

Abstract
A scalable and versatile approach is reported to prepare high paclitaxel (PTX)-loading and low-systemic-toxicity nanoparticles via one-step ring-opening polymerization of a prodrug monomer consisting of PTX that is appended to a cyclic carbonate through a hydrolysable ester linker. Initiating this monomer from a hydrophilic macroinitiator results in an amphiphilic diblock copolymer that spontaneously self-assembles into well-defined nanoparticles with tunable size.
Keywords: Paclitaxel, Ring-opening polymerization, Prodrug, Nanoparticle, Anticancer
Graphical Abstract
Paclitaxel (PTX), a potent chemotherapeutic agent, has demonstrated significant activity against various solid tumours1. However, PTX is highly water insoluble, and consequently has poor bioavailability and significant systemic toxicity2. To address these challenges, two main strategies have been explored3–5. The first strategy involves physical loading of PTX into a delivery system6,7. However, it has been reported that the noncovalent encapsulation of PTX can result in premature release and undesirable systemic toxicity. For instance, hypersensitivity reactions, myelosuppression, neurotoxicity and nephrotoxicity have been caused by the use of a 1:1 blend of Cremophor EL® (polyethoxylated castor oil) and ethanol to dissolve PTX8,9. Another formulation, Abraxane, that is currently used in the clinic involves the physical encapsulation of PTX in human serum albumin nanoparticles, but questions remain about the stability of the drug in the albumin matrix upon intravenously injection10,11. The second approach involves covalent attachment of PTX to a hydrophilic polymer12–18. An example is Xyotax–that is currently in the clinical pipeline–in which PTX is covalently conjugated by an ester linkage to the γ-carboxylic acid side chains in poly-L-glutamic acid19,20. However, a limitation of most current approaches for the synthesis of synthetic polymer-drug conjugates is that they typically require multiple reaction steps that have a limited overall yield, and have limited control of the site and degree of drug-loading. New methods are hence needed to develop efficient PTX-loading formulations that can deposit a therapeutic dose of the drug in solid tumours. Ideally, these methods should: 1) be able to prepare formulations with high PTX-loading and water-solubility, but exhibit low-systemic-toxicity; 2) proceed with simple chemical procedures, allow easy purification and proceed with high yield; 3) enable convenient control over the physicochemical properties of the formulation, such as the size of the carrier and the release behavior of drug.
To address these limitations, we report a scalable and versatile approach to prepare highly water-soluble and low-systemic-toxicity nanoparticles with high paclitaxel (PTX)-loading, via a simple one-step organocatalyzed ring-opening polymerization (ROP) of a prodrug monomer consisting of PTX that is appended to a cyclic carbonate through a hydrolysable ester linker (Scheme 1). Initiating ROP of this PTX prodrug monomer from a poly(ethylene glycol) methyl ether (mPEG) macroinitiator results in an amphiphilic diblock copolymer that spontaneously self-assembles into well-defined nanoparticles with high PTX-loading and tunable size. Nanoparticles with a PTX-loading capacity of 50 wt% and a uniform size of ~90 nm were synthesized with a maximum solubility in buffer of 11.9 mg mL−1 PTX equivalent, which is ~5×104-fold higher than the aqueous solubility of free PTX. These highly water-soluble nanoparticles with high PTX-loading have a 9-fold higher maximum tolerated dose than free drug, and induce significant tumour regression after three doses in an orthoptopic murine cancer model of human triple-negative breast cancer. These data also raise important questions about the design of drug-loaded nanoparticles that are optimized for in vivo efficacy. Answers to these questions have implications broadly for the delivery of chemotherapeutics, and provide a road-map for future optimization of nanomedicines.
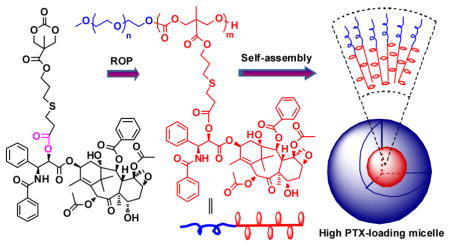
Scheme 1.

