

Abstract
Background
Patients with long-term ulcerative colitis are at risk for developing colorectal cancer.
Methods
Archival formalin-fixed paraffin-embedded tissue from ulcerative colitis patients who underwent a colectomy for high-grade dysplasia or carcinoma was examined for changes in expression of plasminogen activator inhibitor-1 (PAI-1) as well as other mediators of inflammation-associated cancer. Epithelia from areas of colons that showed histologic evidence of carcinoma, high-grade dysplasia, and epithelia that were not dysplastic or malignant but did contain evidence of prior Inflammation (quiescent colitis) was microdissected using laser capture microscopy. mRNA was extracted from the microdissected tissue and PCR array analysis was performed. To extend our findings, PAI-1 protein levels were determined using immunohistochemistry.
Results
The mRNA expression of PAI-1 is increased 6-fold (p=0.02) when comparing the carcinoma group to the quiescent colitis group; increases were also observed in NFKB2, REL, SRC, and VEGFA. The protein levels of PAI-1 are increased by 50% (p<0.001) in high-grade dysplasia and by 60% (p<0.001) in carcinoma when compared to the quiescent colitis group.
Conclusions
The increase in PAI-1 in high-grade dysplasia and carcinoma suggests a functional role for PAI-1 in malignant transformation in colitis-associated cancer. PAI-1 could also prove a useful diagnostic marker to identify patients at risk for neoplasia and it may be a useful therapeutic target to treat colitis-associated cancer.
Keywords: Plasminogen activator, inhibitor-1, IBD, Ulcerative colitis, Dysplasia, Colon cancer
1. Introduction
Although several lines of evidence suggest that chronic inflammation is associated with a higher incidence of cancer in various organs,1 perhaps no association is stronger than that for chronic ulcerative colitis,2 where the risk of colon and rectal cancer is increased by both extent and duration of the disease.3,4 Both the length and severity of disease are associated with cancer risk which suggests that the chronicity of the inflammation is a causative factor in carcinogenesis in ulcerative colitis patients. Hence, there is likely a causal link between chronic inflammation in a tissue and the development of cancer in that tissue. Of particular interest are the molecular mechanisms that underlie the transition from a chronically inflamed tissue to a cancer.
Plasminogen activator inhibitor-1 (PAI-1) is derived from the gene SERPINE1 and belongs to the class of proteins known as serine protease inhibitors (serpins). Unlike many serpins, PAI-1 levels are tightly regulated due to its most characterized role in the coagulation system: a suicide inhibitor of both plasminogen activators, urokinase (uPA) and tissue plasminogen activator (tPA). This process ultimately inhibits activation of plasmin and its subsequent proteolytic activities including fibrinolysis. In addition to their key roles in the coagulation system, increased expression of PAI-1, uPA, and uPAR is associated with poor prognoses in many types of cancer (reviewed in 5). PAI-l is an attractive target to examine in inflammation-associated cancers because it can be upregulated by several signals present in a chronic inflammation. These include inflammatory signaling through nuclear factor-kappa B (NF-κB),6 hypoxia,7,8 and oxidative stress.9 In addition, the oncogene c-src, previously shown to be increased in colitis-associated cancer,10 can also upregulate PAI-1.11 PAI-1 can also cause effects that could promote cancer and worsen prognoses, including angiogenlc,12 anti-apoptotic,13 and metastatic effects.14 While PAI-1 has received significant attention in cancer research, little is known about its role in ulcerative colitis and colitis-associated cancer. This study is the first to examine PAI-1 levels in colitis-associated cancer.
Here we report on a group of patients with colitis-associated cancer who all underwent total colectomy due to the detection of carcinoma or dysplasia during routine surveillance. Because each colon contained tissues in at least three different disease stages, we were able to identify and study the premalignant stage high-grade dysplasia as well as carcinoma and non-neoplastic tissue that had evidence of prior inflammation (quiescent colitis) in each patient. High-grade dysplasia is an intermediate stage, and alterations in gene and protein expression in high-grade dysplasia represent potential targets for early treatment and detection of colitis-associated cancer. Using Laser-capture microdissection and immunohistochemistry, we were able to specifically isolate epithelial cells and to ensure that our results were specific to the affected tissue. This combination of techniques and sample selection has allowed us a powerful means to interrogate changes in PAI-1 and other related genes associated with cancer in the setting of chronic inflammation as we examine the transition from chronic inflammation to carcinoma in the colon tissues of patients with colitis-associated cancer.
