

Abstract
Background
Parenteral nutrition (PN) is a lifesaving therapy but is associated with gut atrophy and cholestasis. While bile acids (BAs) can modulate intestinal growth via gut receptors, the gut microbiome likely influences gut proliferation and inflammation. BAs also regulate the bile salt export pump (BSEP) involved in cholestasis. We hypothesized that the BA receptor agonist oleanolic acid (OA) regulates gut TGR5 receptor and modulates gut microbiota to prevent PN-associated injury.
Materials and Methods
Neonatal piglets were randomized to approximately 2 weeks of isocaloric enteral nutrition (EN), PN, or PN + enteral OA. Serum alanine aminotransferase, bilirubin, BAs, hepatic BSEP, gut TGR5, gut, liver morphology, and fecal microbiome utilizing 16S rRNA sequencing were evaluated. Kruskal-Wallis test, pairwise Mann-Whitney U test, and multilevel logistic regression analysis were performed.
Results
PN support resulted in gut atrophy substantially prevented by OA. The median (interquartile range) for villous/crypt ratio was as follows: EN, 3.37 (2.82–3.80); PN, 1.73 (1.54–2.27); and OA, 2.89 (2.17–3.34; P = .006). Pairwise comparisons yielded P = .002 (EN vs PN), P = .180 (EN vs OA), P = .026 (PN vs OA). OA upregulated TGR5 and BSEP without significant improvement in serum bilirubin (P = .095). A decreased microbial diversity and shift toward proinflammatory phylum Bacteroidetes were seen with PN, which was prevented by OA.
Conclusions
OA prevented PN-associated gut mucosal injury, Bacterioides expansion, and the decreased microbial diversity noted with PN. This study demonstrates a novel relationship among PN-associated gut dysfunction, BA treatment, and gut microbial changes.
Keywords: oleanolic acid, TGR5, gut microbiota, bile acid transporters, parenteral nutrition
Introduction
Parenteral nutrition (PN), which was first described in 1968, is a lifesaving therapy whereby essential nutrients are delivered via intravenous access.1,2 This vehicle is chosen specifically to bypass the enteric system in patients who are unable to tolerate enteral nutrition (EN). While the role of PN in the clinical setting cannot be understated, the benefits of this lifesaving therapy tend to come at the cost of severe gut mucosal atrophy and cholestasis.3–7 The mechanism that drives these pathologic changes has been examined in multiple studies with strong evidence indicating an alteration in the gut-liver axis during PN therapy.8–10
Animal studies have shown an improvement in PN-induced hepatic and gut pathology upon treatment with enterally delivered chenodeoxycholic acid, a dual agonist for the G protein–coupled membrane receptor TGR5 and the nuclear farnesoid X receptor (FXR).11–13 We previously showed decreased levels of the circulating hepatoprotective protein fibroblast growth factor 19 (FGF19) in PN-infused animals,6 and expression of FGF19 by enterocytes is a major response to bile acid–induced FXR signaling. Data suggest that the well-recognized cholestatic liver injury during PN therapy is likely due to impaired FXR-FGF19 signaling.14,15 Studies have also shown additional modulation of cholestasis via the key hepatobiliary transporter bile salt export pump (BSEP).16–19 However mechanisms regulating gut-proliferative responses remain unclear.
Although our studies have shown that bile acid treatment helps preserve PN-induced gut atrophy,6,14 these beneficial effects come at the cost of supraphysiologic upregulation of TGR5, a modulator of downstream gut-proliferative signaling. Thus, it is possible that additional gut trophic factors may be contributors to gut-proliferative responses during regular enteral feeding with an innate redundancy allowing gut proliferation with significant TGR5 upregulation.
In recent years, there has been a growing interest in the roles that gut-derived signals and gut microbiota have in regulating disease pathology. Driving the belief that gut bacteria modulate human disease, several studies have shown an improvement in hepatic and gut pathology upon exogenous modulation of the gut microbiota via enterally delivered bile acids.20–23 This has led to an increased focus on the effects of PN therapy on the gut microbiota, including its role as an adjuvant to the noted improvement seen with bile acid treatment. We thus hypothesized that bile acid agonist upregulates the TGR5 receptor and modulates the gut microbiota, thus preventing PN-associated gut atrophy.
For this study, we utilized the potent bile acid receptor TGR5 agonist oleanolic acid (OA), which is a plant-derived triterpenoid (EC50, 1.42 μmol/L).13,24,25 This study illustrates a novel relationship among bile acid treatment, PN-associated injury, and gut microbial changes in animals receiving PN.
Design and Methods
Animal Procurement
The protocol for bile acid treatment of neonatal pigs (piglets) was approved by the Institutional Animal Care and Use Committee of Saint Louis University (animal use protocol 2346, US Department of Agriculture registration 43-R-011). The study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals.26 Approximately 7-day-old female piglets (n = 18) were procured from an approved class A vendor and immediately placed in heated cages. As described previously, jugular and duodenal catheters were surgically implanted, and preconditioned jackets and ambulatory pumps for PN or EN delivery were placed.27
Animal Grouping
After 24 hours of postsurgical recovery, animals were randomly assigned to receive EN, PN only, or PN plus enterally delivered OA (Cayman Chemical, Ann Arbor, MI) via slow bolus infusions at 50 mg/kg/d (divided into 2 doses).27,28
Nutrition
As previously described, the EN control group received a swine replacement formula (LitterLife; Merrick’s Inc, Middleton, WI). EN was provided via a duodenal catheter at a rate of 260 mL/kg/d with 25 g/kg of lactose, 12.4 g/kg of protein, and 5 g/kg of fat supplemented with electrolytes, trace minerals, and vitamins for a total of 187 kcal/kg/d (Figure 1). PN piglets received a commercially available PN preparation (Clinimix E; Baxter, Deerfield, IL) continuously via a jugular venous catheter, which provided fluids at 260 mL/kg/d with 26 g/kg of dextrose, 11.05 g/kg of protein, and 5 g/kg of fat along with electrolytes, trace minerals, and vitamins for a total of 182 kcal/kg/d (Figure 1).
Figure 1.