Synthetic route of polymer-PTX conjugates by ROP of a PTX-prodrug monomer from a mPEG macroinitiator, and a schematic illustration of self-assembly of the PEG-prodrug diblock copolymer into nanoparticles.
We chose a cyclic carbonate as the monomer group because it can undergo ROP to yield a biodegradable polycarbonate backbone21–23. A hydrolysable ester bond was employed as the linker as it can be hydrolyzed at physiologically relevant conditions and subsequently release PTX in free form24. To encapsulate the prodrug polymer in a long circulating carrier, mPEG was selected as the macroinitiator because the resulting diblock copolymer, consisting of PEG and the polymer prodrug, self-assembles into long circulating nanoparticles by virtue of PEG’s stealth–protein and cell evasive–properties. The detailed synthetic route of the polymer prodrug, and a schematic illustration of its self-assembly into PTX containing nanoparticles is shown in Scheme 1. The PTX prodrug monomer (Carb-PTX) was synthesized with a high yield of 80 wt% by a simple one-step esterification reaction between PTX and a carboxyl functionalized cyclic carbonate (Carb-COOH). Proton nuclear magnetic resonance (1H NMR) spectroscopy demonstrates that the cyclic carbonate is selectively linked to the C-2′-OH of PTX (Figure S2). Details of the synthesis and characterization of Carb-PTX are described in the Supporting Information.
We carried out organocatalyzed ROP of Carb-PTX by using mPEG (5kD) as the macroinitiator and N-(3,5-trifluoromethyl)phenyl-N′-cyclohexylthiourea (TU) in combination with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as co-catalysts. The kinetics of polymerization are linear on a semi-logarithmic kinetic plot (Figure 1a). Gel permeation chromatography (GPC) curves showed monomodal and symmetric elution peaks for mPEG-polyPTX that exhibited a clear shift to a higher molecular weight with increasing polymerization time (Figure 1a, inset). In comparison with mPEG, GPC elution curves of mPEG-polyPTX showed no visible residual mPEG peak, indicating quantitative initiation efficacy of the ROP from the mPEG macroinitiator. The DP of polyPTX could be easily adjusted by tuning the monomer/initiator molar feed ratio (Table 1, entries 1–2). As shown in Figure 1c, the PTX-loading of mPEG-polyPTX was tuned from 43 to 50 wt% by increasing the Carb-PTX/mPEG ratio from 6.0 to 10. To extend the range of PTX loading to lower values, polymer prodrugs were synthesized by copolymerizing Carb-PTX with trimethylene carbonate (TMC) (Table 1, entries 3–4). The drug-loading of the mPEG-poly(TMC-PTX) copolymers could be controlled from 15 to 24 wt% by adjusting the feed ratio of Carb-PTX and TMC. A linear semi-logarithmic kinetic plot was also observed for the Carb-PTX and TMC copolymerization (Figure 1b). The faster consumption of Carb-PTX indicated its higher reactivity compared to TMC, which suggests that gradient polymers may be formed with an enrichment of Carb-PTX units closer to PEG. Similar results have been reported for the copolymerization of TMC with other cyclic carbonate monomers25. The evolution of the molecular mass, as monitored by GPC, was also found to be linear for the copolymerization. It is worth mentioning that the proton signal of C-7′(CH)-OH in PTX remained unchanged during all polymerization reactions, indicating that this hydroxyl group did not participate in the ring-opening reaction of cyclic carbonate under these experimental conditions (Figure S3 and S4), which can be explained by its sterically hindered location26. Highly pure polymer prodrugs were obtained with a high yield of ~80 wt% by repeated precipitation from dichloromethane to diethyl ether and all Mw/Mn values were less than 1.15. Notably, mPEG-polyPTX with 50 wt% drug-loading had a water-solubility as high as 11.9 mg mL−1 PTX equivalent, which is ~5×104-fold higher than the aqueous solubility of free PTX (Figure 1d). Taken together, these results confirm that organocatalyzed ROP of Carb-PTX enables the facile synthesis of polymer-PTX prodrugs with quantitative polymerization initiation efficiency, highly tunable PTX-loading, and excellent water-solubility.
