

Summary
Background
Targeting FVIII expression to platelets is a promising gene therapy approach for hemophilia A and is successful even in the presence of inhibitors. It is well known that platelets not only play important roles in hemostasis, but also in thrombosis and inflammation.
Objective
To evaluate whether platelet-FVIII expression might increase the risk for thrombosis and thereby compromise the safety of this approach.
Methods
In this study, platelet-FVIII expressing transgenic mice were examined either in steady state or under prothrombotic conditions induced by inflammation or the factor V Leiden mutation. Native whole blood thrombin generation assay, ROTEM analysis, and ferric chloride induced vessel injury were used to evaluate the hemostatic properties. Various parameters associated with the thrombosis risk, including D-Dimer, thrombin anti-thrombin complexes, fibrinogen, tissue fibrin deposition, platelet activation status and activatability, and platelet-leukocyte aggregates, were assessed.
Results
We generated a new line of transgenic mice that expressed 30-fold higher platelet-FVIII levels than therapeutically required to restore hemostasis in hemophilic mice. In steady state as well as under prothrombotic conditions induced by LPS-mediated inflammation or the factor V Leiden mutation, supratherapeutic levels of platelet-FVIII did not appear thrombogenic. Furthermore, FVIII-expressing platelets were neither hyper-activated nor hyper-activatable upon agonist activation.
Conclusion
We conclude that in mice, more than 30-fold higher platelet FVIII levels than required for therapeutic efficacy in hemophilia A are not associated with a thrombotic predilection.
Keywords: Hemophilia A, Gene therapy, Platelet, Factor VIII, Thrombosis
Introduction
Factor VIII (FVIII) replacement is the prevailing therapy for hemophilia A (HA) patients. A major complication in protein replacement therapy, however, is the development of inhibitory antibodies against FVIII (inhibitors) [1]. Alternative treatment options for inhibitor patients like immune tolerance induction or treatment with FVIII bypassing agents are available but are very expensive and may reduce quality of life for patients [2]. Due to its monogenic nature, HA is an ideal disease candidate for gene therapy, which might cure the disease if successful. Several gene therapy approaches for the treatment of HA have been described in preclinical and clinical studies [3–5].
Platelet-targeted FVIII gene therapy has been evaluated by several groups and appears promising [6–14]. Our group has previously developed a strategy in which human B-domain deleted FVIII (hBDDFVIII) expression is driven by the αIIb promoter, resulting in FVIII expression in platelets and is hereafter referred to as 2bF8 [7–12]. 2bF8 protects hemophilic mice from lethal blood loss upon vessel injury, does not induce inhibitor development and importantly, is even successful in the treatment of mice with pre-existing inhibitors [7;9;10]. Additionally, 2bF8 gene therapy induces immune tolerance to FVIII allowing the intravenous infusion of recombinant FVIII protein without the formation of inhibitors [12]. Furthermore, 2bF8 has been shown to restore hemostasis in a larger pre-clinical model, the hemophilic dog [15]. Hemostatic efficacy in the absence and in the presence of inhibitors has also been demonstrated by expressing FVIII in platelets using the GPIb promoter [6;13;14]. Thus, targeting FVIII expression to platelets may offer the first permanent haemostatic correction to patients with inhibitors.
While FVIII is normally expressed in endothelial cells and not in platelets [16;17], platelets can express FVIII following platelet-targeted gene therapy. It has been known that platelets not only play important roles in hemostasis, but also in thrombosis and inflammation [18]. Although the platelet-targeted FVIII gene therapy approach is successful in restoring hemostasis, it is warranted to evaluate the safety concerns associated with platelet-targeted FVIII expression before this novel approach can be applied to a clinical trial. A higher embolism rate has been reported in a transgenic mouse model expressing FVIII in platelets driven by the GPIb promoter on an exon 16 disrupted FVIII deficient background [19]. Whether ectopic expression of FVIII to platelets would have a potential thrombotic risk has not been investigated.
