

Abstract
Catalytic dehydrogenation of saturated or partially saturated six-membered carbocycles into aromatic rings represents an appealing strategy for the synthesis of substituted arenes. Particularly effective methods have been developed for the dehydrogenation of cyclohexanones and cyclohexenes into substituted phenol, aniline, and benzene derivatives, respectively. In this Perspective, we present the contributions of our research group to the discovery and development of palladium-based catalysts for aerobic oxidative dehydrogenation methods, including general methods for conversion of cyclohexanones and cyclohexenones into substituted phenols and a complementary method for partial dehydrogenation cyclohexanones to cyclohexenones. The mechanistic basis for chemoselective conversion of cyclohexanones to phenols or enones is presented. These results are presented within the context of recent methods developed by others for the synthesis of aryl ethers, anilines and other substituted arenes. Overall, Pd-catalyzed dehydrogenation methods provide a compelling strategy for selective synthesis of aromatic and related unsaturated molecules.
Keywords: Dehydrogenation, palladium, aerobic, oxidation, catalysis, aromatic
Graphical Abstract
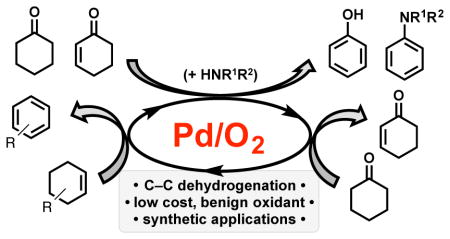
1. Introduction
Aromatic molecules are universal targets of industrial and laboratory chemical synthesis, with applications ranging from pharmaceuticals and agrochemicals to electronic materials and commodity chemicals. New methods for the preparation of substituted aromatic molecules, particularly those that exhibit selectivity patterns different from existing methods, can have a major impact across the chemical industry. This principle is prominently illustrated by the development of metal-catalyzed cross-coupling reactions of aryl halides and related substrates,1 which dramatically expanded the scope of accessible substituted aromatic compounds and bypassed many of the regioselectivity constraints and substrate limitations of classical nucleophilic and electrophilic aromatic substitution reactions (Scheme 1A–C). The requirement of cross-coupling reactions to use an aromatic substrate that is prefunctionalized with a halide, triflate or related leaving group, however, has inspired many contemporary efforts to develop methods for direct functionalization of arene C–H bonds (Scheme 1D).2,3 Many successful examples of arene C–H functionalization reactions have been reported, especially with substrates bearing a chelating directing group to control regioselectivity; however, a common feature among all of the methods in Scheme 1 is the use of an aromatic molecule as the starting material for selective incorporation of substituents to the periphery of the ring.
Scheme 1.

Common Methods for the Synthesis of Substituted Arenes
In this Perspective, we highlight a fundamentally different strategy to prepare selectively substituted aromatic compounds that has been the focus of recent investigation in our group: aerobic dehydrogenation of saturated or partially unsaturated 6-membered carbocyclic rings. The latter starting materials are readily available and easily derivatized by classical synthetic methods. Therefore, they represent appealing precursors to diversely substituted aromatic compounds. The overall strategy relies on a sequence of well established mechanistic steps in the field of Pd catalysis, including PdII-mediated C–H activation to form a Pd-alkyl species, β-H elimination from the Pd-alkyl species to generate the alkene moiety, and reoxidation of the resulting PdII-H intermediate by O2 to complete the catalytic cycle (Scheme 2).4
Scheme 2.

General Catalytic Cycle for Pd-Catalyzed Aerobic Dehydrogenation of C–C Bonds
We first demonstrated the synthetic opportunities available via this strategy in the aerobic dehydrogenation of cyclohexanones and cyclohexenones into substituted phenols (cf. Section 2, below).5 Since this initial report, we and several other groups have expanded upon this concept, significantly broadening the scope of Pd-catalyzed aerobic dehydrogenation methods, including applications to the synthesis of aryl ethers, anilines, substituted arenes and other aromatic molecules that take advantage of the a carbonyl group in the saturated precursor to enter the catalytic cycle (Scheme 3). The groups of Deng, Li and Lemaire have been particularly active in this area, as summarized in a recent account.6 In this Perspective article, we present the concepts that guided our initial discovery of the cyclohexanone-to-phenol reaction, together with the subsequent development of related dehydrogenation reactions in our laboratory, including synthetic applications as well as mechanistic studies. This work is presented in in the context of work by of other groups, with the aim of providing relevant perspective on the utility of dehydrogenation reactions as synthetic methods.
Scheme 3.

Select Examples of Aromatic Products Obtained from Substituted Cyclohexanones or Cyclohexenones.
2. Dehydrogenation of Cyclohexanones and Cyclohexenones to Phenols
The dehydrogenation of cyclohexenones to phenols is typically accomplished with stoichiometric reagents, such as 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ)7 or bromination/dehydrobromination procedures. Catalytic aerobic dehydrogenation methods represent an appealing alternative; however, most precedents were limited to high temperature, gas-phase reactions of the cyclohexanones itself, or methods that employ stoichiometric Pd and proceed in relative low (e.g., <30%) yield.8,9 Liquid-phase reactions would be amenable to the synthesis of substituted phenols, but such applications were not known. Methods of this type could leverage the facile access to substituted cyclohexanones, which may be prepared via numerous routes, including enolate alkylation, 1,4-additions to cyclohexenones, and Robinson annulation and Diels-Alder reactions. Readily accessible 3-substituted cyclohexanones are direct precursors to meta-substituted phenols that are challenging to prepare via traditional aromatic substitution methods (Scheme 4). We envisioned that this transformation could be achieved via Pd-catalyzed aerobic dehydrogenation methods.
Scheme 4.

Synthesis of Meta-Substituted Phenols and Key Dehydrogenation Mechanistic Steps
Our initial studies showed that Pd(TFA)2 (TFA = trifluoroacetate) and dimethyl sulfoxide (DMSO) solvent are important components of the catalyst system. Addition of traditional monodentate and bidentate nitrogen ligands, such as pyridine, 2,2′-bipyridyl (bpy) or 1,10-phenanthroline (phen) did not result in increased yields. We hypothesized that a more electrophilic catalyst system would improve rates of the key organometallic steps and we found that the addition of 2-dimethylaminopyridine (2-Me2Npy) and a strong acid (p-TsOH) provided high yields of the desired product at 80 °C from either cyclohexanones and cyclohexenones. We speculated that the 2-dimethylamino group of the 2-Me2Npy ligand might be protonated by the strong acid and lead to formation of a more-electron-deficient Pd catalyst. Heterogeneous catalysts (e.g., Pd/C), which have precedent as catalysts in related dehydrogenation reactions,10 were ineffective under similar conditions. This catalyst system was used in the formation of diverse substituted phenols (Scheme 5A). The ortho-, meta- and para-cresols, as well as various aryl substituted phenols, were obtained in good yields. The catalyst was also effective for dehydrogenation of substituted cyclohexenones bearing aryl, alkyl and ester groups (Scheme 5B).
Scheme 5.