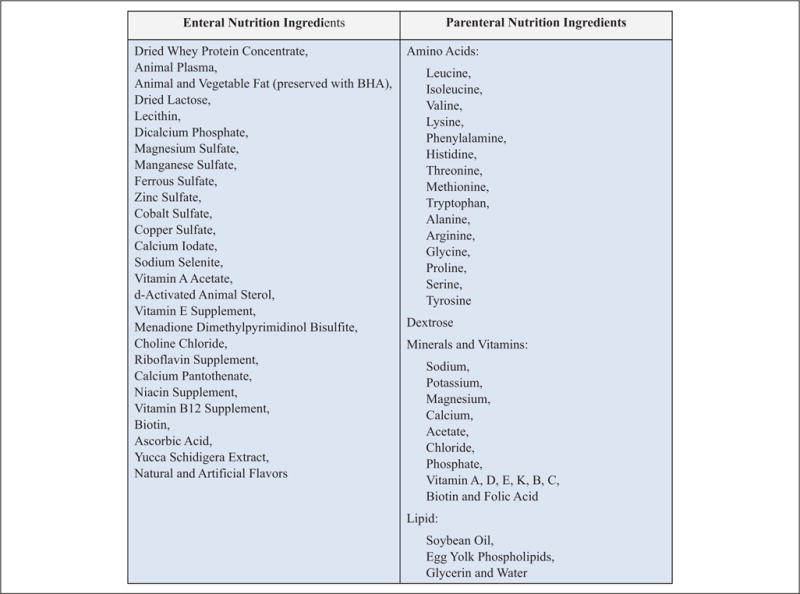
Nutritional composition of the enteral and parenteral nutrition delivered to animals. BHA, butylated hydroxyanisole.
The PN and EN were placed in nutrition bags (EVA, product code 66050; Medtec Medical, Buffalo Grove, IL) and replaced every 12 hours. Isocaloric nutrition was provided to all animals for approximately 2 weeks as previously published.27 Another group of animals received OA delivered enterally in addition to PN.
Animal Care
Animal weights were recorded daily, prior to nutrition bag changes. Animals were continuously monitored through wireless web cameras with remote access and frequent scheduled visits by research personnel in accordance with the Institutional Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals.26
Euthanasia and Tissue Collection
As described previously,6,27 the abdomen was opened posteuthanasia and the liver removed in its entirety. Subsequently, the entire small intestine distal to the stomach up to the ileocecal junction was removed. The small intestine was immediately flushed with cold saline, its contents extruded, and the tissue weighed. Small segments of the liver and the distal small bowel were sliced and weighed. Tissue was then cut into smaller pieces, snap-frozen in liquid nitrogen, and stored at −80°C for future analysis.
Stool Collections and DNA Extraction
Freshly produced stool samples at the start of the study and a colonic fecal effluent collected at the time of animal euthanasia were utilized. Samples were stored in 2.0-mL sample-processing beaded tubes (S6003-50; Zymo Research, Irvine, CA) and transferred to a 4°C refrigerator. All extractions were carried out with the Xpedition Soil/Fecal DNA Miniprep (D6202; Zymo Research). Lysis and storage buffer (1.5 mL) provided with the kit was added within 8 hours of procurement to each 2.0-mL tube and then processed with the Xpedition Sample Processor (Zymo Research). The remaining protocol was carried out according to the manufacturer’s instructions. Endpoint DNA quality and quantity were assessed via a NanoDrop ND2000 Spectrophotometer (Thermo Scientific, Waltham, MA).
Polymerase Chain Reaction, Cloning, and Sequencing
Polymerase chain reaction (PCR) was carried out on 250 ng of purified DNA with New England Biolab Q5 polymerase under the following conditions (M0491L; New England Biolabs, Ipswich, MA): 500 mL each of 16S forward and 16S reverse primer were used per reaction (Table 1). The universal 16S primers as originally described encompass the V1 region of the 16S rRNA.29,30
Table 1.
Primer Sequences Indicating the Forward and Reverse Primers.
| Primer | Sequence |
|---|---|
| 16S ribosomal | |
| Forward | 5′-GAGTTTGATCCTGGCTCAGG |
| Reverse | 5′-GCGTGGACTACCAGGGTATC |
| G protein–coupled receptor: TGR5 | |
| Forward | 5′-CCATGCACCCCTGTTGCT |
| Reverse | 5′- GGTGCTGTTGGGTGTCATCTT |
| Bile salt export pump | |
| Forward | 5′-TTTCATTCAGCGCCTGACCA |
| Reverse | 5′-ACTCCAATGAGAGGGCTGAC |
| FXR: farnesoid X receptor | |
| Forward | 5′-TTTGTGTCGTTTGCGGAGAC |
| Reverse | 5′-GTTGCCCCCATTTTTACACTT |
| Beta actin | |
| Forward | 5′-GGACCTGACCGACTACCTCA |
| Reverse | 5′-GCGACGTAGCAG AGCTTCTC |
An initial 2-minute 98°C complete denaturation step was performed, followed by 30 cycles of denaturing for 8 seconds at 98°C, a joined primer annealing at 64°C for 20 seconds, and an extension step at 72°C for 25 seconds. A final extension step for 2 minutes at 72°C was carried out to ensure complete extension of the template. Eight reactions per sample were run and then combined, purified, and concentrated with the Zymo DNA Clean and Concentrator Kit per the manufacturer’s instructions (D4034; Zymo Research).
Samples were separated on 2% agarose + TAE gel, and a band between 750–800 base pairs was extracted and purified with the Qiagen MinElute Gel Extraction Kit per the manufacturer’s instructions (28604; Qiagen, Hilden, Germany). Endpoint DNA quality and quantity were again assessed via a NanoDrop ND2000 Spectrophotometer (Thermo Scientific). These amplicons were ligated into pCR4-Blunt TOPO vectors and transformed into chemically competent Escherichia coli cells with the Zero Blunt TOPO PCR Cloning Kit (K2800-20; Invitrogen, Life Technologies, Grand Island, NY). E coli were plated in LB-Kanamycin and incubated overnight at 37°C, with clones sequenced at Quintarabio (www.quintarabio.com). Fifty to 60 well-spaced colonies were selected per nestling sample; the M13 forward primer that flanked the insert was used for sequencing. Sequences were first analyzed for quality with Applied Biosystems Sequence Scanner Software (version 1; Thermo Fischer). Vector and primer sequence removal was carried out (version 8.0.5; Biomatters, Auckland, New Zealand). All sequences were individually run through Mega BLAST31 against the entire database, and sequences with high identity to the database were kept. Chimera removal was carried out with DECIPHER.32 All sequences were deposited to the NCBI database.