Figure 1.

(a) Semi-logarithmic kinetic plot for the ROP of Carb-PTX using TU and DBU as co-catalysts, [mPEG]0: DBU: TU: [Carb-PTX]0 = 1.0: 2.5: 2.5: 20. (b) Semi-logarithmic kinetic plot for the copolymerization of TMC and Carb-PTX using TU and DBU as co-catalysts, [mPEG]0: DBU: TU: TMC0: [Carb-PTX]0 = 1.0: 2.5: 2.5: 50: 5. The insets show representative GPC curves after a reaction time of 0 min (black), 30 min (blue) and 1 h (red). (c) Plot of drug-loading versus molar feed ratio of Carb-PTX/mPEG. (d) The maximal concentration of free PTX and conjugated PTX from mPEG-polyPTX8.3 in aqueous solution. (e) Plot of micelle size versus molar feed ratio of Carb-PTX/mPEG. (f) Representative cryo-TEM image of mPEG-poly(TMC-PTX1.4) (upper left), mPEG-poly(TMC-PTX2.7) (upper right), mPEG-polyPTX5.8 (lower left) and mPEG-polyPTX8.7 (lower right), respectively. Scale bar 200 nm.
Table 1.
Summary of synthesized polymer prodrugs [a]
| Entry | Molar feed ratioCarb-PTX: TMC: I | DPCarb-PTX: TMC | Yield% | Mng mol−1 | Mw/Mn |
|---|---|---|---|---|---|
| mPEG-polyPTX8.3 | 10: 0: 1 | 8.7: 0 | 81 | 14900 | 1.05 |
| mPEG-polyPTX5.8 | 6: 0: 1 | 5.8: 0 | 80 | 11600 | 1.08 |
| mPEG-poly(TMC-PTX2.7) | 3: 15: 1 | 2.7: 14.8 | 82 | 9600 | 1.12 |
| mPEG-poly(TMC-PTX1.3) | 2: 15: 1 | 1.4: 14.2 | 80 | 8000 | 1.10 |
I is mPEG (Mn of 5 kDa). The DP was calculated by 1H NMR in CDCl3. The Mn and Mw/Mn were obtained from GPC-MALLS in tetrahydrofuran. The polymerization was carried out in dichloromethane at room temperature using TU and DBU as co-catalysts.
The amphiphilic nature of these polymer prodrugs drives their self-assembly into spherical micelles in aqueous media. The critical micellization concentration (CMC) of polymer prodrugs was characterized using pyrene as a probe27. The CMCs of mPEG-polyPTX and mPEG-poly(TMC-PTX) slightly decreased from 18 to 5 μg mL−1 (2 to 0.3 μM) as the PTX content was varied from 15 to 50 wt%, and scaled inversely with PTX content (Figure S5). The micelle size was measured by dynamic light scattering (DLS) and showed that the average hydrodynamic diameter of mPEG-polyPTX and mPEG-poly(TMC-PTX) micelles increased from ~15 to ~90 nm as the Carb-PTX/mPEG feed ratio increased from 2 to 10 (Figure 1e). The size and morphology of these micelles were further measured by cryogenic transmission electron microscopy (cryo-TEM), which allows direct visualization of self-assembled nanostructures in a near-native, hydrated state. As displayed in Figure 1f, these amphiphilic polymer prodrugs self-assembled into spherical nanoparticles with a size that agreed well with the DLS results. These results confirm that both the size and PTX-loading of these prodrug nanoparticles can be tuned by the DP of polyPTX that is easily controlled by living ROP. Taken together, three independent variables–the size, drug-loading and nanoparticle stability–that control the efficacy of drug-loaded nanoparticles, can be independently tuned in this system by adjusting the feed ratio of Carb-PTX and TMC.