2. Methods
2.1. Sample collection
Cases were identified from ulcerative colitis patients that underwent total colectomy for dysplasia or carcinoma by a single surgeon at Boston Medical Center. Patient specimens were anonymized prior to examination and analysis as approved by the Institutional Review Board at Boston University Medical Center. Cases were selected if the initial diagnosis was histologically reconfirmed by two independent gastrointestinal pathologists (MO, SC) and if the colon contained the two stages of colitis-associated cancer of interest (carcinoma and high-grade dysplasia) as well as quiescent colitis in the same specimen. Histologic examination of each case revealed that quiescent colitis is the most common occurring non-actively inflamed tissue in the ulcerative colitis colon. It was therefore chosen as our reference group because it had been subjected to the same chronic inflammation as the neoplastic groups but had not undergone neoplastic transformation. The major advantage of obtaining blocks representing all three groups from the same patient is that this controls for intra-patient variability: differences in genetic background; disease history, duration, and severity; environmental factors; and therapeutic interventions. Additional cases were selected for immunohistochemistry if blocks were available that contained either quiescent colitis and high-grade dysplasia or quiescent colitis and carcinoma. Representative hematoxylin and eosin stains of these stages and their histologic descriptions are shown In Fig. 1.
Figure 1.

Hematoxylin and eosin stained sections from colitis-associated cancer patients. Bright field photomicrographs from colon section displaying quiescent colitis at (A) 100× and (B) 200× magnification showing branched and elongated crypts typical of reparative processes after inflammation; high-grade dysplasia at (C) 100× and (D)200× magnification; and carcinoma at (E) 40× and (F) 100× magnification. In high-grade dysplasia, note the increased size and disorder of the nuclei (D, indicated by arrows). In carcinoma, note the Invasive pseudo-crypts that extend beyond the mucosal membrane (E, Indicated by arrows).
2.2. Laser capture microscopy, RNA extraction, isolation and purification
RNA extraction from microdissected formalin-fixed, paraffin-embedded tissue is feasible15 and can produce results similar to frozen fixed tissue even though RNA quantity and quality are reduced.16 Five patients were selected who had formalin-fixed, paraffin-embedded blocks representing each of the three stages (carcinoma, high-grade dysplasia, and quiescent colitis). Slices were cut to 7 μm and then mounted on glass slides. One slide was microdissected for each group/patient. Slides were dewaxed, rehydrated, stained with Paradise Plus stain, and then dehydrated. Multiple crypts were micro-dissected using laser capture microscopy (Arcturus Pixcell 2e, Molecular Diagnostics, Inc., Sunnyvale CA) as we have previously described.17 Crypts were microdissected on to CapSure HS LCM Caps (LCM0214, Molecular Diagnostics, Inc., Sunnyvale CA). RNA was then extracted from the microdissected tissue in accordance with the manufacturer’s protocol (Paradise Plus kit, Molecular Diagnostics, Inc., Sunnyvale CA). Briefly, microdissected samples were digested with proteinase K overnight. The next morning, nucleic acids were bound to a column while cellular debris washed through. DNA was then digested on-column, and purified RNA was eluted. The RNA was reverse-transcribed, pre-amplified, and assayed on a custom PCR array (Qiagen, Valencia CA). No genomic DNA was detected using array controls.
2.3. PCR array
Custom PCR arrays (Qiagen, Valencia CA) containing primers for SERPlNE1 transcripts were used. Results were normalized to a housekeeping control, tyrosine-3-monooxygenase/tryptophan-5-monooxygenase activation protein, zeta peptide (YWHAZ). Wells with no amplification were assigned a cycle threshold (Ct) value of 40. Results were calculated utilizing the ΔΔCt method and were expressed as a fold change of the quiescent colitis group as described in the RT2 Profiler™ PCR Array Data Analysis (http://www.sabiosciences.com/pcrarraydataanalysis.php). Statistical comparisons were performed using Bioconductor’s R statistical program (HTqPCR Bioconductor package; http://www.bioconductor.org). All data passed equivariance and normality tests. Results for any gene were discarded if multiple wells did not show amplification.
2.4. Immunohistochemistry
Archived formalin-fixed, paraffin-embedded samples (n = 11 quiescent colitis, n = 10 high-grade dysplasia, n = 9 carcinoma) described as above were cut to 5 μm, mounted upon glass slides, and then deparaffinized and rehydrated. To facilitate maximal binding, antigen retrieval was performed using a BioGenex EZ-Retriever and CITRA-PLUS buffer (BioGenex, Fremont CA) heated to 97 °C for 15 min. Slides incubated in 10% rabbit serum in PBS, and were then probed with an antibody for PAI-1 (ab31280, Abcam, Cambridge MA). Antibody specificity was validated by probing a small group of samples with and without a blocking peptide. After incubation with the primary antibody, samples were probed with a secondary antibody labeled with fluorescein (31509, Thermo Fisher Scientific, Rockford IL), mounted (VECTASHIElD mounting medium, Vector Labs, Burlingame CA), and photographed (Nikon Deconvolution Wide-Field Epifluorescence Microscope, Nikon Instruments, Melville NY). From each slide, five representative fields were randomly selected, and each field’s mean fluorescent intensity was quantified using ImageJ (National Institutes of Health, Bethesda MD). Average mean intensities for each sample/group were analyzed by one way repeated measures ANOVA. All data passed equivariance and normality tests. Post-hoc pairwise means comparisons were performed using the Holm-Sidak method (SigmaStat, Systat Software, San Jose CA).