RDP Classifier
A total of 40–50 sequences were generated, ranging from 35–50 isolates per treatment. Sequences from each treatment were then used as input for the RDP Classifier algorithm33 with a confidence threshold of 95%.
Histology
As previously reported,6,27 2-cm to 3-cm segments of fresh tissue from the small intestine and liver were fixed in 4% buffered formalin for 24 hours and then stored in 70% ethanol at room temperature for 24 hours. The tissue was then processed, embedded in paraffin, and stained for hematoxylin and eosin. Liver tissue was stained with trichrome (for evaluating fibrosis). The mean villous height and crypt depth were quantified in at least 15 vertically well-oriented villous-crypt columns with the slide reviewer blinded to the treatment. The automated upright microscope system with LED illumination for life sciences (Leica DM4000 B LED) was used along with QCapture Pro digital imaging software. Serum analysis was done at the Saint Louis University clinical pathology core laboratory.
Tissue: RNA Extraction and Real-Time PCR Analysis
RNA was extracted with the Sigma GenElute Mammalian Total RNA Miniprep Kit per the manufacturer’s protocol. Isolated RNA was reverse transcribed into cDNA via the Applied Biosystems Verso cDNA Synthesis Kit. Primers for each transcript were validated (Table 1). Real-time quantitative PCR was performed in triplicate with an Applied Biosystems CFX90 touch. Relative mRNA levels were calculated by the comparative threshold cycle method with beta-actin as the internal control.
Statistical Analysis
SPSS (version 23; IBM, Chicago, IL) software was utilized for statistical analysis. Descriptive statistics on the outcomes were calculated as median and interquartile range (IQR). The Kruskal-Wallis test was utilized to determine if there was a difference among the 3 groups (EN, PN, and OA) for the villous-to-crypt (V/C) ratio, serologic markers, and relative mRNA expression of the genes involved in bile acid homeostasis. If a significant difference was found, then pairwise Mann-Whitney U tests were conducted. All test were 2-sided with a significance level of 0.05. Culture-independent identification of fecal bacterial populations was determined by 16S rRNA sequencing. Sequences were subjected to the unweighted UniFrac metric. Additionally, sequences were classified up to the genus level through the RDP Classifier algorithm. Samples in each animal group were analyzed for phylum-level and family-level bacteria. Number and distribution of positive cultures for a given phylum or family were evaluated. A logistic regression analysis was conducted to assess the odds of testing positive for the given bacteria per the treatment group. With the availability of multiple samples for individual animals, logistic regression models were conducted as a multilevel model, adjusting for the subject. Overall data were available for 3–6 animals per group for each outcome of interest.
Results
Improved Gut Morphology and Histology
Severe villous atrophy and a mucosal inflammatory infiltrate were noted in animals receiving PN compared with animals receiving EN (Figure 2A). To quantify morphometric differences, we evaluated the median (IQR) V/C ratio in each group: EN, 3.37 (2.82–3.80); PN, 1.73 (1.54–2.27); and OA, 2.89 (2.17–3.34). A Kruskal-Wallis revealed a P value of .006. The PN group had significantly reduced V/C ratio as compared with the EN group (P = .002). OA-treated animals had a significant preservation of the V/C ratio when compared with the PN-induced reduction (P = .026). There was no statistical difference between EN and OA (P = .180; Figure 2B).
Figure 2.
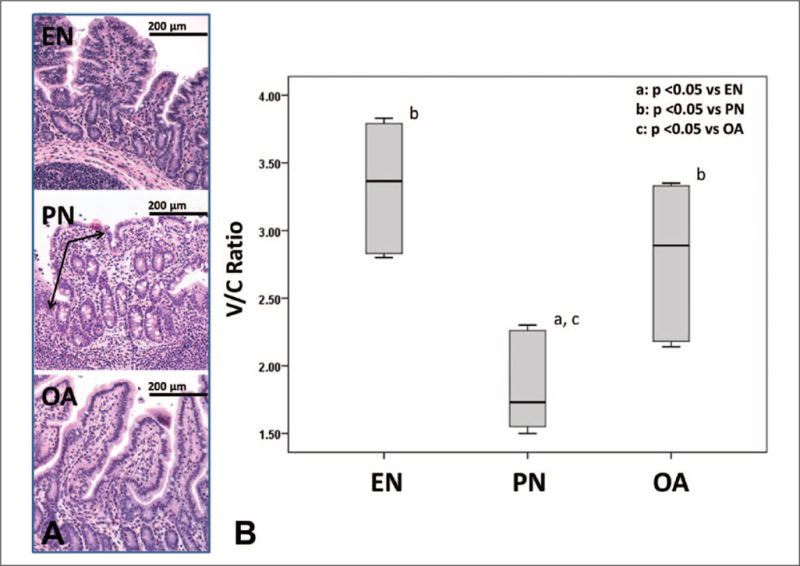
A, Small bowel histology (200× magnification) in animals treated with enteral nutrition (EN), parenteral nutrition (PN), and oleanolic acid (OA). Note villous atrophy and inflammation (arrows) in PN with improvement upon OA treatment. B, Villous/crypt (V/C) ratio is shown as a box and whisker plot. Boxes represent the 25th–75th percentile; central lines represent median values. Whiskers extend to a maximum of 1.5 times the interquartile range. A Kruskal-Wallis test and subsequent pairwise Mann-Whitney U test were conducted to determine P value. All tests were 2-sided with a significance level of 0.05.
Preserved Gut Microbiota
Bacterial sequences were classified with the RDP Classifier algorithm. Significant clonal proliferation of the phylum Bacteroidetes was noted with animals receiving PN versus EN. Bacteroidetes clonal proliferation was prevented by OA treatment (Figure 3).
Figure 3.
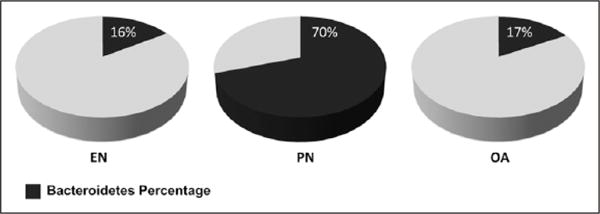
Gut microbiota: percentage of Bacteroidetes phyla shown for each group. Note significant increase in Bacteroidetes with parenteral nutrition (PN). EN, enteral nutrition; OA, oleanolic acid.