Representative Scope of Substrates and Their Respective Yields of Phenols with the Pd(TFA)2/2-NMe2py Catalyst System
The utility of the method was demonstrated in the synthesis of a 3,5-diarylsubstituted phenol precursor to a potent in vitro allosteric inhibitor of the human luteinizing hormone receptor (Scheme 6).11 The original synthetic route to this molecule formed the unsymmetrically substituted phenol via sequential Suzuki coupling reactions that proceed in comparatively low yields, starting from 3,5-dibromophenol, (30%, and 62% respectively). Sequential aldol condensation of benzaldehyde and 4-methylacetophenone, followed by Robinson annulation with acetone provided access to the cyclohexenone precursor, and subsequent Pd-catalyzed aerobic dehydrogenation afforded the substituted phenol in excellent yield.
Scheme 6.

Application of Pd-Catalyzed Dehydrogenation in the Preparation of a 3,5-Diaryl Phenol Derivative En Route to a Terphenyl Derived Allosteric Inhibitor
The success of this catalyst system prompted us to pursue sequential, one-pot aerobic oxidative-coupling/dehydrogenation reactions, beginning with oxidative Heck reactions of cyclohexenone to prepare 3-arylcyclohexenone derivatives.12 Cyclohexenones are often challenging substrates in oxidative Heck reactions relative to acyclic substrates, such as acrylates, because alkene insertion into the Pd-aryl intermediate results in a Pd-enolate intermediate that must undergo isomerization to access an intermediate capable of undergoing syn β-H elimination. A neutral catalyst system was crucial to the success of the one-pot oxidative Heck/dehydrogenation reaction because the original acidic dehydrogenation conditions described above resulted in homocoupling of the boronic acid or non-oxidative conjugate addition to the enone. A new catalyst system was identified, consisting of [Pd(CH3CN)4](BF4)2, with 6,6′-dimethyl-2,2′-bipyridine (6,6′-Me2bpy) as an ancillary ligand and sodium anthraquinone-2-sulfonate (AMS) as a quinone co-catalyst, in DMSO at 80 °C. This catalyst system was effective as a dehydrogenation catalyst with diverse substituted cyclohexenones, and in some cases was superior to the previously catalyst system for this step. For example, 6-phenyl-cyclohexen-2-one underwent dehydrogenation in 96% yield, versus 33% with the original catalyst. The sequential oxidative Heck/dehydrogenation procedure was initiated with [Pd(CH3CN)4](BF4)2/6,6′-Me2bpy) catalyst system in NMP at 50 °C to effect the oxidative Heck reaction. Then DMSO was added as a cosolvent to the crude reaction mixture, followed by heating to 80 °C, to form the desired phenol product. Using this procedure, a number of 3-arylphenols were prepared in good yields from cyclohexenone and a variety of boronic acids (Scheme 7). A flow application of this method was recently developed by Park and co-workers, with sequential addition of 5% TFAH/DMSO after the oxidative Heck was carried out in NMP.13 The activity of the catalyst system was improved, with residence times of 130 min. Important changes from the original batch conditions included increasing the temperature of the reaction to 120 °C and the O2 pressure to 5 atm.
Scheme 7.

Synthesis of 3-Arylphenols via Pd-Catalyzed One-Pot Oxidative Heck/Dehydrogenation
Liu and coworkers recently developed a heterogeneous catalytic method for dehydrogenation of cyclohexanones in the absence of an oxidant or other hydrogen acceptor.14 This transformation uses commercially available Pd/C under an atmosphere of N2 and H2 in N,N-dimethylacetamide (DMA) as a solvent at 150 °C. Inclusion of hydrogen gas in the reaction headspace was found to increase the reaction rate, although it also contributed to small amounts of ketone hydrogenation. A number of substituted cyclohexanones were dehydrogenated, including a substrate bearing a sulfide group, which is typically prone to oxidation or catalyst poisoning under aerobic conditions (Scheme 8A). Dehydrogenation of cyclohexenones was also demonstrated, including substrates containing tertiary amines and heterocycles, including free pyrrole (Scheme 8B). No substrates containing halides were demonstrated, which probably reflects their susceptibility to protodehalogenation with Pd/C and H2. Recycling of the catalyst was not demonstrated.
Scheme 8.

Representative Substrates Scope Using Pd/C Under Oxidant and Acceptor-Free Conditions
Cyclohexanone and cyclohexenone derivatives have been used as precursors to other aromatic compounds containing C–O bonds via dehydrogenation methods. For example, Li and coworkers described a copper-catalyzed method for the preparation of aryl ethers from cyclohexenones.15 The reaction can be accomplished with stoichiometric CuCl2•H2O, but also under aerobic conditions with catalytic Cu(OTf)2 in the presence of co-catalytic NHPI and stoichiometric KI (Scheme 9). The reaction has relatively broad substrate scope with respect to the alcohol coupling partner, while the cyclohexenone scope was explored only with stoichiometric CuCl2, and resulted in low to moderate yields of aromatic ethers.
Scheme 9.

Cu-Catalyzed Formation of Aryl Ethers from Cyclohexenones and Alcohols
Lemaire and co-workers reported a similar reaction using Pd/C as a catalyst, typically with neat substrate and excess alcohol (5 equiv), open to air.16 Both cyclohexanones and cyclohexenones were effective substrates, as well as tetralones (Scheme 10).
Scheme 10.

Pd/C Catalyzed Formation of Aryl Ethers by Lemaire and co-workers
Hong and co-workers reported a Pd-catalyzed method for the preparation of coumarins from cyclohexanones that proceeds via dehydrogenation of cyclohexanones to phenols, followed by ortho-selective oxidative Heck reaction with acrylates and lactonization.17 The reaction is effective with diverse substituted cyclohexanones (Scheme 11). With 3-substituted cyclohexanones, the oxidative Heck proceeded selectively at the less hindered position ortho- to the phenolic –OH moiety.
Scheme 11.

Aerobic Pd-Catalyzed Synthesis of Coumarins via Dehydrogenation/Oxidative Heck
These syntheses of aryl ethers and coumarins via Pd-catalyzed aerobic dehydrogenationim methods highlight the utility of cyclohexanones and cyclohexenones as precursors to functionalized aromatic molecules and showcase the potential to expand the scope of these reactions beyond phenol formation. The recent account by Li and co-workers describes a number of related applications of this type.6
3. Partial Dehydrogenation of Cyclohexanones and Acyclic Ketones to α,β-Unsaturated Carbonyl Compounds
3.1 Dehydrogenation of Cyclohexanones to Cyclohexenones
Cyclohexenones are important intermediates in the synthesis of bioactive molecules, and cyclohexenones were observed as intermediates in our studies of the cyclohexanone dehydrogenation reactions described above. This observation prompted us to consider the prospect of developing a method for “interrupted” dehydrogenation of cyclohexanones to afford cyclohexenone products (Scheme 12). Limited precedents supported the possibility of direct Pd-catalyzed monodehydrogenation reactions.18 Typically, the conversion of cyclohexenones into the corresponding enones involves two or more steps and the use of stoichiometric oxidants,19 such as 2-iodoxybenzoic acid (IBX)20 or DDQ.7 Another common method is the Pd-catalyzed dehydrosilylation of silyl enol ethers, initially reported by Saegusa and Ito.21 A direct dehydrogenation method that would not require the formation of silyl enol ethers has significant appeal.
Scheme 12.