3. Results
3.1. mRNA expression of SERPINE1 is increased in the transition from quiescent colitis to carcinoma
The mRNA of the SERPJNE1 transcript is increased 6.0-fold (p=0.02) in carcinoma when compared to non-neoplastic epithelium from areas of quiescent inflammation (Fig. 2). Samples from patients with high grade dysplasia showed no statistically significant differences compared to either of the other two groups.
Figure 2.
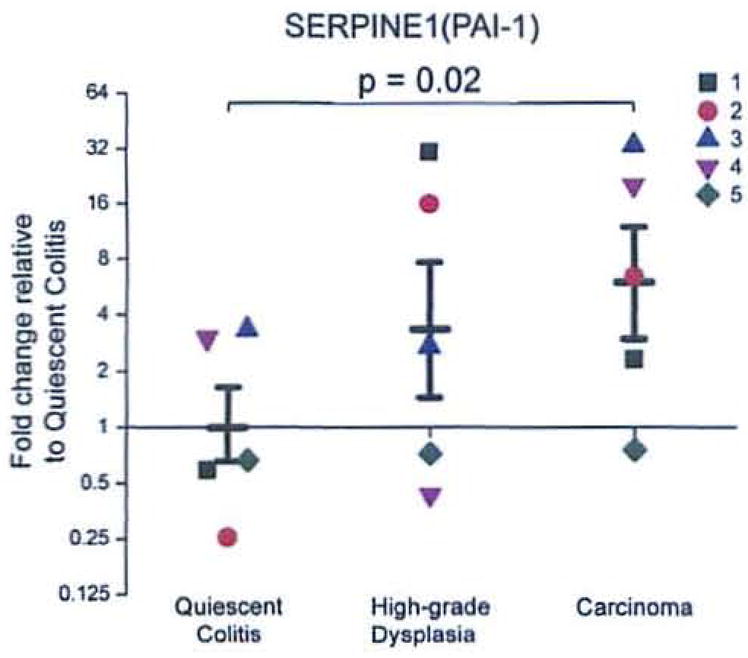
mRNA expression of SERPINE1 is increased in the transition from quiescent colitis to carcinoma. Each group (n = 5) contains individual observations (each individual is a distinct color/shape) and means ± SEM for groups. Results were calculated using the ΔΔCt method and expressed as a fold change of the quiescent colitis group as described in the RT2 Profile™ PCR Array Data Analysis (http://www.sabiosciences.com/pcrarraydataanalysis.php).
3.2. Protein levels of PAI-1 are increased in the transition from quiescent colitis to high grade dysplasia and carcinoma
Immunofluorescent staining with an antibody that selectively recognizes PAI-1 reveals that in quiescent colitis, rare cells in basal crypts show a diffuse cytoplasmic signal. High-grade dysplasia and adenocarcinoma show similar patterns of diffuse reactivity with strong cytoplasmic staining and peripheral or membranous accentuation. One-way repeated measures ANOVA shows a difference in mean fluorescent intensity among the three groups (p<0.001). Multiple mean comparisons show that there is a 50% increase (p<0.001) in high-grade dysplasia and a 60% increase (p<0.001) in carcinoma compared to quiescent colitis (Fig. 3). These data show that there is an increase in PAI-1 protein as the chronically inflamed epithelium transitions from quiescent colitis to neoplasia and malignancy.
Figure 3.

Protein levels of PAI-1, are increased in the transition from quiescent colitis to high grade dysplasia and carcinoma. Representative images immunofluorescent staining of quiescent epithelium (top left), high-grade dysplasia (middle left) and carcinoma (bottom left) using an antibody targeting PAI-1. Exposure time = 300 msec. Top right, mean fluorescent intensities with individual observations (each individual is a distinct color/shape) and mean ± SEM (n = 11 quiescent colitis, n = 10 high-grade dysplasia, n = 9 carcinoma). Each observation represents the average fluorescent intensity from five randomly selected fields.*p<0.05 compared to quiescent colitis. Bottom right, staining without (left) and with (right) neutralizing peptide displays specificity of the antibody.
3.3. mRNA expression of genes that regulate PAI-1 is increased in the transition from non-neoplastic epithelium to carcinoma
The mRNA of the NFKB2 transcript, which encodes the NF-κB subunit p52, is increased 6-fold (p = 0.03) in carcinoma compared to quiescent epithelium, but there are no significant differences between high-grade dysplasia and the other two groups (Fig. 4). The mRNA of the REL transcript, which encodes the NF-κB subunit c-Rel, is increased 8-fold (p = 0.02) in carcinoma compared to quiescent epithelium; there are no significant differences between high-grade dysplasia and the other two groups (Fig. 4). The mRNA of the SRC transcript, which encodes the oncogene c-src, is increased 2-fold (p = 0.04) when comparing the carcinoma group to the high-grade dysplasia group; there are no significant differences between the quiescent colitis group and the other two groups (Fig. 4).