To ascertain the odds of testing positive for Bacteroidetes per the treatment group, a multilevel logistic regression analysis was conducted. EN and OA were protective for Bacteroidetes as compared with PN (odds ratio [OR] = 0.09, 95% confidence interval [95% CI]: 0.03–0.26; OR = 0.1, 95% CI: 0.04–0.27, respectively). EN and OA were also protective for the family Porphyromonadaceae (to phylum Bacteroidetes, class Bacteroidetes, order Bacteroidales) when compared with PN (OR = 0.05, 95% CI: 0.01–0.33; OR = 0.02, 95% CI: 0.003–0.18, respectively). However, convergence was not seen for phyla Actinobacteria and Proteobacteria as well as the families Clostridiaceae and Campylobacteraceae (Table 2).
Table 2.
Gut Microbiota Profile: Odds of a Sample Testing Positive.a
| Gut Bacteria | n | EN vs PN
|
OA vs PN
|
EN vs OA
|
|||
|---|---|---|---|---|---|---|---|
| OR (95% CI) | P Value | OR (95% CI) | P Value | OR (95% CI) | P Value | ||
| Phylum level | |||||||
| Actinobacteria | 4 | DNC | |||||
| Bacteroidetes | 123 | 0.09 (0.03–0.3) | .001 | 0.1 (0.04–0.3) | .001 | 0.9 (0.3–2.6) | .82 |
| Proteobacteria | 3 | DNC | |||||
| Unclassified bacteria | 8 | DNC | |||||
| Family level | |||||||
| Unclassified Actinobacteria | 4 | DNC | |||||
| Bacteroidaceae | 41 | 0.43 (0.04–4.2) | .41 | 1.02 (0.1–8.0) | .98 | 0.42 (0.05–3.6) | .37 |
| Porphyromonadaceae | 59 | 0.05 (0.01–0.3) | .007 | 0.02 (0.003–0.2) | .003 | 2.16 (0.2–20.2) | .44 |
| Unclassified Bacteroidetes | 11 | DNC | |||||
| Clostridiaceae | 7 | DNC | |||||
| Lachnospiraceae | 76 | 1.47 (0.4–5.6) | .52 | 0.72 (0.2–2.6) | .57 | 2.04 (0.6–7.2) | .23 |
| Campylobacteraceae | 3 | DNC | |||||
CI, confidence interval; DNC, did not converge; EN, enteral nutrition; OA, oleanolic acid; OR, odds ratio; PN, parenteral nutrition.
Note increase in phylum Bacteroidetes and family Porphyromonadaceae with parenteral nutrition.
Sequences were subjected to unweighted UniFrac to compare phylogenetic diversity. Alpha diversity was calculated at the phylum level with Shannon diversity index. EN-treated animals had the highest diversity.
Animals receiving PN and OA had significantly more diverse microbial populations as compared with animals receiving PN alone. The mean alpha diversity was significantly different among the groups (Shannon diversity index: 0.6403, 0.1351, and 1.077 for EN, PN, and OA, respectively; P = .049 via Kruskal-Wallis analysis).
Bile Acid Receptors and Transporters
Enhanced TGR5 expression
OA is a known TGR5 agonist. The median (IQR) relative gut TGR5 abundance for EN was 0.42 (0.21–1.26); PN, 0.39 (0.26–1.55); and OA, 3.89 (1.85–4.25). A P value of .036 was noted on Kruskal-Wallis analysis. We noted a significant upregulation of gut TGR5 transcript in the OA group (P = .032 vs EN and P = .032 vs PN). In comparison, no statistically significant differences in TGR5 were noted between EN and PN (P = .841; Figure 4A). To confirm TGR5-specific agonism by OA, we also evaluated FXR expression. As noted in our prior studies, no FXR activation was seen with OA treatment.
Figure 4.
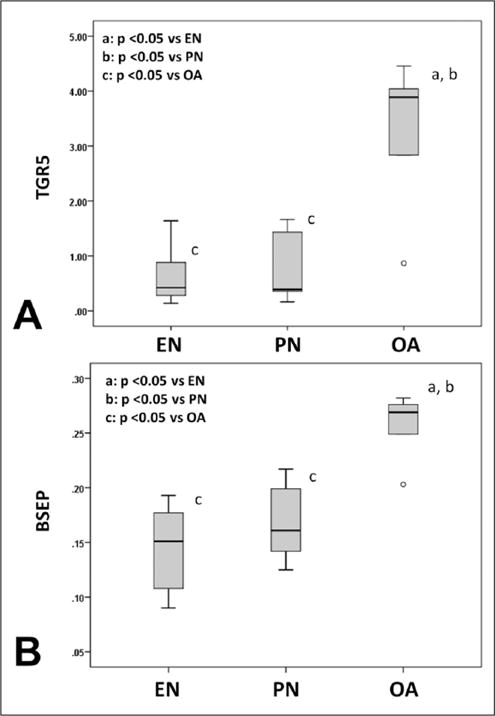
Bile acid receptors and transporters. A, Gut TGR5 expression. B, Hepatic bile salt export pump (BSEP) expression. The figure shows box and whisker plots. Boxes represent the 25th–75th percentile; central lines represent median values. Whiskers extend to a maximum of 1.5 times the interquartile range. Small circles represent outlier observations between 1.5–3 times the interquartile range from the edge of the box. A Kruskal-Wallis test and subsequent pairwise Mann-Whitney U test were conducted to determine P value. All tests were 2-sided with a significance level of 0.05. Note increase in TGR5 and BSEP abundance with oleanolic acid (OA). EN, enteral nutrition; PN, parenteral nutrition.
Bile salt export pump
Upregulation of the major bile salt efflux transporter pump BSEP was noted with OA treatment (P = .008 vs EN and P = .016 vs PN). However, no differences in hepatic BSEP mRNA expression were noted between EN-infused and PN-infused animals (P = .421). The median (IQR) for EN was 0.15 (0.10–0.19); PN, 0.16 (0.13–0.21); and OA, 0.27 (0.23–0.28). A P value of .010 was noted with the Kruskal-Wallis test (Figure 4B).