“Interrupted” Dehydrogenation of Cyclohexanones to Cyclohexenone Derivatives
In our initial test of direct dehydrogenation methods, we evaluated a number of different PdII source/ligand combinations. A Pd(TFA)2/DMSO (1:2) catalyst system was identified that enabled efficient and chemoselective access to the monodehydrogenation products with O2 as the sole oxidant.22 Key differences between the phenol formation catalyst and the newly developed catalyst are the use of DMSO as a catalytic ligand, rather than a solvent, and the use of AcOH, EtOAc or toluene as solvent. 4-Substituted cyclohexanones, N-substituted piperidones, chromanones, as well as 5- and 7-membered cyclic ketones were smoothly converted to the α,β-unsaturated products (Scheme 13A). Application of this catalyst system to substrates that presented regio- or chemoselectivity challenges exhibited mixed results. For example, monodehydrogenation of 2- and 3-phenylcyclohexanones led to mixtures of regioisomers of ~ 3:1 (Scheme 13B). On the other hand, dehydrogenation of a steroid containing a cyclohexanone and a cyclopentanone moiety led to selective dehydrogenation of the 6-membered ring with a single regioisomer to form 5α-androst-1-ene-3,17-dione (Scheme 13C).
Scheme 13.

Monodehydrogenation of Cyclic Ketones with Pd(DMSO)2(TFA)2 and O2
Cyclic enones that are intermediates in the synthesis of natural products were also synthesized using the Pd(TFA)2/DMSO catalyst system. An α,α-disubstituted cyclohexanone en route to (–)-mersicarpine was dehydrogenated in 85% yield, which compares favorably to the previously reported method using 3 equiv of IBX23 (72%) (Scheme 14A). Dehydrogenation to a cyclopentene-α-dione, which previously required stoichiometric Pd(OAc)2,24 was accomplished in 90% yield with an increase in the Pd and DMSO catalyst loading (10 and 30 mol %, respectively (Scheme 14B).
Scheme 14.

Synthesis of Natural Product Precursors Using Pd(DMSO)2(TFA)2 and O2
The examples presented in Schemes 13 and 14 support Pd-catalyzed aerobic methods as a promising alternative to other dehydrogenation reactions that employ multiple steps or require undesirable stoichiometric oxidants.19 On the other hand, the practicality of the methods, especially for large scale applications, would benefit from lower catalyst loading. Additional synthetic opportunities have been realized through the use of Pd-catalyzed α,β-dehydrogenations of carbonyl compounds in tandem reactions, representative examples of which include dehydrogenation/Heck-type reactions of acyclic aldehydes and ketones25, chromanones,26 and cyclic carbonyl compounds.27
3.2 Origin of Chemoselectivity in the Dehydrogenation of Cyclohexanones
Mechanistic studies were carried out to probe the basis for the different chemoselectivity of the Pd(TFA)2/2-Me2Npy and Pd(DMSO)2(TFA)2 catalyst systems for phenol and enone product formation, respectively. Reaction time-course plots revealed notable differences between the two catalysts in their dehydrogenation of 4-tert-butylcyclohexanone (Scheme 15). The time course data were fit to a simple A→B→C kinetic model in order to compare the relative rates of the two steps with the two different catalyst systems.22 With the Pd(TFA)2/2-NMe2py catalyst, the first step is almost two-fold slower than the second step (k1/k2 = 0.61). The reaction also exhibited a significant non-zero concentration of cyclohexenone, however, arising from a kinetic burst of enone formation (see further discussion below). In contrast, the Pd(DMSO)2(TFA)2 catalyst exhibits a rate for the first dehydrogenation step that is 33-fold faster than the second step.
Scheme 15.

Comparison of Kinetic Profiles of Pd(TFA)2/2-Me2Npy/p-TsOH (Catalyst 1) and Pd(DMSO)2(TFA)2 (Catalyst 2) for Dehydrogenation of 4-tert-Butylcyclohexanone
Kinetic studies of the Pd(DMSO)2(TFA)2-catalyzed reaction shed light on several important features of the reaction and highlighted the role of the catalytic DMSO ligand in controlling the reaction selectivity.28 The cyclohexanone-to-cyclohexenone step exhibited only minor rate dependence on DMSO (Scheme 16A); however, high product yield was obtained only in the presence of DMSO. Rapid formation of Pd black and catalyst deactivation occurred in the absence of DMSO. These features contrast those associated with the cyclohexenone-to-phenol step. Studies of the second step required higher catalyst loading and substrate concentration, owing to the slow rate of this step with the Pd(DMSO)2(TFA)2 catalyst. Analysis of the effect of DMSO on this reaction revealed an induction period when DMSO was present, and DMSO had a significant inhibitory effect on initial rate of the reaction (Scheme 16B). The length of the induction period increased at higher DMSO concentration. These and other mechanistic data revealed that dehydrogenation of cyclohexanone to cyclohexenone is promoted by a homogeneous Pd catalyst, supported by the DMSO ligand, whereas the cyclohexenone-to-phenol step involves nanoparticles that form more rapidly at lower DMSO concentration. These collective observations indicated that the active catalyst for the second step consists of soluble Pd nanoparticles that form under the reaction conditions. The inhibitory effect of DMSO on phenol formation in EtOAc as the solvent was explained by its ability to stabilize homogeneous Pd and hinder formation of Pd nanoparticles.
Scheme 16.