Burst release is one of the long-standing formulation challenges of nanoparticle drug delivery systems in which the drug is physically encapsulated, and leads to undesirable side effects and reduced therapeutic efficacy28. Physically encapsulated drug-loaded nanoparticles typically show burst release of the majority of their payload within a few hours, because the release of drug is controlled solely by diffusion29. Because the drug release kinetics of mPEG-polyPTX and mPEG-poly(TMC-PTX) prodrug nanoparticles is primarily controlled by hydrolysis of the ester linker, the release of free PTX from these prodrug nanoparticles is more controllable and shows a significantly reduced burst release effect. It is reported that the ester bond between PTX and polymer is more hydrolysable than the carbonate bond in main chain.21 The prodrug nanoparticles were reasonably stable at pH 7.4, as <15% release of drug was observed over 48 h (Figure S6). In contrast, a faster release rate was observed at pH 5.5 and a plateau (≥ 60%) was reached at 48 h. These data demonstrate that the covalent conjugate of PTX is reasonably stable at the pH of blood, but that the drug is likely to be cleaved intracellularly at an much faster rate at the lower pH encountered in late endosomes.
We next evaluated the anticancer effect of these prodrug nanoparticles in vitro by a cell viability assay in human HT-29 colon, MDA-MB-231 breast and PANC-1 pancreatic cancer cell lines. These cell lines were chosen because they have been reported to be sensitive to PTX30. All prodrugs nanoparticles exhibited dose-dependent inhibition against these cancer cells after 72 h incubation (Figure 2a–c). The half-maximal inhibitory concentration (IC50) of these nanoparticles was 1.5 to 45 fold higher than that of free PTX, depending on the cell line and the specific nanoparticle. In general, however, in all of the cells, larger nanoparticles with higher drug-loading had a lower IC50 (Table S1). This finding also correlates with the stability of the nanoparticles, as the CMC decreased with increased drug-loading content (Figure S5), indicating that larger and more stable nanoparticles have a somewhat greater cytotoxic effect on tumour cells. Despite these differences between the different nanoparticles, these results clearly show that all the prodrug nanoparticles inhibit the in vitro proliferation of all HT-29, MDA-MB-231 and PANC-1 cancer cells, and that conjugation of PTX to the polymer does not markedly decrease the activity of the drug, especially for nanoparticles with the highest levels of drug-loading.
Figure 2.

Cell viability of free PTX (■), mPEG-poly(TMC-PTX1.4) (
 ), mPEG-poly(TMC-PTX2.7) (
), mPEG-poly(TMC-PTX2.7) (
 ), mPEG-polyPTX5.8 (
), mPEG-polyPTX5.8 (
 ) and mPEG-polyPTX8.7 (
) and mPEG-polyPTX8.7 (
 ) against (a) HT-29, (b) MDA-MB-231 and (c) PANC-1 cells, respectively. The cells were incubated for 72 h and the cell viability (in %) is normalized against untreated cells in the same experiment. (d) Plot of mean body weight change of mice with a dose escalation trial of mPEG-PPTX8.7 as a function of time. Points represent the mean ± SD (n=3 to 4). (e) Tumour volume up to day 100 (mean ± SD; n = 5). PBS (■), 25 mg kg−1 of free PTX (
), and mPEG-PPTX8.7 at dose of 25 (
), 75 (
), and 225 (
) mg PTX equivalent per kilogram BW were systemically administered via intravenous tail vein injection on day 0, 6 and 12, respectively. * indicates P < 0.001 (One-tailed heteroscedastic t-test). (f) Cumulative survival of mice (Kaplan–Meier).
) against (a) HT-29, (b) MDA-MB-231 and (c) PANC-1 cells, respectively. The cells were incubated for 72 h and the cell viability (in %) is normalized against untreated cells in the same experiment. (d) Plot of mean body weight change of mice with a dose escalation trial of mPEG-PPTX8.7 as a function of time. Points represent the mean ± SD (n=3 to 4). (e) Tumour volume up to day 100 (mean ± SD; n = 5). PBS (■), 25 mg kg−1 of free PTX (
), and mPEG-PPTX8.7 at dose of 25 (
), 75 (
), and 225 (
) mg PTX equivalent per kilogram BW were systemically administered via intravenous tail vein injection on day 0, 6 and 12, respectively. * indicates P < 0.001 (One-tailed heteroscedastic t-test). (f) Cumulative survival of mice (Kaplan–Meier).