Serum Bilirubin and Histology
There was a significant elevation in serum bilirubin level with animals treated with PN versus EN (P = .008). Histologically, intraparenchymal hepatic bile deposition was noted in animals receiving PN. While we noted an improvement in hepatic cholestatic deposits as well as a reduction in serum bilirubin with OA treatment, this did not reach statistical significance (P = .095 vs EN and P = .31 vs PN). The median (IQR) serum bilirubin value for EN was 0.13 (0.10–0.16); PN, 0.35 (0.27–0.45); and OA, 0.25 (0.14–0.37; P = .020, Kruskal-Wallis test; Figure 5). No significant fibrosis was seen in either group. No statistical differences in serum alanine aminotransferase levels were noted among the groups.
Figure 5.
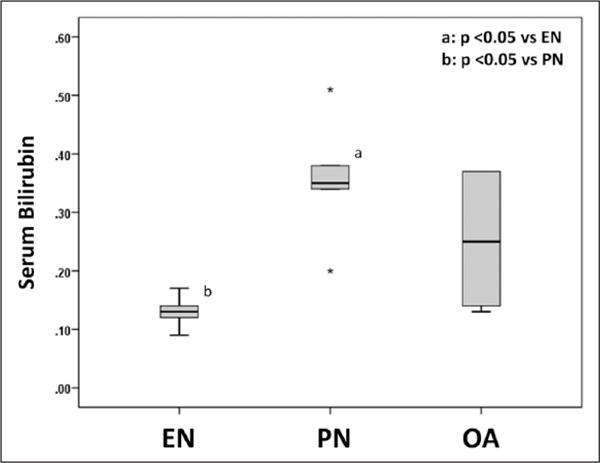
Serum bilirubin level is shown as a box and whisker plot. Boxes represent the 25th–75th percentile; central lines represent median values. Whiskers extend to a maximum of 1.5 times the interquartile range. If there are no whiskers, then the data point is within the interquartile range. Stars represent outlier observations, beyond 3 times the interquartile range from the edge of the box. A Kruskal-Wallis test and subsequent pairwise Mann-Whitney U test were conducted to determine P value. All tests were 2-sided with a significance level of 0.05. Note significant elevation in bilirubin with parenteral nutrition (PN). EN, enteral nutrition; OA, oleanolic acid.
Discussion
Intravenously delivered PN in patient populations unable to tolerate EN remains a critical therapeutic intervention. While it has the ability to provide all essential nutrients and is lifesaving,5,34,35 paradoxically, a known complication is the development of cholestasis and gut injury.7,36–38 The mechanistic basis of such injury is not fully understood.
In the clinical setting, PN-associated injury is mitigated upon providing at least some EN,9,10 thus driving the idea that there are gut-derived signals capable of preserving gut and liver health during regular EN.
Recent studies have demonstrated that bile acids, which have generally been thought of as toxic emulsifying agents, appear to play a major role as key signaling molecules20,21 in the context of this gut-liver crosstalk.39,40
Several studies have exhibited a convincingly strong case for the hepatic-protective and gut-protective effect of bile acids against PN-related injury.6,14,41 Data suggest that a hepatoprotective effect upon bile acid treatment in PN-infused animals is contributed via gut FXR activation leading to FGF19 secretion, thus influencing the FXR-FGF19 axis.6 Recent data from cell culture and animal studies show that bile acids can also directly influence cholestasis via the key hepatic bile acid transporter BSEP.18,42–45
Additionally, gut proliferation can be induced via activation of the G protein–coupled receptor TGR5 by bile acids, as shown in animal studies.46,47 In fact, TGR5 is highly localized in crypts and is known to regulate trophic effects.48,49 Although the exact mechanisms of this effect remain incompletely defined, TGR5 has been linked to the c-Jun N-terminal kinase signaling pathways in gut and hepatic cell culture models, where modulation of cell proliferation and apoptosis has been demonstrated.11,50–53
This study builds on our previously published data describing improved gut outcomes upon regulation of TGR5. Of particular note, there has been much interest in the bile acid receptor agonist OA, which is a naturally occurring triterpenoid that targets TGR513,24,54 without inducing FXR signaling.
Despite the correlation between OA treatment and gut mucosal proliferation, the exact mechanism by which this protective effective is conferred is still not fully defined. Although we noted that supraphysiologic upregulation of TGR5 confers gut protection, this finding highlights a gap in current understanding, indicating possible redundancy in TGR5 regulation as well as the existence of complementary pathways regulating gut growth.
Recent studies have established the existence of a crosstalk between the gut and the liver, with additional modulation of disease pathology by the gut microbiota.20,21 With >100 trillion commensal organisms and thousands of species, the intestinal microbiota genome represents 200,000–300,000 genes, 10 times that of the aggregate host genome.55,56 Although humans on average have only a small percentage of body weight attributable to gut bacteria,57 the microbial genome exceeds the human genome by 2 orders of magnitude, making us genetically 99% bacterial and 1% human.58,59 When viewed as a whole, this “super organism” of the gut can perform critical physiologic functions that benefit the host, including education of the mucosal immune system, extraction of nutrients, production of enterocyte essential short-chain fatty acids, production of vitamins, and metabolism of bile salts—all critical to gut growth and proliferation.60,61 The results from such studies have driven the belief that gut bacteria can modulate disease.
Recent evidence in rat models also suggests a relationship between PN-related changes in endothelial gut integrity and gut microbial composition.62–64 These studies have shown that virulent bacteria within the opportunistic proinflammatory phylum Bacteroidetes and its Porphyromonadaceae family tend to be more resilient than normal flora in the gut starvation state that PN induces.65,66 Data also suggest that colonization with such bacteria can lead to intestinal inflammation,67,68 which can impair regulation of critical bile acid transporters69–71 as well as gut-protective and gut-proliferative factors.50,72
Our current study demonstrates a clonal proliferation of phylum Bacteroidetes and its Porphyromonadaceae family along with decreased microbial diversity upon PN infusion and marked improvement with concomitant OA treatment. It has thus been theorized that such shifts in gut microbiota as noted with PN therapy may be additionally responsible for PN-induced injury. Given the protective effect that OA appears to demonstrate on the gut mucosa, it may be hypothesized that bile acids might additionally confer this effect by promoting a less virulent microbial composition in animals undergoing PN. A limitation of this study is the lack of knowledge of the relative contributions of microbial changes versus direct TGR5-regulated gut-proliferative responses influencing gut growth with OA treatment.