DMSO Dependence on Dehydrogenations with Pd(DMSO)2(TFA)2. A. Cyclohexanone to cyclohexenone. B. Cyclohexenone to phenol
A subsequent mechanistic investigation of the Pd(TFA)2/2-NMe2py/p-TsOH catalyst led to complementary observations about the speciation of the active Pd catalysts involved, but in this case controlled formation of Pd nanoparticles under the reaction conditions leads to formation of the phenol product.29 Time course data showing a burst in cyclohexenone formation corresponding to one turnover of PdII, followed by steady-state kinetics (cf. Scheme 15), implicated a change in the nature of the active catalyst. When cyclohexenone was used as the substrate, rather than cyclohexanone, an induction period was observed for phenol formation. Several classical tests to distinguish between homogeneous and heterogeneous catalysis were conducted. Hot filtration tests, as well as testing commercial Pd/C and Pd/Al2O3, eliminated heterogeneous Pd as the active species for cyclohexenone or phenol formation. Hg-metal30 and poly(4-vinylpyridine) poisoning tests were consistent with rapid cyclohexenone formation with a molecular PdII catalyst. In situ evolution of the molecular catalyst into Pd nanoparticles, however, led to quenching of catalytic activity by these poisons. Transmission electron microscopy (an ex situ technique) and dynamic light scattering (DLS; an in situ technique) data provided evidence for the formation of Pd nanoparticles during catalytic turnover of cyclohexanone. Collectively, the results indicate that the combination of Pd(TFA)2 with 2-NMe2py and p-TsOH in DMSO31 promotes very rapid monodehydrogenation of cyclohexanone, but the catalyst immediately begins to aggregate into Pd nanoparticles after a single turnover, and the Pd nanoparticles are responsible for the steady state dehydrogenation activity that leads to phenol formation.
These studies of these two catalyst systems demonstrate that molecular and nanoparticle Pd catalysts exhibit different chemoselectivity (Scheme 17). The former promote rapid monodehydrogenation of cyclohexanones to afford cyclohexenones, whereas the latter exhibit moderate activity for the dehydrogenation of both cyclohexanones and cyclohexenones, resulting in the net conversion of cyclohexanones to phenols. Thus, control over the Pd catalyst speciation provides the basis for chemoselective reactivity. This approach to selectivity control in catalytic reactions has limited precedent, but it represents an exciting opportunity for future developments in the field of transitional metal catalysis.32,33
Scheme 17.

Activity of Palladium Species in the Catalytic Dehydrogenation of Cyclohexanone and Cyclohexenone
3.3 Dehydrogenation of Acyclic Carbonyl Compounds and Other More-Challenging Substrates
The development of the Pd-catalyzed aerobic monodehydrogenation reaction of cylohexanones prompted us to investigate the dehydrogenation of acyclic substrates. This application would significantly expand the scope of this mode of reactivity and include other carbonyl compounds such as aldehydes, esters and amides. Analogous to cyclohexanone monodehydrogenations, classic reactions include the use of stoichiometric selenium-based reagents, IBX or DDQ.19 Prior examples of Pd-catalyzed dehydrogenation of aldehydes utilized the formation of enamine intermediates with a catalytic amine and Pd(OAc)2 (Scheme 18).34,35 The scope was limited to hydrocinnamaldehyde derivatives, as analogous ketones were inactive.
Scheme 18.

Pd-Catalyzed Dehydrogenations of Enamines of Hydrocinnamaldehydes
We targeted direct α-C–H activation by PdII salts, and systematic evaluation of PdII sources and ligands led to the identification of an aerobic Pd(TFA)2/4,5-diazafluorenone (DAF) catalyst in DMSO.36 This catalyst system enabled the efficient dehydrogenation of hydrocinnamaldehydes, as well as β-arylketone derivatives (Scheme 19). Dehydrogenation of a 1,4-dicarbonyl compound also proceeded in good yield. Acyclic ketones lacking an aryl group in the β-position undergo further dehydrogenation to the dienone product in poor yield and are not good substrates for this reaction. Esters and amides are also ineffective substrates, presumably owing to the lower acidity of the alpha C–H bond proton.
Scheme 19.

Aerobic Pd-catalyzed dehydrogenations of acyclic aldehydes and ketones developed by Stahl and co-workers
A similar Pd(OAc)2/4,5-diazafluorenone catalyst system consisting of was reported simultaneously by Huang et al., using catalytic K2CO3 as a base and DMF as a solvent.37 Similar substrate scope was observed for acyclic carbonyl compounds. In addition, it was shown that the dehydrogenation of β-arylbutanone derivatives can take place the absence of the diazafluorenone ligand, albeit at 110 °C (Scheme 20).
Scheme 20.

Acyclic substrate dehydrogenations developed by Huang and co-workers
Cyclopentanones are more challenging substrates for dehydrogenation relative to cyclohexenones, but variations of the above methods have proven effective in a series of useful synthetic applications (Schemes 21 and 22). Our group demonstrated dehydrogenation of a bicyclic cyclopentanone precursor to a metatrobic glutamate receptor (mGluR) antagonist.38 The Pd(TFA)2/DAF catalyst system proved to be the most effective, enabling dehydrogenation of the ketone in 79% yield (Scheme 21A). Our previous Pd-based catalyst systems were less effective (≤37% yield). Magauer and co-workers employed the same method in gram-scale dehydrogenation of an indanone substrate in their route to the anticancer natural product chartarin (Scheme 21B).39 And, Minnaard and co-workers employed Pd(TFA)2/DAF-catalyzed aerobic dehydrogenation of substituted cyclopentanone in synthetic routes to the sesquiterpenes herbertenediol, enokipodin A and enokipodin B (Scheme 22).40
Scheme 21.

Aerobic Dehydrogenation of Substituted Cyclopentanone Precursors to Phamaceutical and Natural Product Targets by Stahl (A) and Magauer (B)
Scheme 22.

Sesquiterpenes Synthesized Using Pd(TFA)2/DAF-Catalyzed Aerobic Dehydrogenation by Minnaard and Coworkers
While these results are promising, new catalyst systems will be needed to achieve selective monodehydrogenation of carbonyl compounds beyond the substrates in Schemes 18–20. Key challenges include unsymmetrical ketones, which can form two different enone products, and esters and amides and related substrates, which are less reactive. Important complementary developments have been reported recently by Newhouse and coworkers, who described the one-pot dehydrogenation of esters, nitriles and amides using Pd catalysts (Scheme 23).41 Use of bulky, strong Li bases in the presence of ZnCl2 generate zinc enolates or cyanoalkylzinc species. Subsequent addition of an allyl-PdII catalyst precursor, together with allyl acetate or pivalate as the oxidant, results in effective dehydrogenation of these challenging substrates. The conditions are not compatible with of O2 as an oxidant.
Scheme 23.