In this study we attempted to define the breadth of the therapeutic window and ensure safety by exploring whether supratherapeutic levels of 2bF8 induce a prothrombotic state. We generated a new line of 2bF8 transgenic mice with high levels of platelet-FVIII expression. We found that in steady state as well as under prothrombotic conditions induced by LPS-mediated inflammation or the factor V Leiden mutation, supratherapeutic levels of platelet-FVIII did not appear thrombogenic. Furthermore, FVIII-expressing platelets were neither hyper-activated nor hyper-activatable upon agonist activation.
Materials and methods
Mice
FVIIInull mice (FVIII exon 17 disrupted) [20] in C57BL6/129S mixed background were a kind gift from H. Kazazian (University of Pennsylvania School of Medicine) C57BL6/129S mice served as wild type (WT) controls. Transgenic mice which were on a C57BL6/129S background and expressed high levels of hBDDFVIII in platelets (LV17/18tg mice) were generated by crossing our previously described LV17tg and LV18tg mice [21;22]. Factor V Leiden (FVL) mice were bred in house and derived from the originally described colony [23]. LV18tg mice were crossed with FVL mice for studies of 2bF8 on the FVL background in addition to normal levels of mouse FVIII. Animal studies complied with a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Lipopolysaccharide (LPS) challenge
Mice were injected intraperitoneally with LPS (40 mg kg−1, E coli serotype O55:B5; Sigma Aldrich, St. Louis, MO, USA) or phosphate buffered saline (PBS) as control, followed by subcutaneous injection of 700 μL Lympholyte A (Baxter Healthcare, Deerfield, IL, USA) for supportive care. Blood and tissue samples were obtained 16 hours after LPS challenge. One hour before sample harvest, LPS challenged mice were intraperitoneally injected with 700 μL Lympholyte A for supportive care.
Whole blood, plasma and platelet lysate collection
For all coagulation studies blood of anesthetized mice was drawn from the inferior vena cava (IVC) into 3.8% sodium-citrate at a 9:1 ratio. To determine platelet counts and hematocrit (HCT) blood was counted on a scil Vet ABC hematologic analyzer (scil animal care company, Gurnee, IL, USA). For genotyping and isolation of platelets, blood was drawn from the retro-orbital plexus into 3.8% sodium-citrate at a 9:1 ratio. Platelet poor plasma and platelet lysates were obtained by processing of blood collected from the IVC or retro-orbital bleed as previously described [9].
FVIII activity assays
FVIII activity (FVIII:C) in platelet lysates was determined with a modified chromogenic assay using Coatest SP4 FVIII kit (DiaPharma Group, West Chester, OH, USA) as previously described [7]. Dilutions of hBDDFVIII (rhFVIII, Xyntha, Pfizer Inc, New York, NY, USA) were used to create a standard curve for measuring FVIII:C levels.
Measurement of thrombin-antithrombin-III complexes (TAT), D-Dimer, and fibrinogen
Blood was drawn from the IVC and platelet poor plasma was prepared for determination of TAT, D-Dimer and fibrinogen levels. Commercially available assay kits were used to assess levels of TAT (Enzygnost TAT micro, Siemens, Marburg, Germany) and D-Dimer (Asserachrom D-Di, Diagnostica Stago, Parsippany, NJ, USA). Fibrinogen levels were detected by ELISA as previously described [24].
Immunohistochemistry
Anesthetized mice were perfused with 20 mL PBS through the left ventricle. The liver was removed, fixed in 10% formalin and embedded in paraffin. After rehydration, antigen retrieval, peroxidase block, and protein block, sections were incubated overnight with goat anti-mouse fibrinogen antibody (Nordic Immunological Laboratories) in a humidified chamber. Anti-goat IgG peroxidase (Vector Laboratories, Burlingame, CA, USA) was used as secondary antibody for 30 minutes. Antigen-antibody complexes were visualized with ImmPACT DAB Peroxidase Substrate (Vector Laboratories) and counterstained with hematoxylin and eosin.