To determine the systemic toxicity of these prodrug nanoparticles, their maximum tolerated doses (MTD) were evaluated by a dose escalation study. mPEG-polyPTX8.7 nanoparticle with drug-loading of 50 wt% and the highest in vitro potency was chosen because its high drug-loading allows a wide range of doses to be administered via intravenous tail vein injection. As shown in Figure 2d, no mortality and significant body weight (BW) loss was observed for mPEG-polyPTX8.7 even at the highest dose of 225 mg PTX equivalent per kilogram BW. We believe that the true MTD of mPEG-polyPTX8.7 nanoparticles is even higher than 225 mg kg−1, but we were unable to increase the dose beyond this point since this is the maximal volume that can be injected into a mouse in a single day according to the animal protocol. In a previous study, we found that the MTD of free PTX is 25 mg kg−1 in the same animal model31.
We next chose a murine orthotopic tumour model of MDA-MB-231, a human triple-negative breast cancer (TNBC), to test the in vivo therapeutic effect of the prodrug nanoparticles, because PTX is used to treat patients with TNBC32. TNBC presents a difficult clinical challenge as it is ultimately refractory to chemotherapy, and displays a shorter median time to relapse and death than other subtypes of breast cancer33,34. Treatment of MDA-MB-231 with the prodrug nanoparticles hence provides a useful test of the potential clinical utility of these formulations. Mice with MDA-MB-231 tumours were treated via intravenous tail vein injection with PBS, free PTX at its MTD of 25 mg kg−1, mPEG-polyPTX8.7 nanoparticles at 25, 75 and 225 mg kg−1 of PTX equivalent on day 0, 6 and 12, respectively. The mPEG-polyPTX8.7 nanoparticles exhibited dose-dependent inhibition of MDA-MB-231 tumours after three injections (Figure 2e). At 6 weeks after treatment, mice treated with 75 mg kg−1 of mPEG-polyPTX8.7 had a mean tumour volume of 314 mm3 (n = 5) versus 1200 mm3 (n = 5) for mice treated with a dose of 25 mg kg−1 (P < 0.001). Ten weeks after treatment, the 225 mg kg−1 treated mice had a mean tumour volume of 35 mm3 (n = 5) versus 245 mm3 for 75 mg kg−1 (P < 0.001). The mPEG-polyPTX8.7 nanoparticles at the highest high dose of 225 mg kg−1 of PTX equivalent induced significant tumour regression, which correlated with a substantial increase in animal survival (Figure 2f).
The median survival time for mice treated with PBS (n = 5) was 26 days, and treatment with the 25 mg kg−1 slightly increased this survival to 38 days. Mice treated with 75 and 225 mg kg−1 showed median survival time of 105 and 115 days, respectively. While free PTX at its MTD showed good antitumour efficacy in the first two months, 100% oft he treated mice had tumour recurrence and no mice survived more than 100 days. In contrast, 40% mice in both groups treated with nanoparticles at a dose of 75 and 225 mg/kg showed complete regression of tumours and long-term survival. Thus, these polymer prodrug nanoparticles provide a substantial curative effect at the two highest doses, albeit at fairly high doses, though we note that these doses do not result in any overt systemic toxicity, as judged by the change in body weight of the mice (Figure S7).
While these in vivo results are encouraging, much remains to be done to improve the potency before these nanoparticles are ready for a clinical trial in humans. The impact of critical parameters including nanoparticle size, stability, drug-loading, and drug release that are likely to control their efficacy need to be systematically investigated to optimize their in vivo efficacy. The size and stability of the nanoparticles control their in vivo half-life and their tissue distribution35,36. In contrast, the drug-loading is important, in our view to minimize the amount of carrier that must be injected and may also have some impact on the in vivo stability of the nanoparticles though we note that their CMC, a measure of thermodynamic stability but not necessarily of their stability in vivo, does not appear to be related to their drug-loading37,38. An important and unintended consequence of increased drug-loading is that it also increases the size of the nanoparticle, which is known to have an impact on the efficacy of nanoparticle drug carriers. The release of the dug is the final variable that is likely critical to their efficacy39,40. We believe that optimization of the linker using alternative chemistries such as hydrazone or disulfide may further improve the efficacy of these nanoparticles.
