev Oncol Hematol. 2015;95(1):1–11. doi: 10.1016/j.critrevonc.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700–2703. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 3.Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim–Chester disease. J Clin Oncol. 2015;33(5):411–418. doi: 10.1200/JCO.2014.57.1950. [DOI] [PubMed] [Google Scholar]
- 4.Campochiaro C, Tomelleri A, Cavalli G, Berti A, Dagna L. Erdheim–Chester disease. Eur J Intern Med. 2015;26(4):223–229. doi: 10.1016/j.ejim.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Tran TA, Fabre M, Pariente D, et al. Erdheim–Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol Oncol. 2009;31(10):782–786. doi: 10.1097/MPH.0b013e3181b76827. [DOI] [PubMed] [Google Scholar]
- 6.Globerman H, Burstein S, Girardina PJ, Winchester P, Frankel S. A xanthogranulomatous histiocytosis in a child presenting with short stature. Am J Pediatr Hematol Oncol. 1991;13(1):42–46. doi: 10.1097/00043426-199121000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Song SY, Lee SW, Ryu KH, Sung SH. Erdheim–Chester disease with multisystem involvement in a 4-year-old. Pediatr Radiol. 2012;42(5):632–635. doi: 10.1007/s00247-011-2235-8. [DOI] [PubMed] [Google Scholar]
- 8.Sohn MH, Kim MW, Kang YH, Jeong HJ. Tc-99m MDP bone and Ga-67 citrate scintigraphy of Erdheim–Chester disease in a child. Clin Nucl Med. 2006;31(2):90–92. doi: 10.1097/01.rlu.0000196413.47370.c9. [DOI] [PubMed] [Google Scholar]
- 9.Joo CU, Go YS, Kim IH, Kim CS, Lee SY. Erdheim–Chester disease in a child with MR imaging showing regression of marrow changes. Skeletal Radiol. 2005;34(5):299–302. doi: 10.1007/s00256-004-0846-y. [DOI] [PubMed] [Google Scholar]
- 10.Tran TA, Pariente D, Lecron JC, Delwail A, Taoufik Y, Meinzer U. Treatment of pediatric Erdheim–Chester disease with interleukin-1-targeting drugs. Arthritis Rheum. 2011;63(12):4031–4032. doi: 10.1002/art.30638. [DOI] [PubMed] [Google Scholar]
- 11.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim–Chester disease. Blood. 2014;124(4):483–492. doi: 10.1182/blood-2014-03-561381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hervier B, Haroche J, Arnaud L, et al. Association of both Langerhans cell histiocytosis and Erdheim–Chester disease linked to the BRAFV600E mutation. Blood. 2014;124(7):1119–1126. doi: 10.1182/blood-2013-12-543793. [DOI] [PubMed] [Google Scholar]
- 14.Cangi MG, Biavasco R, Cavalli G, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim–Chester disease. Ann Rheum Dis. 2015;74(8):1596–1602. doi: 10.1136/annrheumdis-2013-204924. [DOI] [PubMed] [Google Scholar]
- 15.Cavalli G, Biavasco R, Borgiani B, Dagna L. Oncogene-induced senescence as a new mechanism of disease: the paradigm of Erdheim–Chester disease. Front Immunol. 2014;5:281. doi: 10.3389/fimmu.2014.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. doi: 10.1182/blood-2016-01-690636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hervier B, Haroche J, Arnaud L, French Histiocytoses Study Group Association of both Langerhans cell histiocytosis and Erdheim–Chester disease linked to the BRAFV600E mutation. Blood. 2014;124(7):1119–1126. doi: 10.1182/blood-2013-12-543793. [DOI] [PubMed] [Google Scholar]
- 18.Kim S, Lee M, Shin HJ, Lee J, Suh YL. Coexistence of intracranial Langerhans cell histiocytosis and Erdheim-Chester disease in a pediatric patient: a case report. Childs Nerv Syst. 2016;32(5):893–896. doi: 10.1007/s00381-015-2929-6. [DOI] [PubMed] [Google Scholar]
- 19.Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495–1500. doi: 10.1182/blood-2012-07-446286. [DOI] [PubMed] [Google Scholar]
- 20.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373(8):726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaspar N, Boudou P, Haroche J, et al. High-dose chemotherapy followed by autologous hematopoietic stem cell transplantation for adult histiocytic disorders with central nervous system involvement. Haematologica. 2006;91(8):1121–1125. [PubMed] [Google Scholar]
- 22.Boissel N, Wechsler B, Leblond V. Treatment of refractory Erdheim–Chester disease with double autologous hematopoietic stem-cell transplantation. Ann Intern Med. 2001;135(9):844–845. doi: 10.7326/0003-4819-135-9-200111060-00027. [DOI] [PubMed] [Google Scholar]
- 23.Abdellateef EE, Abdelhai AR, Gawish HH, Abdulmonaem GA, Abdelbary EH, Ahmed AI. The first reported case of Erdheim–Chester disease in Egypt with bilateral exophthalmos, loss of vision, and multi-organ involvement in a young woman. Am J Case Rep. 2016;17:360–370. doi: 10.12659/AJCR.897479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim J, Kim KH, Suh KJ, et al. A unique case of Erdheim–Chester disease with axial skeleton, lymph node, and bone marrow involvement. Cancer Res Treat. 2016;48(1):415–421. doi: 10.4143/crt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]