Figure 4.

mRNA expression of genes that regulate PAI-1 is increased in the transition from non-neoplastic epithelium to carcinoma. Graphs for NKFB2, REL, SRC and VEGFA with individual observations (each patient is a distinct color/shape) with means±SEM. Results were calculated using the ΔΔCt method and expressed as a fold change of the quiescent colitis group as described in the RT2 Profiler™ PCR Array Data Analysis (http://www.sabiosciences.com/pcrarraydataanalysis.php). *p<0.05 difference between groups.
3.4. mRNA expression of VEGFA, a gene that is regulated by PAI-1. is increased in the transition from non-neoplastic epithelium to carcinoma
The mRNA of the VEGFA transcript, which encodes vascular endothelial growth factor A, is increased by 4-fold (p = 0.03) in carcinoma compared to quiescent epithelium (Fig. 4), and the comparison between high-grade dysplasia and carcinoma is increased 8-fold, (p = 0.06).
4. Discussion
This is the first study to examine the expression of PAI-1 in colitis-associated cancer. A unique group of ulcerative colitis patients that had transitional stages of colitis-associated cancer (high-grade dysplasia and carcinoma), as well as quiescent colitis, present in the same colectomy specimen was utilized to demonstrate that there are significant increases in both the mRNA and protein levels of PAI-1 as this disease transitions from quiescent colitis to dysplasia and carcinoma. By specifically isolating colonic epithelia using laser capture microdissection for PCR analysis, we were able to analyze SERPINE1 expression, along with other genes up and downstream of PAI-1. We were also able to localize increased immunostaining to the colonic epithelium using immunofluorescence. The mRNA and protein increases follow a similar pattern and suggest that the changes are controlled at the transcriptional level. The finding that PAI-1 is increased in colitis-associated cancer is pathologically significant because it is a key mediator induced by chronic inflammation that can promote the development of dysplasia and malignancy. PAI-1 can be increased by a variety of signals present in chronic inflammatory conditions such as ulcerative colitis, and it can initiate angiogenic,12 anti-apoptotic,13 and metastatic14 effects that promote malignancy.
We also demonstrated in this study that NFKB2 (p52) mRNA and REL (c-Rel) mRNA, members of the NF-κB family, were significantly increased in the transition to colitis-associated cancer (Fig. 4). These results implicate chronic inflammatory signaling as a mediator of increased SERPINE1 gene expression. It has been reported that pro-inflammatory signals including LPS,18 TNF,19 IL-1,20,21 and IL-622 increase SERPINE1 gene expression and PAI-1 protein levels in vitro. The observation that LPS induces SERPINE1 gene expression is especially interesting in light of recent work showing that mice taking toll-like receptor 4 (TLR4), which binds LPS and initiates a pro-inflammatory program, have reduced incidence of colitis-associated cancer induced by dextran sodium sulfate/azoxymethane.23 Conversely, mice with constitutively active TLR4 have increased tumor number and mortality in the same model.24 This may be in part due to increased PAI-1 expression in wild-type mice as opposed to TLR4-deficlent mice. IL-6 protein levels and signaling have also been shown to be increased in mouse models of colitis-associated cancer25 where they have a pro-survival effect26; again, this could be partially explained by increased PAI-1 expression. Studies also show that LPS and TNF upregulate PAI-1 through an NF-κB-dependent pathway.6 In ulcerative colitis, there are likely to be many factors in addition to those already discussed that are produced by chronic inflammation and can cause NF-κB activation. In light of our results regarding the NFKB2 and REL genes, it Is interesting to speculate that cancerous cells may be hyper-responsive to these signals, producing elevated levels of PAI-1 and other cancer promoting molecules.
We also show an increase of the oncogene c-src, which was previously reported by Cartwright et al.10 in malignant epithelia from patients with ulcerative colitis. This result represents another mechanism by which PAI-1 may be elevated in colitis-associated cancer. c-src has been shown to mediate the increased expression of PAI-1 through an ERK-dependent pathway.11 ERK activates a pro-proliferative program and is responsive to many signaling pathways including c-src; several of these pathways have already been shown to be dysregulated in colitis-associated cancer. Increases In oncogenic k-ras in high-grade dysplasia27 as well as the epidermal growth factor receptor28 and the truncated neuroklnln-1 receptor17 in carcinoma may play a role in activating ERK and increasing expression of PAI-1 as well.