Thus, while there is need for establishing further mechanistic links, we present novel data linking restorative alterations in gut microbiota upon bile acid treatment during PN infusion. Assessing cytokines, their influence on hepatobiliary and gut transporters, and the gut barrier function remains a high priority for further studies.
While our results show a gut-protective effect with OA treatment, it is imperative to note that some other studies have indicated the development of a detrimental effect upon OA treatment that may seem to contradict our results.73,74 While such studies have indicated hepatic toxicity with OA at higher doses, our central purpose of using OA was to serve as a TGR5 receptor agonist and not to change the innate bile acid pool. Thus, our chosen OA dose is several folds lower than the minimum toxic dose reported in the earlier studies.
We noted an improvement in PN-induced gut atrophy, evidenced by higher V/C ratio upon OA treatment at 50 mg/kg/d. While we did not find a statistically significant improvement in serum bilirubin in the OA group, there appeared to be a trend toward a lower bilirubin level with OA treatment. We compared FXR expression among the various groups and did not find its upregulation, nor was there any significant elevation in FGF19 with OA treatment. Thus, these hepatic responses are unlikely mediated through modulation of the FXR-FGF19 axis.
We did note a significant upregulation of hepatic BSEP, which is the major regulator of bile acid efflux out of hepatocytes.75–77 Interestingly, hepatoprotection conferred at low doses of OA has been reported.55,78 It is possible that with our enteral OA treatment, low OA doses may have been delivered to the hepatocytes via direct gut absorption. While we assayed total bile acid levels in different groups, we did not find a statistically significant difference in OA-treated animals. Although this result may be due to technical limitations or possibly to our relatively low number of animals, it may suggest that additional pathways other than the FXR-FGF19 axis are involved in bile acid–mediated hepatic improvement during PN infusion.
Additionally, we were intrigued by a lack of transaminase elevation in PN-treated animals. In fact, no statistically significant differences in alanine aminotransferase were noted among any group. This result, however, is consistent with our previously published literature and likely due to shorter duration of PN therapy.6 Evaluating PN-associated changes upon longer duration of PN therapy is worthwhile and may be possible by using our recently published ambulatory PN infusion model.27
Conclusion
This study presents data indicating that OA treatment helps to prevent PN-associated injury. There is upregulation of gut TGR5 as well as modulation of key bile acid homeostatic transporters and regulators influencing cholestasis in the liver, possibly via a pathway distinct from the classical bile acid–regulated FXR-FGF19 axis. We also note that microbial communities diverge with PN infusion and that a statistically significant expansion of the proinflammatory phylum Bacteroidetes and its Porphyromonadaceae family occurs in animals receiving PN, along with decreased microbial diversity. This study thus explores a potential novel relationship between gut microbiota and PN-induced injury with promising preventive effects with OA treatment. Studies to explore further mechanistic links could help advance knowledge that may ultimately prove critical in mitigating PN-associated gut and liver injury.
Clinical Relevancy Statement.
Parenteral nutrition (PN) remains a lifesaving therapy where essential nutrients are delivered via intravenous access. This therapy bypasses the gut in patients unable to tolerate enteral nutrition, but it results in significant complications. Our study, in a clinically relevant pig model, illustrates a novel mechanism of preventing PN-associated injury with enteral bile acid treatment modulating the gut microbiota and key hepatobiliary receptors and transporters. Thus, strategies targeting the gut-liver crosstalk may hold significant potential in preventing complication resulting from PN therapy.
Acknowledgments
Financial disclosure: This work was partly supported by the ASPEN Rhoads Research Foundation grant to A.K.J. Additional funding was provided to A.K.J. via the Saint Louis University internal grant mechanism, the North American Society for Pediatric Gastroenterology Hepatology and Nutrition Foundation Young Investigator Award, and funding from the National Institutes of Health (1K08DK098623-01A1).
Footnotes
Conflicts of interest: None declared.
Statement of Authorship
A. K. Jain contributed to the conception/design of the research; all authors contributed to acquisition, analysis, or interpretation of the data, as well as drafting of the manuscript; and A. K. Jain, J. Long, B. A. Neuschwander-Tetri, and J. Teckman critically revised the manuscript. All authors agree to be fully accountable for ensuring the integrity and accuracy of the work and approved the final manuscript.
References
- 1.Wilmore DW, Dudrick SJ. Growth and development of an infant receiving all nutrients exclusively by vein. JAMA. 1968;203:860–864. [PubMed] [Google Scholar]
- 2.Dudrick SJ. Early developments and clinical applications of total parenteral nutrition. JPEN J Parenter Enteral Nutr. 2003;27:291–299. doi: 10.1177/0148607103027004291. [DOI] [PubMed] [Google Scholar]
- 3.Buchman A. Total parenteral nutrition-associated liver disease. JPEN J Parenter Enteral Nutr. 2002;26:S43–S48. doi: 10.1177/014860710202600512. [DOI] [PubMed] [Google Scholar]
- 4.Teitelbaum DH. Parenteral nutrition-associated cholestasis. Curr Opin Pediatr. 1997;9:270–275. doi: 10.1097/00008480-199706000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Kumpf VJ. Parenteral nutrition-associated liver disease in adult and pediatric patients. Nutr Clin Pract. 2006;21:279–290. doi: 10.1177/0115426506021003279. [DOI] [PubMed] [Google Scholar]
- 6.Jain AK, Stoll B, Burrin DG, Holst JJ, Moore DD. Enteral bile acid treatment improves parenteral nutrition-related liver disease and intestinal mucosal atrophy in neonatal pigs. Am J Physiol Gastrointest Liver Physiol. 2012;302:G218–G224. doi: 10.1152/ajpgi.00280.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niinikoski H, Stoll B, Guan X, et al. Onset of small intestinal atrophy is associated with reduced intestinal blood flow in TPN-fed neonatal piglets. J Nutr. 2004;134:1467–1474. doi: 10.1093/jn/134.6.1467. [DOI] [PubMed] [Google Scholar]