Select Scope for the Pd-Catalyzed Direct Dehydrogenation of Unactivated Esters, Nitriles and Tertiary Amides
Using TMPLi (TMPH = 2,2,6,6-tetramethylpiperidine) as the base, a number of α,β-unsaturated esters and nitriles were obtained in moderate to good yields, including acyclic substrates, as well as lactones (Scheme 23).42 Dehydrogenation of amides required further optimization, and a bulkier secondary amide lithium base, LiCyan (CyanH = N-Cyclohexyl-2,6-diisoproylaniline), was used.43 Acyclic amides and lactams were dehydrogenated in good yields, including in the presence of free –OH and –NH functional groups. Only tertiary amide substrates were reactive, even in the presence of secondary amides.
4. Dehydrogenative Coupling of Cyclohexanones/Cyclohexenones and Amines to Afford Anilines
The condensation of amines onto cyclohexanones and cyclohexenones affords imines and/or enamines that can undergo dehydrogenation to afford substituted anilines (Schemes 24). Our preliminary attempts to implement this strategy were unsuccesful due to competing phenol formation; however, the groups of Deng/Li44 and Yoshikai45 developed aerobic Pd-catalyzed methods for the synthesis of secondary and tertiary anilines, as well as aromatic sulfonamides44c via this approach. The catalyst developed by Yoshikai and co-workers was primarily active when primary amines or electron-rich primary anilines were used as substrates in combination with cyclohexanones (Scheme 25A). The Deng/Li catalyst system was employed with primary anilines as nucleophilic partners (Scheme 25B). The authors demonstrated a much broader amine scope, including secondary and aliphatic amines, was successful when cyclohexenones were used as substrates. In addition, Lemaire and co-workers showed that various cyclohexanones could be used as neat substrates to achieve dehydrogenative coupling with amines using Pd/C as the catalyst (Scheme 25C).46 This method was typically more efficient and selective with 1-octene as a hydrogen acceptor, rather than O2, although tetralones underwent coupling in the absence of an acceptor to afford 1-aminonaphthalenes. Collectively, these results represent a valuable application of oxidative dehydrogenation catalysis.
Scheme 24.

General Strategy for the Synthesis of Anilines via Pd-Catalyzed Dehydrogenation
Scheme 25.

Substituted Anilines from Cyclohexanone via Pd-Catalyzed Dehydrogenation
Primary anilines represent an appealing targets because they are often difficult to access via the traditional C-N coupling methods;47,48 however, synthesis of primary anilines via dehydrogenative coupling is challenging because condensation of the ketone and ammonia to form the primary imine via is challenging. A classic method for the synthesis of primary anilines is the Semmler-Wolff reaction,49–51 which involves high-temperature dehydration of cyclohexenone oximes promoted by strong acid (Scheme 26A). The harsh conditions (e.g., refluxing Ac2O/AcOH with anhydrous HCl) limit its synthetic utility, and the reactions are complicated by competing formation of Beckmann rearrangement products and acylation of the aniline products.52 Inspired by recent publications that make use of N–O bonds as an internal oxidant,53 we developed a Pd-catalyzed variant of the Semmler-Wolff reaction, in which O-acyloximes of cyclohexenone and tetralone derivatives undergo conversion to the corresponding primary aniline, with the N–O bond of the substrate serving as the oxidant (Scheme 26B).54 Oxidative addition of N–O bond to Pd0 generates an imino-PdII intermediate, which was independently prepared at low temperature and characterized by X-ray crystallography. This species was then shown to generate the desired aniline at elevated temperatures, presumably via tautomerization, conversion to an α-palladated imine species, and β-H elimination. Tautomerization of the resulting dienimine affords the desired primary aniline (Scheme 26B).
Scheme 26.

Primary Aniline Synthesis via Classic Semmler-Wolff Reaction (A) and the Recently Developed Pd-Catalyzed Variant (B)
The optimal oxygen activating group was pivalate and a catalyst system employing Pd(OAc)2 and PCy3 as a ligand was developed. The addition of catalytic pivalic acid (PivOH, 30 mol %) was found to boost yields as well. With this catalyst system, a broad range of primary anilines and 1-aminonaphthalenes were synthesized in moderate to good yields (Scheme 27). In the synthesis of two 1-aminonaphthalene precursors to bioactive molecules, the new protocol led to higher yields than classic Semmler-Wolff conditions (for HKI 0231B)50d or an alternative nitration/nitro-reduction route (for norallonitidine)55. The Pd-catalyzed method also proved effective for the preparation of meta-substituted anilines and a tetrasubstituted anilines, although the latter was obtained in relatively low yield (46%) and required higher temperature (120 °C). Although this method is not aerobic, it represents a useful route to anilines from partially saturated precursors and exhibits many features in common with the dehydrogenation methods described above.
Scheme 27.

Pd-Catalyzed Semmler-Wolff Reaction of Cyclohexenone Oxime Pivalates
5. Pd-Catalyzed Dehydrogenation of Cyclohexenes to Substituted Arenes
The majority of dehydrogenation reactions utilize cyclohexanone or cyclohexenone derivatives as starting materials to access phenols, aryl ethers and anilines. In an effort to generalize the aerobic dehydrogenative strategy for the preparation of substituted aromatics, we targeted the preparation of arenes that lack heteroatom substituents by using cyclohexenes as precursors. Cyclohexenes are readily available substrates, most notably via Diels-Alder cycloadditions. The dehydrogenation of cyclohexenes could proceed via a PdII-mediated allylic C–H activation, followed by β-H elimination and aerobic regeneration of Pd0 (Scheme 28). Pd-mediated disproportionation of cyclohexene into cyclohexane and benzene was precedented and represented a possible undesirable side-reaction.56,57a,b,f
Scheme 28.

Mechanistic Model for the Pd-Catalyzed Dehydrogenation of Cyclohexenes and the Disproportionation Reaction
Precedents for cyclohexene aromatization were primarily limited to the parent cyclohexene or related simple derivatives;57 a synthetically useful example was demonstrated by Oestreich and co-workers (Scheme 29).58 Aerobic dehydrogenation of substituted cyclohexenes was observed in the study of oxidative C–C coupling reactions with indole substrates, although the yields were typically rather modest (36–55%, 71% for one example) with 10 mol % Pd catalyst.
Scheme 29.

Aerobic Pd-Catalyzed Dehydrogenation of Indole Substrates Bearing Cyclohexenes Reported by Oestreich and Co-workers
Using 3-cyclohexene-1-carboxylic acid as a substrate, we optimized a catalyst system that consists of Pd(TFA)2 and sodium anthraquinone-2-sulfonate (AMS) as a co-catalyst. Although the precise role of AMS is not known, it enabled efficient aerobic turnover and avoided disproportionation of the cyclohexene.59,60 This catalyst showed substantially increased substrate scope and higher yields than previously reported dehydrogenation catalysts. A variety of functional groups are tolerated under the reaction conditions (Scheme 30). This catalyst system showed lower activity with more sterically hindered substrates. An alternative catalyst, generated in situ from Pd(OAc)2 and p-TsOH, allowed the synthesis of tri- and tetrasubstituted aromatic rings. We speculated that this more electrophilic catalyst should be more active for generation of the PdII-allyl intermediate. This catalyst also show some activity for catalytic dehydrogenations initiated by benzylic C–H activation (Scheme 30B). Several limitations were identified in this study. For example, substrates with aryl bromides resulted in low yields, exocyclic alkenes underwent isomerization prior to dehydrogenation in some cases, and substrates with nitrogen-and sulfur-containing heterocycles resulted in diminished yields.
Scheme 30.