Another mechanism by which SERPlNE1 expression could be increased in ulcerative colitis is the mucosal hypoxia that typifies the inflamed colitic lesions.29 Patients with ulcerative colitis have elevated levels of the hypoxia-inducible factor 1-alpha (HIF-1α) and HIF 2-alpha focally expressed in colonic epithelial cells.30 These transcription factors can remain elevated during remission of colitis, suggesting that hypoxia can persist during quiescent stages of disease.31 In response to hypoxia, inflamed colonic mucosa have increased vascularity compared to normal tissue, in part due to increased levels of vascular endothelial growth factor (VEGF). a pro-angiogenic downstream target of HIF-1α.30 In our study, we show an increase in the VEGFA gene between the quiescent colitis and carcinoma groups; this may be an effect of both PAI-1 and hypoxia, a result corroborated by in vitro studies.32 Hypoxia and HIF-1α activation are also capable of upregulating PAI-1,7 and such increases can persist after HIF-1α levels return to baseline.8 Thus, inflammation-induced hypoxia is another mechanism by which chronic ulcerative colitis could upregulate expression of SERPINE1.
PAI-1 activation may occur due to inflammation-associated increases in oxidative stress. Inflammation causes increases in reactive oxygen and nitrogen species (RONS), which serve to destroy pathogens and signal changes in the inflammatory process. RONS have long been considered to play a significant role in inflammation-induced carcinogenesis because of their ability to oxidize DNA, proteins and lipids which leads to dysfunction and importantly, DNA mutation. Constant exposure to oxidative stress is a source of genetic instability that drives carcinogenesis. In addition to DNA damage, production of superoxide in vitro can upregulate PAI-19; therefore, oxidative stress represents another mechanism that could upregulate PAI-1.
PAI-1 initiates several effects that could promote cancer or increase malignancy in the chronically inflamed colon (Fig. 5). Clinical data shows that increased PAI-1 levels in colonic cancer epithella are associated with increased metastases.33 It is estimated that in colon cancer, one quarter of patients will present with synchronous liver metastases and one half will develop liver metastases.34 For patients with liver metastases, the progression of the disease in the liver determines the life expectancy of the patient.35 This is surprising in light of the fact that the most characterized function of PAI-1 is to reduce plasmin-mediated proteolysis through direct inhibition of tPA and uPA which reduces matrix degradation, a process linked to metastasis. However, PAI-1 mediates several other functions that could explain its metastatic effects: it can increase cell motility by causing de-adhesion by competing for binding Interactions between the uPA:uPAR complex and integrins36 and vitronectin.37–39 These observations suggest that cells may migrate away from vitronectin-containing matrices, as was reported in vitro that neuroblastoma cells move from vitronectin to fibronectln after treatment with PAI-1.40 Another mechanism by which PAI-1 increases cell migration is by binding to the LDL receptor-related protein and stimulating the Jak/Stat pathway.41 Thus, PAI-1 is likely capable of increasing cell motility and tumor cell metastasis, which would contribute to malignancy and worsen prognosis.
Figure 5.

Pathways by which PAI-1 may contribute to colitis-associated cancer. Here we propose a mechanism by which an epithelial cell responds to the stimuli present in chronic inflammation by upregulating PAI-1 and some pro-malignant sequelae of that upregulation. Inflammation produces a variety of signals that can upregulate PAI-1 in colitis-associated cancer; these signals include oxidative stress, inflammatory mediators, hypoxia, and substance P. We have shown that both isoforms of the neurokinin-1 receptor are expressed in colon epithelial cells and that the truncated form is Increased in dysplasia and colitis-associated cancer.17 The upregulation of PAI-1 has direct consequences for the epithelial cell that promote its transformation to dysplasia or malignancy, including anti-apoptotic signaling and the promotion of anglogenesis and cell motility and metastasis. Additionally, transformed cells may be capable of producing signals that upregulate PAI-1. Genes depicted in bold/red were also identified as increased in this study. Abbreviations: RONS, reactive oxygen and nitrogen species; SP, substance P; HIF, hypoxia inducible factor; NF-κB, nuclear factor kappa B; PAI, plasminogen activator inhibitor; VEGF, vascular endothelial growth factor.
Several studies confirm that PAI-1 plays a significant role in angiogenesis that can support tumor growth.42–44 One possible explanation can be found in our result that VEGFA is increased in carcinoma and the increase in PAI-1 may be partially responsible for this result.32 The mechanism by which PAI-1 controls angiogenesis is not well-understood, but in vitro studies have shown that it is capable of promoting migration and proliferation of vascular smooth muscle cells45 as well as inhibiting apoptosis.46 Overall, PAI-1 appears to have pro-angiogenic effects.