Ferric chloride induced carotid artery injury model
To assess in vivo clot formation we modified a previously reported protocol [25]. Briefly, the right carotid artery of anesthetized mice was exposed. A 1×2 mm filter paper (Whatman #1, GE Healthcare, Pittsburgh, PA, USA) soaked in 20% ferric chloride (Sigma Aldrich, St. Louis, MO, USA) was applied to the carotid artery, removed after 3 minutes and the surface of the artery carefully washed 3-times with warm PBS to remove residual ferric chloride. A Doppler ultrasound flow probe (Model MA0.5PSB, Transonic Systems, Ithaca, NY, USA) was placed on the artery to monitor blood flow after injury. Time to occlusion (TTO) of the carotid artery was defined as the time from removal of the filter paper to a lack of blood flow for 3 consecutive minutes. The maximum observation time was 45 minutes.
Whole blood coagulation assays
Thrombin generation in whole blood was determined using our recently reported native whole blood thrombin generation assay (nWB-TGA) [21]. Briefly, 15 μL whole blood drawn from the IVC was recalcified without the addition of tissue factor in the presence of a rhodamine-based, thrombin-cleavable, fluorescent substrate (Invitrogen, Carlsbad, CA, USA). The reaction mix was added to a filter paper disk placed in a 96 dark well plate. Increase of fluorescence was monitored over time and thrombin generation was calculated with Technothrombin TGA evaluation software (Technoclone, Vienna, Austria) based on a calibration experiment using a thrombin standard. Whole blood clot formation was determined by rotational thromboelastometry (ROTEM) (TEM Systems, Muenchen, Germany). ROTEM mini cups were preloaded with 7 μL of 0.2 M CaCl2, 105 μL of whole blood was added and clot formation was recorded over 90 minutes using the NATEM assay function.
Assessment of platelet activation by whole blood flow cytometry
Whole blood drawn from the IVC (2 μL) was carefully combined with indicated concentrations of ADP (Chronolog, Havertown, PA,USA), mouse PAR4 ligand (Gly-Tyr-Pro-Gly-Lys-Phe-NH2, GYPGKF-NH2, made in house by our protein core facilities) or Tyrode buffer as control and the following antibodies: Dylight 649-conjugated anti-mouse GPIb, PE-conjugated anti-mouse CD41 (clone: JON/A), FITC-conjugated anti-mouse P-selectin (all antibodies were from Emfret, Eibelstadt, Germany). To determine frequency of platelet leukocyte aggregates (PLA), whole blood was combined with 20 μM ADP, 1 mM PAR4 or Tyrode buffer as control and the following antibodies: APC-eFluor 780-conjugated anti-mouse CD45.2 (eBioscience, San Diego, CA, USA), PE-conjugated anti-mouse integrin αIIb (GPIIb, Santa Cruz Biotechnology Inc, Dallas, TX, USA). Reaction mixtures (40 μL final volume) were incubated at room temperature in the dark for 20 minutes and quenched with a 10-fold excess volume of Tyrode buffer. Data was acquired on an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and evaluated with FlowJo X10 software (FlowJo, Ashland, OR, USA).
Statistical analysis
Statistical analysis was performed with GraphPadPrism 4 software (GraphPad Software, La Jolla, CA, USA). Statistical differences between groups were determined by the nonparametric Mann-Whitney test or the Student’s t-test. Chi-square test was used to assess Mendelian distribution of mouse genotypes. All data are presented as mean plus or minus standard deviation (SD). A p-value of P < 0.05 was considered statistically significant.