In summary, we have developed a one-pot approach to synthesize PTX-conjugated, PEG decorated biodegradable polymer nanoparticles with tunable loading, high water-solubility and low systemic toxicity. The synthesis is a one-step organocatalyzed ring-opening polymerization of a prodrug monomer consisting of PTX that is appended to a polymerizable cyclic carbonate through a cleavable ester linker. Initiating ROP of the PTX prodrug monomer from a mPEG macroinitiator results in an amphiphilic diblock copolymer with narrow polydispersity and high yield that spontaneously self-assembles into well-defined nanoparticles with tunable properties. The PTX-loading can be easily tuned from 15 to 50 wt% and the size can similarly be tuned from 15 to 90 nm. Nanoparticles with a PTX-loading capacity of 50 wt% and a diameter of ~90 nm exhibited a 9-fold higher maximum tolerated dose than free PTX, and induced significant tumour regression after three doses in a murine cancer model of human triple-negative breast cancer. Future work will optimize the linker chemistry and explore the impact of nanoparticle size and drug-loading on in vivo biodistribution and tumour regression, with the goal of further improving the in vivo potency of these nanoparticles.
Supplementary Material
Acknowledgments
This work was supported by funding from the NIH (R01-EB 000188 and R01 EB-007205) to A.C. Supporting Information is available online from Wiley InterScience or from the author.
References
- 1.Spencer CM, Faulds D. Drugs. 1994;48:794–847. doi: 10.2165/00003495-199448050-00009. [DOI] [PubMed] [Google Scholar]
- 2.Rowinsky EK, Donehower RC. N Engl J Med. 1995;332:1004–1014. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 3.Singla AK, Garg A, Aggarwal D. Int J Pharmaceutics. 2002;235:179–192. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 4.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Proc Natl Acad Sci USA. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong R, Cheng JJ. Angew Chem Int Ed. 2008;47:4830–4834. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
- 6.Zhang FW, Zhang SY, Pollack SF, Li RC, Gonzalez AM, Fan JW, Zou J, Leininger SE, Pavia-Sanders A, Johnson R, Nelson LD, Raymond JE, Elsabahy M, Hughes DMP, Lenox MW, Gustafson TP, Wooley KL. J Am Chem Soc. 2015;137:2056–2066. doi: 10.1021/ja512616s. [DOI] [PubMed] [Google Scholar]
- 7.Fonseca C, Simoes S, Gaspar R. J Controlled Release. 2002;83:273–286. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 8.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Eur J Cancer. 2001;37:1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 9.Rowinsky EK, Chaudhry V, Cornblath DR, Donehower RC. J Natl Cancer Inst Monogr. 1993:107–115. [PubMed] [Google Scholar]
- 10.Gradishar WJ. Expert Opin Pharmacother. 2006;7:1041–1053. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 11.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Int J Nanomed. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Yu DF, Inoue T, Yang DJ, Milas L, Hunter NR, Kim EE, Wallace S. Anti-Cancer Drugs. 1996;7:642–648. doi: 10.1097/00001813-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Xie ZG, Guan HL, Chen XS, Lu CH, Chen L, Hu XL, Shi Q, Jing XB. J Controlled Release. 2007;117:210–216. doi: 10.1016/j.jconrel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Lee H, Lee K, Park TG. Bioconjugate Chem. 2008;19:1319–1325. doi: 10.1021/bc8000485. [DOI] [PubMed] [Google Scholar]
- 15.Lin R, Cheetham AG, Zhang PC, Lin YA, Cui HG. Chem Commun. 2013;49:4968–4970. doi: 10.1039/c3cc41896k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu YD, Zhong YN, Meng FH, Cheng R, Deng C, Zhong ZY. Biomacromolecules. 2013;14:2772–2780. doi: 10.1021/bm400615n. [DOI] [PubMed] [Google Scholar]