Studies have also suggested that PAI-1 has anti-apoptotic and pro-proliferative effects as well. One study showed that addition of PAI-1 to cancer cells causes decreased spontaneous and induced apoptosis.47 Furthermore, fibrosarcomas derived from PAI-1 deficient mice are more sensitive to apoptotic stimuli in vitro and have a longer lag time to tumor growth in vivo.48 PAI-1 regulates apoptosis by several mechanisms: it inhibits plasminogen activation, which has been shown to be pro-apoptotic49–51; it directly inhibits caspase-346; it activates the anti-apoptotic signaling molecule Akt52; and it prolongs ERK1/2 phosphorylation, either directly45 or in conjunction with uPA.53 Thus, there are clearly several mechanisms by which PAI-1 might promote malignancy in colitis-associated cancer.
In the setting of chronic colitis, much of the colon is often affected at the cellular level despite appearing grossly normal or quiescent. As inflammatory episodes recur, the resulting tissue damage initiates a continuous process of tissue remodeling, healing and repair. In the design of this study where patient’s colonic tissues are used as their own controls, this phenomenon can create a dilemma as to which tissue to use as a reference standard. Since histologically unaffected colon tissue is uncommon in patients with chronic inflammation, we chose quiescent tissue since our pathologists noted that this was the predominant non-inflamed tissue in the majority of specimens. In addition, quiescent tissue has been exposed to the stresses of chronic inflammation that initiate carcinogenesis; consequently, there will be pre-neoplastic changes in tissue that appears to be non-neoplastic. One such change is the mutations to the tumor suppressor gene p53 that have been observed in histologically normal epithelium from patients with CAC.54 Thus, quiescent cells may be undergoing early neoplastic events and may be tumor precursors. Further examination of pre-neoplastic quiescent epithelium may provide key functional mechanisms of tumor promotion and suppression that may be important pharmacological targets.
The colon specimens utilized in this study were obtained anonymously, although all cases were confirmed histologically by two pathologists. We therefore have no access to information such as the duration and severity of the disease or details regarding the patient’s treatment history. These represent potential confounding factors that may have influenced this study; however, by including only patients that had areas of quiescent disease and non-neoplastic epithelium and by using patient matched samples, we feel we have controlled for this potential variability. Thus, PAI-1 could be a useful diagnostic marker to assist in identification of patients with neoplasia. In addition, further examination of the pathways by which PAI-1 influences cancer may allow for the discovery of novel targets and treatments for colitis-associated cancer. These findings merit validation in a larger study, and PAI-1 should be examined as a potential marker for inflammation-associated cancers in other organs.
Acknowledgments
We thank the Robert and Dana Smith Family Foundation; the Smithwick Endowment Fund from the Department of Surgery; The Neuropeptide Laboratory; and, the Department of Pharmacology, NIH Training Grant 5T32GM008541-14, Boston University School of Medicine for their financial support of this work. We thank Jeremy Hetzel for his assistance in identifying human samples and in performing data analysis. We thank Dr. Shi Yang and Charline Mack for their training in laser capture microscopy and fluorescence microscopy.
Abbreviations
- PAI-1
plasminogen activator inhibitor-1
- uPA
urokinase
- tPA
tissue plasminogen activator
- NF-κB
nuclear factor kappa-B
- LPS
lipopolysaccharide
- TNF
tumor necrosis factor
- IL-1
interleukin-1
- IL-6
interleukin-6
- TLR4
toll-like receptor 4
- ERK
extracellular signal-responsive kinase
- HIF
hypoxia-Inducible factor
- VEGF
vascular endothelial growth factor
- RONS
reactive oxygen and nitrogen species
- uPAR
urokinase receptor
Footnotes
Presented in part at the annual meeting of the American Gastroenterological Association in New Orleans, LA; May 1–4, 2010 and published in abstract form in Gastroenterology 138(5), Suppl1: S446, 2010.
Conflict of interest
All Authors have no conflict of interest.
Statement of authorship
EG carried out studies and data analyses and drafted the manuscript. SEL and AFS participated in the design of study, assisted in data analysis and edited the manuscript. LAW, MJO, and SRC obtained, validated and identified human specimens. JAC performed the statistical analyses. FAF and MJO participated in the design of the study. All authors read and approved the final manuscript.