Results
Characterization of transgenic mouse model expressing supratherapeutic levels of FVIII in platelets
In order to evaluate a potential risk for thrombosis involved with platelet expressed FVIII (2bF8) we developed a transgenic mouse model expressing higher 2bF8 levels than we were previously able to achieve. To accomplish this, we crossed two founder lines, LV17tg and LV18tg, which we obtained by lentivirus mediated transgenesis. We previously described LV18tg mice to have a single transgene insert into the deoxyribonuclease 1 like 2 gene on chromosome 17 [21]. LV17tg mice also carried a single insert of the transgene, but in a different chromosomal location than LV18tg mice. In LV17tg mice, the transgene integrated into the oxysterol binding protein-like protein 7 gene on chromosome 11. Both lines were on a FVIII deficient background. LV18tg mice expressed FVIII at 11.7 ± 1.1 mU per 108 platelets when homozygous [21]. LV17tg mice expressed 6.9 ± 1.7 mU per 108 platelets when homozygous. When crossing the two lines, the resulting LV17/18tg mice expressed 23.4 ± 2.4 mU per 108 platelets when homozygous for both traits (Fig. 1). This 2bF8 level was approximately 30-fold higher than therapeutically required to restore hemostasis in our originally described 2bF8trans mouse line (0.75 mU per 108 platelets) [7], and allowed evaluating the thrombosis risk of supratherapeutic levels of platelet-expressed FVIII.
Fig. 1. Characterization of novel transgenic mouse model expressing supratherapeutic levels of 2bF8 on FVIIInull background (LV17/18tg mice).
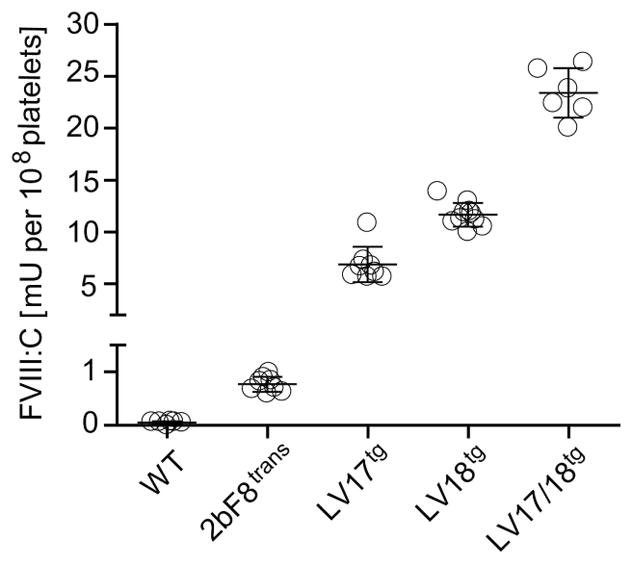
FVIII:C in platelet lysates from WT mice (n = 6), and homozygous LV17tg (n = 8), LV18tg (n = 10), and LV17/18tg (n = 6) mice, and from 2bF8trans mice (n = 8) determined by chromogenic FVIII:C assay. LV17/18tg mice expressed 30-fold greater levels of FVIII in platelets than 2bF8trans mice in which hemostasis on an endogenous FVIII-deficient background was restored.
Mice expressing high levels of 2bF8 do not show signs of increased thrombosis risk under steady state
To evaluate the risk of thrombosis in LV17/18tg mice we first determined levels of plasma parameters which have been associated with increased thrombosis risk and compared them to WT control mice. Fibrinogen and D-Dimer levels were similar in WT and LV17/18tg mice (Fig. 2A and 2B). While TAT levels were significantly elevated in LV17/18tg mice compared with WT mice, FVIII deficient mice also had similarly elevated levels of TAT as LV17/18tg mice. This would suggest that the FVIII deficient background rather than 2bF8 might be the cause for this increase (Fig. 2C). Next, we determined with whole blood assays if thrombin generation or ex vivo clot formation were increased in LV17/18tg mice. Using our previously reported [21] native whole blood thrombin generation assay (nWB-TGA) we found that thrombin generation was not accelerated above WT levels in the transgenic mice. In WT and LV17/18tg, respectively, peak thrombin was 216 ± 41 and 195 ± 32 nM and endogenous thrombin potential was 1718 ± 110 and 1642 ± 139 nM. Lag and peak time were somewhat longer in LV17/18tg compared with WT mice. In comparison, thrombin generation in FVIIInull mice was dramatically lower than in LV17/18tg or WT mice (Table 1). Clotting time determined by rotational thromboelastometry (ROTEM) in WT and LV17/18tg, respectively, was 424 ± 54 and 705 ± 91 seconds and maximum clot firmness was 51.3 ± 3.7 and 54.0 ± 4.1 mm (Table 1). Thus, under steady state, platelet-FVIII did not appear prothrombotic using either approach.