Select Scope for the Aerobic Pd-Catalyzed Dehydrogenation of Cyclohexenes
The utility of this aromatization strategy was highlighted in a 3-step synthetic route toward phthalimide-based modulators of TRPA1, an ion channel that has been implicated in pain response (Scheme 31). The methyl sorbate and N-methylmaleimide starting materials are very inexpensive, and the carbocyclic core with the desired functionality may be accessed in a single step via a Diels-Alder cycloaddition. Pd-catalyzed aerobic dehydrogenation of the resulting cyclohexene provide efficient access to the desired phthalimide product. The previously reported route61 starts from 3-nitrophthalimide, featured a series of functional group interconversions, including a Suzuki cross-coupling step and Ru-catalyzed oxidative cleavage of an alkene, to access the desired precursor in 5 steps (Scheme 31B). This comparison, similar to the synthesis of a 3,5-diarylphenol in Scheme 6,5 highlights the strategic opportunity and synthetic appeal of dehydrogenation methods relative to metal-mediated cross-coupling methods to access substituted aromatic compounds.
Scheme 31.

Routes to TRPA1 Modulators Intermediates. Diels-Alder/Pd-Catalyzed Dehydrogenation Sequence (A) and Previous Route Starting From 3-Nitrophthalimide (B)
Recently, Riveira, La-Venia and Mischneapplied a modified version of this catalyst system in the last step of the synthesis of benzosimulin, an alkaloid with cytotoxic and antiplatelet aggregation activity.62 The natural product was obtained in relatively low yield (34%), but this approach proved superior to more classical dehydrogenation methods, using chloranil or Pd/C (Scheme 32). This creative application highlights both the utility and the need for continued catalyst development efforts.
Scheme 32.

Synthesis of Benzosimuline via Pd-Catalyzed Dehydrogenation
Deng and co-workers demonstrated a compelling cyclohexanone condensation route to 3-aryl indoles that proceeds via aerobic dehydrogenation of cyclohexene intermediate (Scheme 33).63 The cyclohexene intermediate was independently synthesized and shown to convert into the aryl-substituted indole product. The cyclohexanones represent arene surrogates, similar to the aryl ether and aniline synthetic routes described above. The catalyst system, consisting of Pd(TFA)2 and bis[2-(diphenylphophino)phenyl]ether (DPEPhos), was effective for the formation of a number of different 3-aryl indoles utilizing substituted indoles and 3- and 4-substituted cyclohexanones.
Scheme 33.