References
- 1.O’Connor PM, Lapointe TK, Beck PL, Buret AG. Mechanisms by which inflammation may increase intestinal cancer risk in inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1411–20. doi: 10.1002/ibd.21217. [DOI] [PubMed] [Google Scholar]
- 2.Farraye FA, Odze RD, Eaden J, Itzkowitz SH, McCabe RP, Dassopoulos T, Lewis JD, Ullman TA, James T, III, McLeod R, Burgart LJ, Allen J, Brill JV. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:738–45. doi: 10.1053/j.gastro.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 3.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–33. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 5.Binder BR, Mihaly J. The plasminogen activator inhibitor “paradox” in cancer. Immunol Lett. 2008;118:116–24. doi: 10.1016/j.imlet.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Kong J, Sun T, Li G, Szeto FL, Liu W, et al. 1,25-Dihydroxyvitamin D3 suppresses inflammation-induced expression of plasminogen activator inhibitor-1 by blocking nuclear factor-κB activation. Arch Biochem Biophys. 2011;507:241–7. doi: 10.1016/j.abb.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fink T, Kazlauskas A, Poellinger L, Ebbesen P, Zachar V. Identification of a tightly regulated hypoxfa-response element in the promoter of human plasminogen activator inhibitor-1. Blood. 2002;99:2077–83. doi: 10.1182/blood.v99.6.2077. [DOI] [PubMed] [Google Scholar]
- 8.Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–52. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- 9.Du X-L, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–6. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cartwright CA, Coad CA, Egbert BM. Elevated c-Src tyrosine kinase activity in premalignant epithelia of ulcerative colitis. J Clin Invest. 1994;93:509–15. doi: 10.1172/JCI117000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samarakoon R, Higgins PJ. Pp60c-src mediates ERK activation/nuclear localization and PAI-1 gene expression in response to cellular deformation. J Cell Physiol. 2003;195:411–20. doi: 10.1002/jcp.10247. [DOI] [PubMed] [Google Scholar]
- 12.Stefansson S, McMahon GA, Petitclerc E, Lawrence DA. Plasminogen activator inhibitor-1 in tumor growth, angiogenesis and vascular remodeling. Curr Pharm Des. 2003;9:1545–64. doi: 10.2174/1381612033454621. [DOI] [PubMed] [Google Scholar]
- 13.Schneider DJ, Chen Y, Sobel BE. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb Haemost. 2008;100:1037–40. [PubMed] [Google Scholar]
- 14.Dellas C, Loskutoff DJ. Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb Haemost. 2005;93:631–40. doi: 10.1160/TH05-01-0033. [DOI] [PubMed] [Google Scholar]
- 15.Pagedar NA, Wang W, Chen DHC, Davis RR, Lopez I, Wright CG, et al. Gene expression analysis of distinct populations of cells isolated from mouse and human inner ear FFPE tissue using laser capture microdissection – a technical report based on preliminary findings. Brain Res. 2006;1091:289–99. doi: 10.1016/j.brainres.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 16.Nonn L, Vaishnav A, Gallagher L, Gann PH. mRNA and micro-RNA expression analysis in laser-capture microdissected prostate biopsies: valuable tool for risk assessment and prevention trials. Exp Mol Pathol. 2010;88:45–51. doi: 10.1016/j.yexmp.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillespie E, Leeman SE, Watts LA, Coukos JA, O’Brien MJ, Cerda SR, et al. Truncated neurokinin-1 receptor is increased in colonic epithelial cells from patients with colitis-associated cancer. Proc Natl Acad Sci U S A. 2011;108:17420–5. doi: 10.1073/pnas.1114275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quax PH, van den Hoogen CM, Verheijen JH, Padro T, Zeheb R, Gelehrter TD, et al. Endotoxin induction of plasminogen activator and plasminogen activator inhibitor type 1 mRNA in rat tissues in vivo. J Biol Chem. 1990;265:15560–3. [PubMed] [Google Scholar]
- 19.Sawdey MS, Loskutoff DJ. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J Clin Invest. 1991;88:1346–53. doi: 10.1172/JCI115440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van den Berg EA, Sprengers ED, Jaye M, Burgess W, Maciag T, van Hinsbergh VW. Regulation of plasminogen activator inhibitor-1 mRNA in human endothelial cells. Thromb Haemost. 1988;60:63–7. [PubMed] [Google Scholar]
- 21.Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA, Loskutoff DJ. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem. 1988;263:5797–803. [PubMed] [Google Scholar]
- 22.Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243–50. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–81. e1814. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, España C, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis. 2011;17:1464–73. doi: 10.1002/ibd.21527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble–IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543–51. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- 26.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Compton C, Cheng E, Fromowitz F, Viola MV. c-Ki-ras mutations in dysplastic fields and cancers in ulcerative colitis. Gastroenterology. 1992;102:1983–7. doi: 10.1016/0016-5085(92)90323-q. [DOI] [PubMed] [Google Scholar]