Fig. 2. Impact of supratherapeutic levels of 2bF8 on thrombosis risk factors in steady state.

Levels of (A) fibrinogen, (B) D-Dimer, and (C) TAT were evaluated by ELISA in plasma from WT, LV17/18tg and FVIIInull mice as indicated. ***P < 0.001(Student’s t-test).
Table 1.
Comparison of whole blood thrombin generation, in vivo and ex vivo clot formation in WT, LV17/18tg and FVIIInull mice.
| WT | LV17/18tg | FVIIInull | ||
|---|---|---|---|---|
| nWB-TGA | Lag time (min) | 9.1 ± 1.4 * | 13.8 ± 1.5 | 55.6 ± 27.0 ** |
| Peak time (min) | 15.1 ± 2.6 * | 23.5 ± 2.0 | 61.7 ± 22.8 ** | |
| Peak thrombin (nM) | 216 ± 41 | 195 ± 32 | 9.9 ± 10.7 ** | |
| ETP (nM) | 1718 ± 110 | 1642 ± 139 | 115.7 ± 155.4 ** | |
| TGR (nM/min) | 38.3 ± 15.7 | 29.0 ± 7.7 | 1.3 ± 1.2 ** | |
| N (#) | 7 | 6 | 15 | |
| Ex vivo thrombus formation | CT (sec.) | 424 ± 54 * | 705 ± 91 | 6292 ± 709 ** |
| CFT (sec.) | 180 ± 45 | 225 ± 54 | 950, 995, X, X | |
| MCF | 51.3 ± 3.7 | 54.0 ± 4.1 | X | |
| alpha angle (°) | 58.1 ± 5.9 | 52.0 ± 6.5 | X | |
| N (#) | 7 | 6 | 4 | |
| In vivo thrombus formation | TTO (min) | 6.7 ± 1.6 | 10.6 ± 3.0 | >45 ** |
| N (#) | 4 | 4 | 3 |
Thrombin generation in whole blood of WT, LV17/18tg and FVIIInull mice was assessed using nWB-TGA. The five thrombin generation parameters lag time, peak time, peak thrombin, endogenous thrombin potential (ETP) and thrombin generation rate (TGR) are listed for indicated mice. Ex vivo clot formation in whole blood from WT, LV17/18tg and FVIIInull mice was evaluated with ROTEM. Clotting time (CT), clot formation time (CFT), maximum clot firmness (MCF) and the alpha angle are listed. Data from single animals are shown for CFT of FVIIInull mice because 2 mice had not determinable (X) MCF in the 90 min assay. Time to occlusion (TTO) of carotid artery was determined upon ferric chloride induced vessel injury in indicated mice using a Doppler ultrasound flow probe.
P < 0.05 for comparison of WT and LV17/18tg mice.
P < 0.001 for comparison of FVIIInull and LV17/18tg mice.
The Mann-Whitney test, which allows analysis of data with infinite read times scored at the maximum observation period (90 min for lag and peak time, 45 min for TTO) was used for comparisons of nWB-TGA lag and peak time and in vivo clot formation TTO between FVIIInull and LV17/18tg mice. For all other comparisons the Student’s t-test was used.
Supratherapeutic platelet-FVIII does not enhance in vivo clot formation
To determine if 2bF8 accelerates in vivo thrombus formation above WT levels, we exposed mice to ferric chloride induced carotid artery injury. Time to occlusion (TTO) after vessel injury in LV17/18tg mice was 10.6 ± 3.0 minutes and was not statistically different from that obtained in WT mice (6.7 ± 1.6 minutes). In contrast, vessels in F8null mice did not occlude throughout the entire 45 minute observation period (Table 1). Thus, 2bF8 does not increase in vivo clot formation above that observed in WT mice and does correct the clotting time in the ferric chloride injury model.