3-Aryl Indole Synthesis via Aerobic Pd-Dehydrogenation
6. Conclusions
The efficient and selective synthesis of aromatic molecules with non-conventional substitution patterns remains a challenging and active area of research. The Pd-catalyzed aerobic dehydrogenation methods described herein provide a compelling approach for the conversion of readily accessible cyclohexanone and cyclohexene derivatives into substituted phenols, anilines and arenes. The (partially) saturated substrates for these methods are often readily assembled or derivatized by straightforward synthetic methods, and thereby provide a means to access aromatic compounds with substitution patterns not readily available by existing cross-coupling or aromatic C–H functionalization methods. Prominent examples include meta-substituted phenols and anilines and polysubstituted arene derivatives, which can serve as versatile building blocks for the preparation of more-complex molecules. Furthermore, applications of these methods to the preparation of various pharmaceutical and natural product intermediates and targets show that Pd-catalyzed aerobic dehydrogenation reaction can provide compelling solutions to synthetic challenges.
These catalytic reactions exploit well established reaction steps in homogeneous Pd catalysis, including C–H activation to afford PdII-enolates and PdII-allyl complexes, β-hydride elimination to introduce sites of unsaturation in the organic molecules, and oxidation of Pd0 by O2 to regenerate the active PdII catalyst. Nevertheless, the success of the methods has relied on the discovery of new catalyst systems, including new ligands and co-catalytic additives to effect the desired substrate oxidations and achieve efficient aerobic catalytic turnover of Pd. Mechanistic studies further show that complementary opportunities are available from homogeneous, nanoparticle and heterogeneous catalysis. For example, our results show that homogeneous and nanoparticle catalysts show different selectivity for the conversion of cyclohexanones to cyclohexenones versus phenols.28,29 The work of Lemaire,16,46 together with non-oxidative catalytic applications by Liu and Li,64 demonstrates the utility of heterogeneous Pd catalysts in related applications. The development of new catalyst systems in future studies will undoubtedly play an important role in continued expansion of this field.
Acknowledgments
We thank the numerous members of our research group who made contributions to the dehydrogenation reactions described herein. Financial support for our research was provided by by the NIH (R01-GM100143) and the Dow Chemical Company.
References
- 1.de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 2.(a) Yu J-Q, Shi Z, editors. C–H Activation. Topics in Current Chemistry. Springer; New York: 2010. [PubMed] [Google Scholar]; (b) Ribas X, editor. C–H and C–X Bonds Functionalization: Transition Metal Mediation. RSC Publishing; London: 2013. [Google Scholar]
- 3.For select reviews see: Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Ackermann L. Chem Rev. 2011;111:1315. doi: 10.1021/cr100412j.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem, Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666.Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788. doi: 10.1021/ar200185g.
- 4.For context, see the following reviews: Campbell AN, Stahl SS. Acc Chem Res. 2012;45:851–863. doi: 10.1021/ar2002045.Stahl SS. Angew Chem Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.
- 5.Izawa Y, Pun D, Stahl SS. Science. 2011;333:209. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Girard SA, Huang H, Zhou F, Deng GJ, Li CJ. Org Chem Front. 2015;2:279. [Google Scholar]
- 7.(a) Walker D, Hiebert JD. Chem Rev. 1967;67:153. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]; (b) Buckle DR. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Inc; New York: 2010. [Google Scholar]
- 8.Muzart J. Eur J Org Chem. 2010:3779. [Google Scholar]
- 9.(a) Yi CS, Lee DW. Organometallics. 2009;28:947. doi: 10.1021/om8010883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Muzart J, Pete JP. J Mol Catal. 1982;15:373. [Google Scholar]; (c) Wenzel TT. J Chem Soc Chem Commun. 1989:932. [Google Scholar]
- 10.Fu PP, Harvey RG. Chem Rev. 1978;78:317. [Google Scholar]
- 11.Heitman LH, Narlawar R, de Vries H, Willemsen MN, Wolfram D, Brussee J, IJzerman AP. J Med Chem. 2009;52:2036. doi: 10.1021/jm801561h. [DOI] [PubMed] [Google Scholar]
- 12.Izawa Y, Zheng C, Stahl SS. Angew Chem, Int Ed. 2013;52:3672. doi: 10.1002/anie.201209457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JH, Park CY, Kim MJ, Kim MU, Kim YJ, Kim GH, Park CP. Org Process Res Dev. 2015;19:812. [Google Scholar]
- 14.Zhang J, Jiang Q, Yang D, Zhao X, Dong Y, Liu R. Chem Sci. 2015;6:4674. doi: 10.1039/c5sc01044f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon MO, Girard SA, Li CJ. Angew Chem Int Ed. 2012;51:7537. doi: 10.1002/anie.201200698. [DOI] [PubMed] [Google Scholar]
- 16.Sutter M, Sotto N, Raoul Y, Métay E, Lemaire M. Green Chem. 2013;15:347. [Google Scholar]
- 17.Kim D, Min M, Hong S. Chem Commun. 2013;49:4021. doi: 10.1039/c3cc41296b. [DOI] [PubMed] [Google Scholar]
- 18.(a) Theissen RJ. J Org Chem. 1971;36:752. [Google Scholar]; (b) Park YW, Oh HH. Bull Korean Chem Soc. 1997;18:1123. [Google Scholar]; (c) Tokunaga M, Harada S, Iwasawa T, Obora Y, Tsuji Y. Tetrahedron Lett. 2007;48:6860. [Google Scholar]
- 19.For a review on oxidation methods adjacent to carbonyl compounds see: Stahl SS, Diao T. In: Comprehensive Organic Synthesis II. 2. Knochel P, Molander GA, editors. Elsevier; Amsterdam: 2014. pp. 178–212.
- 20.(a) Nicolaou KC, Zhong YL, Baran PS. J Am Chem Soc. 2000;122:7596. [Google Scholar]; (b) Nicolaou KC, Montagnon T, Baran PS, Zhong YL. J Am Chem Soc. 2002;124:2245. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Montagnon T, Baran PS. Angew Chem, Int Ed. 2002;41:993. doi: 10.1002/1521-3773(20020315)41:6<993::aid-anie993>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Angew Chem, Int Ed. 2002;41:996. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 21.(a) Ito Y, Hirao T, Saegusa T. J Org Chem. 1978;43:1011. [Google Scholar]; (b) Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423. [Google Scholar]; (c) Yu JQ, Wu HC, Corey EJ. Org Lett. 2005;7:1415. doi: 10.1021/ol050284y. [DOI] [PubMed] [Google Scholar]; (d) Lu Y, Nguyen PL, Lévaray N, Lebel H. J Org Chem. 2013;78:776. doi: 10.1021/jo302465v. [DOI] [PubMed] [Google Scholar]
- 22.Diao T, Stahl SS. J Am Chem Soc. 2011;133:14566. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakajima R, Ogino T, Yokoshima S, Fukuyama T. J Am Chem Soc. 2010;132:1236. doi: 10.1021/ja9103233. [DOI] [PubMed] [Google Scholar]
- 24.Trost BM, Dong G, Vance JA. Chem—Eur J. 2010;16:6265. doi: 10.1002/chem.200903356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Zhou J, Wu G, Zhang M, Jie X, Su W. Chem—Eur J. 2012;18:8032. doi: 10.1002/chem.201200829. [DOI] [PubMed] [Google Scholar]; (b) Shang Y, Jie X, Zhou J, Hu P, Huang S, Su W. Angew Chem, Int Ed. 2013;52:1299. doi: 10.1002/anie.201208627. [DOI] [PubMed] [Google Scholar]
- 26.Moon Y, Kwon D, Hong S. Angew Chem, Int Ed. 2012;51:11333. doi: 10.1002/anie.201206610. [DOI] [PubMed] [Google Scholar]