- 8.Carter BA, Shulman RJ. Mechanisms of disease: update on the molecular etiology and fundamentals of parenteral nutrition associated cholestasis. Nat Clin Pract Gastroenterol Hepatol. 2007;4:277–287. doi: 10.1038/ncpgasthep0796. [DOI] [PubMed] [Google Scholar]
- 9.Forchielli ML, Walker WA. Nutritional factors contributing to the development of cholestasis during total parenteral nutrition. Adv Pediatr. 2003;50:245–267. [PubMed] [Google Scholar]
- 10.Javid PJ, Collier S, Richardson D, et al. The role of enteral nutrition in the reversal of parenteral nutrition-associated liver dysfunction in infants. J Pediatr Surg. 2005;40:1015–1018. doi: 10.1016/j.jpedsurg.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 11.Yang JI, Yoon JH, Myung SJ, et al. Bile acid-induced TGR5-dependent c-Jun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007;361:156–161. doi: 10.1016/j.bbrc.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Pellicciari R, Sato H, Gioiello A, et al. Nongenomic actions of bile acids: synthesis and preliminary characterization of 23- and 6,23-alkyl-substituted bile acid derivatives as selective modulators for the G-protein coupled receptor TGR5. J Med Chem. 2007;50:4265–4268. doi: 10.1021/jm070633p. [DOI] [PubMed] [Google Scholar]
- 13.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54:1263–1272. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain AK, Wen JX, Blomenkamp KS, et al. Oleanolic acid improves gut atrophy induced by parenteral nutrition [published online April 28, 2015] JPEN J Parenter Enteral Nutr. doi: 10.1177/0148607115583536. [DOI] [PubMed] [Google Scholar]
- 15.Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trauner M, Wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol. 2005;39:S111–S124. doi: 10.1097/01.mcg.0000155551.37266.26. [DOI] [PubMed] [Google Scholar]
- 17.Vaz FM, Paulusma CC, Huidekoper H, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology. 2015;61:260–267. doi: 10.1002/hep.27240. [DOI] [PubMed] [Google Scholar]
- 18.Soroka CJ, Boyer JL. Biosynthesis and trafficking of the bile salt export pump, BSEP: therapeutic implications of BSEP mutations. Mol Aspects Med. 2014;37:3–14. doi: 10.1016/j.mam.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kis E, Ioja E, Rajnai Z, et al. BSEP inhibition: in vitro screens to assess cholestatic potential of drugs. Toxicol In Vitro. 2012;26:1294–1299. doi: 10.1016/j.tiv.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Ferolla SM, Armiliato GN, Couto CA, Ferrari TC. The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients. 2014;6:5583–5599. doi: 10.3390/nu6125583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodin CM, Visschers RG, Rensen SS, et al. Total parenteral nutrition induces a shift in the Firmicutes to Bacteroidetes ratio in association with Paneth cell activation in rats. J Nutr. 2012;142:2141–2147. doi: 10.3945/jn.112.162388. [DOI] [PubMed] [Google Scholar]
- 22.Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:5286–5296. doi: 10.3748/wjg.v16.i42.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singer C, Stancu P, Cosoveanu S, Botu A. Non-alcoholic fatty liver disease in children. Curr Health Sci J. 2014;40:170–176. doi: 10.12865/CHSJ.40.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato H, Genet C, Strehle A, et al. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun. 2007;362:793–798. doi: 10.1016/j.bbrc.2007.06.130. [DOI] [PubMed] [Google Scholar]
- 25.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28:220–224. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- 26.National Research Council. Guide for the Care and Use of Laboratory Animals. 8th. Washington, DC: National Research Council; 2011. [Google Scholar]
- 27.Jain AK, Wen JX, Arora S, et al. Validating hyperbilirubinemia and gut mucosal atrophy with a novel ultramobile ambulatory total parenteral nutrition piglet model. Nutr Res. 2015;35:169–174. doi: 10.1016/j.nutres.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Jeong DW, Kim YH, Kim HH, et al. Dose-linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm Drug Dispos. 2007;28:51–57. doi: 10.1002/bdd.530. [DOI] [PubMed] [Google Scholar]
- 29.Chakravorty S, Helb D, Burday M, Connell N, Alland D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods. 2007;69:330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Davey ME, Figueras JB, Rivkina E, Gilichinsky D, Tiedje JM. Phylogenetic diversity of a bacterial community determined from Siberian tundra soil DNA. Microbiology. 1997;143(Pt 12):3913–3919. doi: 10.1099/00221287-143-12-3913. [DOI] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Wright ES, Yilmaz LS, Noguera DR. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol. 2012;78:717–725. doi: 10.1128/AEM.06516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rager R, Finegold MJ. Cholestasis in immature newborn infants: is parenteral alimentation responsible? J Pediatr. 1975;86:264–269. doi: 10.1016/s0022-3476(75)80486-0. [DOI] [PubMed] [Google Scholar]
- 35.Tomar BS. Hepatobiliary abnormalities and parenteral nutrition. Indian J Pediatr. 2000;67:695–701. doi: 10.1007/BF02762189. [DOI] [PubMed] [Google Scholar]
- 36.Kubota A, Yonekura T, Hoki M, et al. Total parenteral nutrition-associated intrahepatic cholestasis in infants: 25 years’ experience. J Pediatr Surg. 2000;35:1049–1051. doi: 10.1053/jpsu.2000.7769. [DOI] [PubMed] [Google Scholar]
- 37.Ekelund M, Kristensson E, Ekelund M, Ekblad E. Total parenteral nutrition causes circumferential intestinal atrophy, remodeling of the intestinal wall, and redistribution of eosinophils in the rat gastrointestinal tract. Dig Dis Sci. 2007;52:1833–1839. doi: 10.1007/s10620-006-9678-z. [DOI] [PubMed] [Google Scholar]