Inflammation does not trigger abnormal thrombosis risk in mice with supratherapeutic levels of platelet FVIII
Up-regulation of acute phase cytokines during inflammation has been shown to induce a prothrombotic state [26–29]. To determine if 2bF8 elevated the risk for thrombosis under prothrombotic conditions we subjected LV17/18tg and WT mice to LPS challenge. Fluid injections were provided as supportive care to normalize the hematocrit in LPS challenged mice (Fig. 3A). The drop in platelet counts upon LPS challenge, indicating beginning signs of disseminated intravascular coagulopathy, was similar in WT and LV17/18tg mice (Fig. 3B). Fibrinogen, D-Dimer and TAT levels were similarly elevated in WT and LV17/18tg mice during inflammation. Fibrinogen levels were 1.6-fold increased in WT mice and 1.3-fold increased in LV17/18tg mice upon LPS injections (Fig. 3C), D-Dimer levels were 14.7-fold and 8-fold elevated in WT and LV17/18tg mice, respectively (Fig. 3D), and TAT levels were 3.3-fold increased in WT and 2.2-fold increased in LV17/18tg mice after LPS challenge (Fig. 3E). Fibrinogen, D-Dimer and TAT levels were not statistically significantly different between LV17/18tg and WT mice after LPS challenge (Fig. 3C–E). Fibrin deposition upon LPS challenge appeared similar in livers of WT and LV17/18tg mice, while no fibrin deposition was observed in PBS treated control animals (Fig. 3F). In summary, 2bF8 did not induce an increased thrombosis risk under LPS-induced inflammatory conditions.
Fig. 3. Impact of supratherapeutic levels of 2bF8 on thrombosis risk during inflammation.

WT or LV17/18tg mice were challenged intraperitoneally with 40 mg kg−1 LPS or injected with PBS as control. Samples were collected after 16 hours and (A) hematocrit (HCT) and (B) platelet counts were assessed on an animal blood counter. (C) Fibrinogen levels, (D) D-Dimer levels, and (E) TAT levels were determined by ELISA. ***P < 0.001(Student’s t-test). Compared with PBS treated animals, LPS challenge increased fibrinogen levels 1.6 and 1.3-fold, D-Dimer levels 14.7 and 8.0-fold and TAT levels 3.3 and 2.2-fold in WT and LV17/18tg mice, respectively. (F) Fibrin deposition in the livers of indicated mice was evaluated by immunohistochemistry. Representative images from 1 of 3 mice per group are shown.
Platelet expressed FVIII does not increase factor V Leiden (FVL) prothrombotic phenotype
Activated protein C-resistance due to the Leiden mutation in coagulation factor V (FV) (human: R506Q; mouse R504Q) substantially increases the thrombosis risk in homozygous carriers in humans and mice [23;30]. To evaluate the effect of supra-therapeutic platelet FVIII levels in the thrombosis-sensitizing background, LV18tg mice that expressed FVIII at approximately 12 mU per 108 platelets were crossed onto the prothrombotic FVL background in addition to 100% of mouse plasma FVIII. In striking contrast to heterozygous TFPI-deficiency, which results in lethal intra-uterine thrombosis of homozygous FVL mice already before birth [31], we obtained a normal Mendalian distribution of 23.3% LV18+/+ FVL+/+, 50% LV18+/− FVL+/+ and 26.7% LV18−/− FVL+/+ mice (P = 0.97, Chi-squared test, n = 30) indicating that 2bF8 does not impact survival on the FVL background. Fibrinogen levels were similar among all 3 groups of mice with an average of approximately 2500 μg mL−1 (Fig. 4A). D-Dimer and TAT levels were significantly elevated in FVL (LV18−/−FVL+/+) compared with WT mice (D-Dimer: 5.2 ± 5.5 vs 22.8 ± 19.1 ng mL−1; TAT: 7.0 ± 1.3 vs 15.6 ± 6.2 ng mL−1). Importantly, 2bF8 in LV18+/+FVL+/+ mice did not contribute to a further increase in D-Dimer (13.1 ± 10.5 ng mL−1) or TAT (10.5 ± 2.4 ng mL−1) levels (Fig. 4B and 4C). These data demonstrate that 2bF8 does not induce an increased thrombosis risk in a pre-existing thrombosis-prone background.