- 27.(a) Leskinen MV, Yip KT, Valkonen A, Pihko PM. J Am Chem Soc. 2012;134:5750. doi: 10.1021/ja300684r. [DOI] [PubMed] [Google Scholar]; (b) Yip KT, Nimje RY, Leskinen MV, Pihko PM. Chem—Eur J. 2012;18:12590. doi: 10.1002/chem.201201988. [DOI] [PubMed] [Google Scholar]; (c) Nimje RY, Leskinen MV, Pihko PM. Angew Chem, Int Ed. 2013;52:4818. doi: 10.1002/anie.201300833. [DOI] [PubMed] [Google Scholar]; (d) Huang Z, Dong G. J Am Chem Soc. 2013;135:17747. doi: 10.1021/ja410389a. [DOI] [PubMed] [Google Scholar]; (e) Gandeepan P, Rajamalli P, Cheng CH. ACS Catal. 2014;4:4485. [Google Scholar]; (f) Gigant N, Bäckvall JE. Chem—Eur J. 2014;20:5890. doi: 10.1002/chem.201402063. [DOI] [PubMed] [Google Scholar]; (g) Huang Z, Sam QP, Dong G. Chem Sci. 2015;6:5491. doi: 10.1039/c5sc01636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diao T, Pun D, Stahl SS. J Am Chem Soc. 2013;135:8205. doi: 10.1021/ja4031648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pun D, Diao T, Stahl SS. J Am Chem Soc. 2013;135:8213. doi: 10.1021/ja403165u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mercury (Hg) poisoning test can be problematic with homogeneous Pd-catalyzed reactions as Pd0 intermediates can react with Hg and poison the homogeneous catalyst, thereby having the opposite intended effect. This possibility was addressed in the original study; see ref. 29 for details.
- 31.For reactions in which DMSO was shown to have a special ability to stabilize Pd nanoparticles, see: van Bentham RATM, Hiemstra H, Michels JJ, Speckamp WN. J Chem Soc, Chem Commun. 1994:357.van Benthem RATM, Hiemstra H, van Leeuwen PWNM, Geus JW, Speckamp WN. Angew Chem, Int Ed. 1995;34:457.
- 32.Mifsud M, Parkhomenko KV, Arends IWCE, Sheldon RA. Tetrahedron. 2010;66:1040. [Google Scholar]
- 33.Richter C, Schaepe K, Glorius F, Ravoo BJ. Chem Commun. 2014;50:3204. doi: 10.1039/c4cc00654b. [DOI] [PubMed] [Google Scholar]
- 34.(a) Zhu J, Liu J, Ma RQ, Xie H, Li J, Jiang H, Wang W. Adv Synth Catal. 2009;351:1229–1232. [Google Scholar]; (b) Liu J, Zhu J, Jiang H, Wang W, Li J. Chem Asian J. 2009;4:1712. doi: 10.1002/asia.200900238. [DOI] [PubMed] [Google Scholar]
- 35.For non-aerobic enamine dehydrogenation see: Hayashi Y, Itoh T, Ishikawa H. Angew Chem, Int Ed. 2011;50:3920. doi: 10.1002/anie.201006885.Zhang SL, Xie HX, Zhu J, Li H, Zhang XS, Li J, Wang W. Nat Commun. 2011;2:211. doi: 10.1038/ncomms1214.
- 36.Diao T, Wadzinski TJ, Stahl SS. Chem Sci. 2012;3:887. doi: 10.1039/C1SC00724F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao W, He Z, Qian Y, Zhao J, Huang Y. Chem Sci. 2012;3:883. [Google Scholar]
- 38.Zhang F, Moher ED, Zhang TY. Tetrahedron Lett. 2007;48:3277. [Google Scholar]
- 39.Unzner TA, Grossmann AS, Magauer T. Angew Chem, Int Ed. 2016;55:9763. doi: 10.1002/anie.201605071. [DOI] [PubMed] [Google Scholar]
- 40.Buter J, Moezelaar R, Minnaard AJ. Org Biomol Chem. 2014;12:5883. doi: 10.1039/c4ob01085j. [DOI] [PubMed] [Google Scholar]
- 41.Turlik A, Chen Y, Newhouse TR. Synlett. 2016;27:331. [Google Scholar]
- 42.Chen Y, Romaire JP, Hewhouse TR. J Am Chem Soc. 2015;137:5875. doi: 10.1021/jacs.5b02243. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Turlik A, Newhouse TR. J Am Chem Soc. 2016;138:1166. doi: 10.1021/jacs.5b12924. [DOI] [PubMed] [Google Scholar]
- 44.(a) Girard SA, Hu X, Knauber T, Zhou F, Simon MO, Deng GJ, Li CJ. Org Lett. 2012;14:5606. doi: 10.1021/ol3027279. [DOI] [PubMed] [Google Scholar]; (b) Xie Y, Liu S, Liu Y, Wen Y, Deng GJ. Org Lett. 2012;14:1692. doi: 10.1021/ol3002442. [DOI] [PubMed] [Google Scholar]; (c) Cao X, Bai Y, Xie Y, Deng GJ. J Mol Catal A: Chem. 2014;383–384:94. [Google Scholar]
- 45.Hajra A, Wei Y, Yoshikai N. Org Lett. 2012;14:5488. doi: 10.1021/ol302568b. [DOI] [PubMed] [Google Scholar]
- 46.Sutter M, Duclos MC, Guicheret B, Raoul Y, Métay E, Lemaire M. ACS Sustainable Chem Eng. 2013;1:1463. [Google Scholar]
- 47.For examples, see: Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:10028. doi: 10.1021/ja064005t.Xia N, Taillefer M. Angew Chem Int Ed. 2009;48:337. doi: 10.1002/anie.200802569.Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049. doi: 10.1021/ja903049z.Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. Angew Chem Int Ed. 2010;49:4071. doi: 10.1002/anie.201000526.Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J.Ji PJ, Atherton JH, Page MI. J Org Chem. 2012;77:7471. doi: 10.1021/jo301204t.
- 48.For representative coupling methods that use ammonia surrogates, see: Wolfe JP, Åhman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron Lett. 1997;38:6367.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575. doi: 10.1021/jo990408i.Shen QL, Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:6586. doi: 10.1021/ja077074w.
- 49.(a) Semmler W. Ber Dtsch Chem Ges. 1892;25:3352. [Google Scholar]; (b) Wolff L. Justus Liebigs Ann Chem. 1902;322:351. [Google Scholar]
- 50.For more recently developed conditions, see: Newman MS, Hung WM. J Org Chem. 1973;38:4073.Tamura Y, Yoshimoto Y, Sakai K, Kita Y. Synthesis. 1980:483.Tamura Y, Yoshimoto Y, Sakai K, Haruta J, Kita Y. Synthesis. 1980:887.Scopton A, Kelly TR. J Org Chem. 2005;70:10004. doi: 10.1021/jo051762l.
- 51.Heterogeneous Pd catalysts (Pd/C) can promote the Semmler-Wolff reaction; however, very high temperatures (e.g., 200 °C) are required. Horning EC, Horning MG. J Am Chem Soc. 1947;27:1907.Matsumoto M, Tomizuka J, Suzuki M. Synth Commun. 1994;24:1441.Matsumoto M, Tomizuka J. ITE Letters. 2000;1:127.
- 52.Kelly TR, Chandrakumar NS, Saha JK. J Org Chem. 1989;54:980. [Google Scholar]
- 53.For a highlight, see: Patureau FW, Glorius F. Angew Chem, Int Ed. 2011;50:1977. doi: 10.1002/anie.201007241.
- 54.Hong WP, Iosub AV, Stahl SS. J Am Chem Soc. 2013;135:13664. doi: 10.1021/ja4073172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.(a) Geen GR, Mann IS, Mullane MV, McKillop A. J Chem Soc Perkin Trans 1. 1996:1647. [Google Scholar]; (b) Stermitz FR, Gillespie JP, Amoros LG, Romero R, Stermitz TA, Larson KA, Earl S, Ogg JE. J Med Chem. 1975;18:708. doi: 10.1021/jm00241a014. [DOI] [PubMed] [Google Scholar]
- 56.Wolfe S, Campbell PGC. J Am Chem Soc. 1971;93:1499. [Google Scholar]
- 57.(a) Trost BM, Metzner PJ. J Am Chem Soc. 1980;102:3572. [Google Scholar]; (b) Sheldon RA, Sobczak JM. J Mol Catal. 1991;68:1. [Google Scholar]; (c) Neumann R, Debruyn M. Adv Synth Catal. 2007;349:1624. [Google Scholar]; (d) Williams TJ, Caffyn AJM, Hazari N, Oblad PF, Labinger JA, Bercaw JE. J Am Chem Soc. 2008;130:2418. doi: 10.1021/ja076740q. [DOI] [PubMed] [Google Scholar]; (e) Bercaw JE, Hazari N, Labinger JA, Oblad PF. Angew Chem, Int Ed. 2008;47:9941. doi: 10.1002/anie.200804455. [DOI] [PubMed] [Google Scholar]; (f) Bercaw JE, Hazari N, Labinger JA. J Org Chem. 2008;73:8654. doi: 10.1021/jo8016296. [DOI] [PubMed] [Google Scholar]; (g) Jaekel C, Horillo-Martinez P, Virolleaud MA. ChemCatChem. 2010;2:175. [Google Scholar]
- 58.Kandukuri SR, Oestreich M. J Org Chem. 2012;77:8750. doi: 10.1021/jo301088f. [DOI] [PubMed] [Google Scholar]
- 59.Iosub AV, Stahl SS. J Am Chem Soc. 2015;137:3454. doi: 10.1021/ja512770u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.AMS was previously used as a co-catalyst by Sheldon and Sobczak for parent cyclohexene dehydrogenation. See ref. 57b.
- 61.Muthuppal-Niappan M, Kumar S, Thomas A, Khairatkar-Joshi N, Mukhopadhyay I. 09/118596. World Patent WO. 2009 Oct 1;
- 62.Riveira MJ, La-Venia A, Mischne MP. J Org Chem. 2016;81:7977. doi: 10.1021/acs.joc.6b01278. [DOI] [PubMed] [Google Scholar]
- 63.Chen S, Liao Y, Zhao F, Qi H, Liu S, Deng GJ. Org Lett. 2014;16:1618. doi: 10.1021/ol500231c. [DOI] [PubMed] [Google Scholar]
- 64.Chen Z, Zeng H, Girard SA, Wang F, Chen N, Li C-J. Angew Chem, Int Ed. 2015:14487. doi: 10.1002/anie.201506751. [DOI] [PubMed] [Google Scholar]