- 38.Kelly DA. Preventing parenteral nutrition liver disease. Early Hum Dev. 2010;86:683–687. doi: 10.1016/j.earlhumdev.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Lee YK, Bundman D, et al. Redundant pathways for negative feedback regulation of bile acid production. Develop Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 41.Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 42.Chen HL, Chen HL, Yuan RH, et al. Hepatocyte transplantation in bile salt export pump-deficient mice: selective growth advantage of donor hepatocytes under bile acid stress. J Cell Mol Med. 2012;16:2679–2689. doi: 10.1111/j.1582-4934.2012.01586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen RR, Li YJ, Zhou XM, et al. The association between bile salt export pump single-nucleotide polymorphisms and primary biliary cirrhosis susceptibility and ursodeoxycholic acid response. Dis Markers. 2014;2014:350690. doi: 10.1155/2014/350690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sommerfeld A, Mayer PG, Cantore M, Haussinger D. Regulation of plasma membrane localization of the Na+-taurocholate cotransporting polypeptide (Ntcp) by hyperosmolarity and tauroursodeoxycholate. J Biol Chem. 2015;290:24237–24254. doi: 10.1074/jbc.M115.666883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baghdasaryan A, Chiba P, Trauner M. Clinical application of transcriptional activators of bile salt transporters. Mol Aspects Med. 2014;37:57–76. doi: 10.1016/j.mam.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Augustin O, Sanchez de Medina F. Intestinal bile acid physiology and pathophysiology. World J Gastroenterol. 2008;14:5630–5640. doi: 10.3748/wjg.14.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boesjes M, Brufau G. Metabolic effects of bile acids in the gut in health and disease. Curr Med Chem. 2014;21:2822–2829. doi: 10.2174/0929867321666140303142053. [DOI] [PubMed] [Google Scholar]
- 48.Burrin D, Stoll B, Moore D. Digestive physiology of the pig symposium: intestinal bile acid sensing is linked to key endocrine and metabolic signaling pathways. J Anim Sci. 2013;91:1991–2000. doi: 10.2527/jas.2013-6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunnett NW. Neuro-humoral signalling by bile acids and the TGR5 receptor in the gastrointestinal tract. J Physiol. 2014;592:2943–2950. doi: 10.1113/jphysiol.2014.271155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007;354:154–159. doi: 10.1016/j.bbrc.2006.12.168. [DOI] [PubMed] [Google Scholar]
- 51.Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 53.Cao W, Tian W, Hong J, et al. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol Gastrointest Liver Physiol. 2013;304:G322–G327. doi: 10.1152/ajpgi.00263.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis. 2011;29:37–44. doi: 10.1159/000324126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guinane CM, Cotter PD. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap Adv Gastroenterol. 2013;6:295–308. doi: 10.1177/1756283X13482996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cenit MC, Matzaraki V, Tigchelaar EF, Zhernakova A. Rapidly expanding knowledge on the role of the gut microbiome in health and disease. Biochim Biophys Acta. 2014;1842:1981–1992. doi: 10.1016/j.bbadis.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 57.Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci U S A. 2003;100:10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sonnenburg JL, Angenent LT, Gordon JI. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol. 2004;5:569–573. doi: 10.1038/ni1079. [DOI] [PubMed] [Google Scholar]
- 60.Esteve E, Ricart W, Fernandez-Real JM. Gut microbiota interactions with obesity, insulin resistance and type 2 diabetes: did gut microbiote co-evolve with insulin resistance? Curr Opin Clin Nutr Metab Care. 2011;14:483–490. doi: 10.1097/MCO.0b013e328348c06d. [DOI] [PubMed] [Google Scholar]
- 61.Hur KY, Lee MS. Gut microbiota and metabolic disorders. Diabetes Metab J. 2015;39:198–203. doi: 10.4093/dmj.2015.39.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lichtman SN, Keku J, Schwab JH, Sartor RB. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology. 1991;100:513–519. doi: 10.1016/0016-5085(91)90224-9. [DOI] [PubMed] [Google Scholar]
- 63.Freund HR, Muggia-Sullam M, LaFrance R, Enrione EB, Popp MB, Bjornson HS. A possible beneficial effect of metronidazole in reducing TPN-associated liver function derangements. J Surg Res. 1985;38:356–363. doi: 10.1016/0022-4804(85)90049-6. [DOI] [PubMed] [Google Scholar]
- 64.Koga H, Sakisaka S, Yoshitake M, et al. Abnormal accumulation in lipopolysaccharide in biliary epithelial cells of rats with self-filling blind loop. Int J Mol Med. 2002;9:621–626. [PubMed] [Google Scholar]
- 65.Giannelli V, Di Gregorio V, Iebba V, et al. Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol. 2014;20:16795–16810. doi: 10.3748/wjg.v20.i45.16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3:637–682. doi: 10.3390/nu3060637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-kappaB pathway. Infect Immun. 2004;72:5832–5839. doi: 10.1128/IAI.72.10.5832-5839.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon YM, Lee JY, Yoo D, et al. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infect Immun. 2010;78:2024–2033. doi: 10.1128/IAI.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Green RM, Beier D, Gollan JL. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology. 1996;111:193–198. doi: 10.1053/gast.1996.v111.pm8698199. [DOI] [PubMed] [Google Scholar]
- 70.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 71.Trauner M, Arrese M, Lee H, Boyer JL, Karpen SJ. Endotoxin down-regulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J Clin Invest. 1998;101:2092–2100. doi: 10.1172/JCI1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–C10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- 73.Lu YF, Wan XL, Xu Y, Liu J. Repeated oral administration of oleanolic acid produces cholestatic liver injury in mice. Molecules. 2013;18:3060–3071. doi: 10.3390/molecules18033060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu J, Lu YF, Zhang Y, Wu KC, Fan F, Klaassen CD. Oleanolic acid alters bile acid metabolism and produces cholestatic liver injury in mice. Toxicol Appl Pharmacol. 2013;272:816–824. doi: 10.1016/j.taap.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kosters A, Karpen SJ. Bile acid transporters in health and disease. Xenobiotica. 2008;38:1043–1071. doi: 10.1080/00498250802040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Pircher PC, Schulman IG, Westin SK. Regulation of complement C3 expression by the bile acid receptor FXR. J Biol Chem. 2005;280:7427–7434. doi: 10.1074/jbc.M411473200. [DOI] [PubMed] [Google Scholar]
- 77.Stieger B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb Exp Pharmacol. 2011;201:205–259. doi: 10.1007/978-3-642-14541-4_5. [DOI] [PubMed] [Google Scholar]
- 78.Chen P, Zeng H, Wang Y, et al. Low dose of oleanolic acid protects against lithocholic acid-induced cholestasis in mice: potential involvement of nuclear factor-E2-related factor 2-mediated upregulation of multidrug resistance-associated proteins. Drug Metab Dispos. 2014;42:844–852. doi: 10.1124/dmd.113.056549. [DOI] [PubMed] [Google Scholar]