Fig. 4. Thrombosis risk of 2bF8 on FVL prothrombotic background.

Levels of (A) fibrinogen, (B) D-Dimer, and (C) TAT were evaluated by ELISA in plasma of WT mice, and FVL mice that do not (LV18tg−/−) or do (LV18tg +/+) express platelet FVIII. All mice also expressed 100% endogenous plasma FVIII. *P < 0.05, ***P < 0.001 (Student’s t-test).
Normal platelet function in mice with supra-therapeutic platelet FVIII levels
We finally asked if platelets expressing FVIII were hyper-activated or hyper-activatable upon agonist stimulation compared to WT platelets. We employed a whole blood flow cytometry based assay to evaluate activation of the αIIbβ3 integrin using the PE-conjugated JON/A antibody, which specifically binds to the activated form of the integrin [32] and P-selectin expression on platelets to evaluate the α-granule exocytosis pathway. Without stimulation, frequencies and MFI of JON/A binding and P-selectin staining showed similar background levels on WT and LV17/18tg platelets. Stimulation with increasing concentrations of PAR4 agonist dose-dependently elevated αIIbβ3 integrin activation and P-selectin expression. ADP stimulation on the other hand induced αIIbβ3 integrin activation but not P-selectin expression (Fig. 5A). Significantly higher frequencies and MFI of JON/A binding upon ADP stimulation were observed in LV17/18tg compared with WT platelets, which were of mixed C57BL6/129S background (Fig. 5B and 5C). A similar trend for JON/A binding was found after stimulation with PAR4 agonist (Fig. 5B). P-selectin expression upon PAR4 agonist stimulation was overall comparable between LV17/18tg and WT platelets, with the exception that 0.5 mM PAR4 agonist induced a higher frequency of P-selectin expressing platelets (Fig. 5D and 5E). Although LV17/18tg platelets appeared hyper-activatable compared with mixed background (C57BL6/129S) WT platelets, the activation level of LV17/18tg platelets under all stimulation conditions was similar or lower than the activation level of WT platelets from a different colony of mice that were on a pure C57BL6 background (Fig. 5B, 5C and 5E).
Fig. 5. Comparison of platelet activation and activatability in WT and 2bF8 expressing mice.

Activation status and activatability of platelet from WT (mixed C57BL6/129S background, n = 5 – 8), LV17/18tg (n = 4 – 7) or pure C57BL6 background WT (n = 3 – 4) mice were assessed using whole blood flow cytometry. (A) Representative dot plots show gating strategy to assess activation status of platelets. Values shown indicate the percentage of cell population. (B) MFI (mean fluorescence intensity) and (C) frequency of JON/A binding platelets are shown. (D) MFI and (E) frequency of surface P-selectin expressing platelets are shown. Indicated concentrations of ADP or PAR4 agonist were used in vitro to activate platelets. Although LV17/18tg platelets appeared hyper-activated compared with platelets from mixed 129S/C57BL6 background WT mice, platelet activation in LV17/18tg mice under all stimulation conditions was similar or lower than in WT mice on a pure C57BL6 background. Significance levels between WT and L17/18tg samples are indicated. *P < 0.05, **P < 0.01, ***P < 0.001(Student’s t-test).
Activated platelets bind to leukocytes and form platelet leukocytes aggregates (PLA) [33;34]. Monitoring of PLA has been suggested for assessment of thrombosis [35]. The frequency of PLA was approximately 7% in normal murine whole blood and similar in LV17/18tg and WT mice in steady state. While stimulation with ADP did not increase PLA, PAR4 agonist significantly elevated PLA levels to the same extent in LV17/18tg and WT mice (Fig. 6A and 6B). Together, our data show that platelets expressing supratherapeutic levels of FVIII are not hyper-activated or hyper-activateable compared with WT platelets.