- 28.Alexander RJ, Panja A, Kaplan-Liss E, Mayer L, Raicht RF. Expression of growth factor receptor-encoded mRNA by colonic epithelial cells is altered in inflammatory bowel disease. Dig Dis Sci. 1995;40:485–94. doi: 10.1007/BF02064355. [DOI] [PubMed] [Google Scholar]
- 29.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–13. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okuda T, Azuma T, Ohtani M, Matsunaga S, Masaki R, Satomi S, et al. Hypoxia-inducible factor-1 alpha and vascular endothelial growth factor expression in ischaemic colitis and ulcerative colitis. Aliment Pharmacol Ther S. 2006;2:182–8. [Google Scholar]
- 32.Hjortland GO, Lillehammer T, Somme S, Wang J, Halvorsen T, Juell S, et al. Plasminogen activator inhibitor-1 increases the expression of VEGF in human glioma cells. Exp Cell Res. 2004;294:130–9. doi: 10.1016/j.yexcr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Seetoo D-Q, Crowe PJ, Russell PJ, Yang J-L. Quantitative expression of protein markers of plasminogen activation system in prognosis of colorectal cancer. J Surg Oncol. 2003;82:184–93. doi: 10.1002/jso.10210. [DOI] [PubMed] [Google Scholar]
- 34.McMillan DC, McArdle CS. Epidemiology of colorectal liver metastases. Surg Oncol. 2007;16:3–5. doi: 10.1016/j.suronc.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Wood CB, Gillis CR, Blumgart LH. A retrospective study of the natural history of patients with liver metastases from colorectal cancer. Clin Oncol. 1976;2:285–8. [PubMed] [Google Scholar]
- 36.Czekay R-P, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160:781–91. doi: 10.1083/jcb.200208117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ. Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J Cell Biol. 1996;134:1563–71. doi: 10.1083/jcb.134.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng G, Curriden SA, Hu G, Czekay R-P, Loskutoff DJ. Plasminogen activator inhibitor-1 regulates cell adhesion by binding to the somatomedin B domain of vitronectin. J Cell Physiol. 2001;189:23–33. doi: 10.1002/jcp.1133. [DOI] [PubMed] [Google Scholar]
- 39.Waltz DA, Natkin LR, Fujita RM, Wei Y, Chapman HA. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest. 1997;100:58–67. doi: 10.1172/JCI119521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugiura Y, Ma L, Sun B, Shimada H, Laug WE, Seeger RC, et al. The plasminogen–plasminogen activator (PA) system in neuroblastoma. Cancer Res. 1999;59:1327–36. [PubMed] [Google Scholar]
- 41.Degryse B, Neels JG, Czekay R-P, Aertgeerts K, Kamikubo Y-i, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279:22595–604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- 42.Bajou K, Noel A, Gerard RD, Masson V, Brunner N, Holst-Hansen C, et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4:923–8. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 43.Devy L, Blacher S, Grignet-Debrus C, Bajou K, Masson V, Gerard RD, et al. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 2002;16:147–54. doi: 10.1096/fj.01-0552com. [DOI] [PubMed] [Google Scholar]
- 44.McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MKK, Westrick RJ, et al. Plasminogen activator regulates tumor growth and angiogenesis. J Biol Chem. 2001;276:33964–8. doi: 10.1074/jbc.M105980200. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Budd RC, Kelm RJ, Sobel BE, Schneider DJ. Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. 2006;26:1777–83. doi: 10.1161/01.ATV.0000227514.50065.2a. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Kelm RJ, Budd RC, Sobel BE, Schneider DJ. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem. 2004;92:178–88. doi: 10.1002/jcb.20058. [DOI] [PubMed] [Google Scholar]
- 47.Kwaan HC, Wang J, Svoboda K, Declerck PJ. Plasminogen activator inhibitor 1 may promote tumour growth through inhibition of apoptosis. Br J Cancer. 2000;82:1702–8. doi: 10.1054/bjoc.2000.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romer MU, Kirkebjerg Due A, Knud Larsen J, Hofland KF, Christensen IJ, Buhl-Jensen P, et al. Indication of a role of plasminogen activator inhibitor type I in protecting murine fibrosarcoma cells against apoptosis. Thromb Haemost. 2005;94:859–66. [PubMed] [Google Scholar]
- 49.Tonner E, Allan G, Shkreta L, Webster J, Whitelaw CB, Flint DJ. Insulin-like growth factor binding protein-5 (IGFBP-5) potentially regulates programmed cell death and plasminogen activation in the mammary gland. Adv Exp Med Biol. 2000;480:45–53. doi: 10.1007/0-306-46832-8_5. [DOI] [PubMed] [Google Scholar]
- 50.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. Glia. 2000;29:347–54. [PubMed] [Google Scholar]
- 51.Zhang X, Chaudhry A, Chintala SK. Inhibition of plasminogen activation protects against ganglion cell loss in a mouse model of retinal damage. Mol Vis. 2003;9:238–48. [PubMed] [Google Scholar]
- 52.Romer MU, Larsen L, Offenberg H, Brunner N, Lademann UA. Plasminogen activator inhibitor 1 protects fibrosarcoma cells from etoposide-induced apoptosis through activation of the PI3K/Akt cell survival pathway. Neoplasia. 2008;10:1083–91. doi: 10.1593/neo.08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webb DJ, Thomas KS, Gonias SL. Plasminogen activator inhibitor 1 functions as a urokinase response modifier at the level of cell signaling and thereby promotes Mcf-7 cell growth. J Cell Biol. 2001;152:741–52. doi: 10.1083/jcb.152.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107:369–78. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]