- 17.Zhang SY, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, Dorshowdf RB, Wooley KL. Chem Sci. 2013;4:2122–2126. doi: 10.1039/C3SC50252J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Zou J, Yu Y, Li Yk, Ji W, Chen CK, Law WC, Prasad PN, Cheng C. Biomater Sci. 2015;3:1078–1084. doi: 10.1039/c4bm00458b. [DOI] [PubMed] [Google Scholar]; b) Ding Y, Chen WL, Hu JH, Du M, Yang D. Mater Sci Eng, C. 2014;44:368–390. doi: 10.1016/j.msec.2014.08.046. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Yu DF, Newman RA, Cabral F, Stephens LC, Hunter N, Milas L, Wallace S. Cancer Res. 1998;58:2404–2409. [PubMed] [Google Scholar]
- 20.Feng ZL, Zhao G, Yu L, Gough D, Howell SB. Cancer Chemother Pharmacol. 2010;65:923–930. doi: 10.1007/s00280-009-1099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu JY, Liu WG, Weitzhandler I, Bhattacharyya J, Li XH, Wang J, Qi YZ, Bhattacharjee S, Chilkoti A. Angew Chem Int Ed. 2015;54:1002–1006. doi: 10.1002/anie.201409293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Suriano F, Pratt R, Tan JPK, Wiradharma N, Nelson A, Yang Y-Y, Dubois P, Hedrick JL. Biomaterials. 2010;31:2637–2645. doi: 10.1016/j.biomaterials.2009.12.022. [DOI] [PubMed] [Google Scholar]; b) Yang C, Ong ZY, Yang Y-Y, Ee P-LR, Hedrick JL. Macromol Rapid Commun. 2011;32:1826–1833. doi: 10.1002/marc.201100350. [DOI] [PubMed] [Google Scholar]; c) Li Y, Yang C, Khan M, Liu SQ, Hedrick JL, Yang Y-Y, Ee P-LR. Biomaterials. 2012;33:6533–6541. doi: 10.1016/j.biomaterials.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Kiesewetter MK, Shin EJ, Hedrick JL, Waymouth RM. Macromolecules. 2010;43:2093–2107. [Google Scholar]
- 24.Bensaid F, du Boullay OT, Amgoune A, Pradel C, Reddy LH, Didier E, Sablé S, Louit G, Bazile D, Bourissou D. Biomacromolecules. 2013;14:1189–1198. doi: 10.1021/bm400161g. [DOI] [PubMed] [Google Scholar]
- 25.Pratt RC, Nederberg F, Waymouth RM, Hedrick JL. Chem Commun. 2008:114–116. doi: 10.1039/b713925j. [DOI] [PubMed] [Google Scholar]
- 26.Zhang GY, Zhang MZ, He JL, Ni PH. Polym Chem. 2013;4:4515–4525. [Google Scholar]
- 27.Wilhelm M, Zhao CL, Wang YC, Xu RL, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991;24:1033–1040. [Google Scholar]
- 28.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 29.Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, Puglisi G. Int J Pharm. 2006;325:172–179. doi: 10.1016/j.ijpharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 30.Martinez EJ, Corey EJ, Owa T. Chemistry & Biology. 2001;8:1151–1160. doi: 10.1016/s1074-5521(01)00082-5. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li XH, Lin CC, Chi JTA, Chilkoti A. Nat Commun. 2015;6:7939–7950. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bombuwala K, Kinstle T, Popik V, Uppal SO, Olesen JB, Viña J, Heckman CA. Beilstein J Org Chem. 2006;2:13–21. doi: 10.1186/1860-5397-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Cancer Treat Rev. 2010;36:206–215. doi: 10.1016/j.ctrv.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Hudis CA, Gianni L. Oncologist. 2011;16:1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 35.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 36.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 37.Kataoka K, Harada A, Nagasaki Y. Adv Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 38.Zhigaltsev IV, Maurer N, Akhong QF, Leone R, Leng E, Wang JF, Semple SC, Cullis PR. J Controlled Release. 2005;104:103–111. doi: 10.1016/j.jconrel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Bioconjugate Chem. 2005;16:122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- 40.Lim HJ, Masin D, McIntosh NL, Madden TD, Bally MB. J Pharmacol Exp Ther. 2000;292:337–345. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.