Fig. 6. Assessment of platelet-leukocyte aggregates (PLA) in WT and 2bF8 expressing mice.
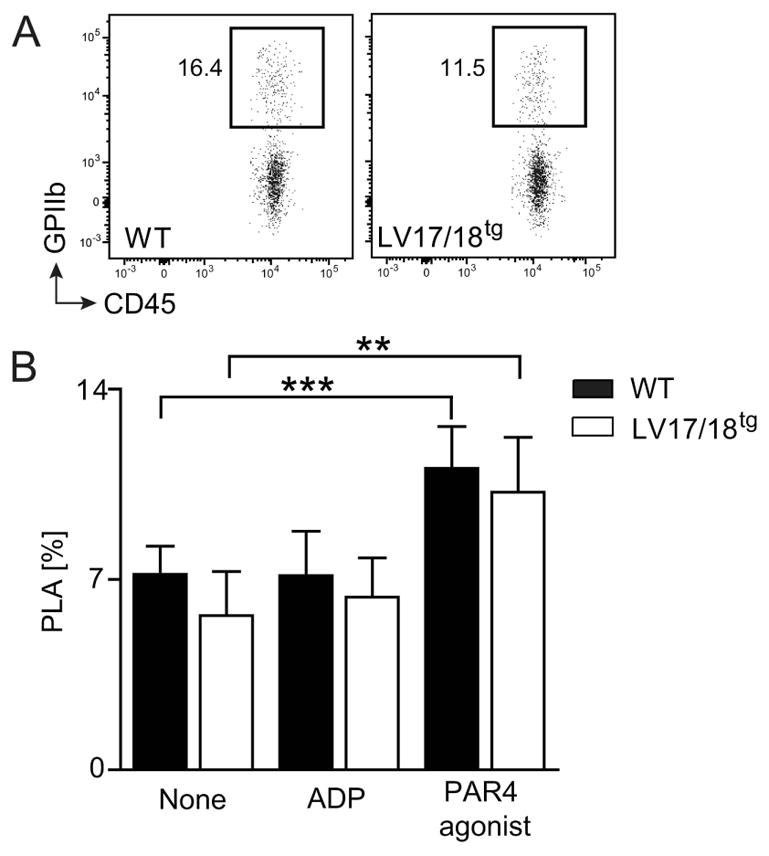
GPIIb+CD45+ double positive PLA in WT and LV17/18tg mice were assessed using whole blood flow cytometry. (A) Representative dot plots of PLA pre-gated on CD45+ cells in whole blood of WT and LV17/18tg mice are shown. Values shown indicate the percentage of cell population. (B) Frequency of PLA among CD45+ events in WT (n = 7) and LV17/18tg (n = 6) blood without stimulation or upon in vitro stimulation with 20 μM ADP or 1 mM PAR4 agonist are depicted. *P< 0.05 (Student’s t-test).
Discussion
Platelet-targeted FVIII gene therapy has been proven to restore hemostasis in pre-clinical hemophilia A models [6–14]. Since platelets play fundamental roles not only in hemostasis but also in thrombosis, it is important to evaluate whether there are potential pathological consequences associated with platelet-targeted FVIII expression as a means of gene therapy for hemophilia A before considering this approach for clinical application. In this study we demonstrate that supratherapeutic levels of FVIII expressed in platelets by the αIIb promoter appear non-thrombogenic in steady state as well as under prothrombotic conditions induced by LPS-mediated inflammation or the factor V Leiden mutation. Furthermore, targeting FVIII expression to platelets does not induce platelet hyper-activation or hyper-responsiveness.
We have previously reported that in a transgenic mouse model the FVIII expression of 0.75 mU per 108 platelets restored hemostasis on a hemophilic background [7]. For the clinically applicable gene therapy protocol, ex vivo transduction of bone marrow with a lentivirus carrying the 2bF8 gene cassette and transplantation of transduced bone marrow into preconditioned hosts is required. Due to transduction and gene expression efficiency, expression levels of 2bF8 between individuals may vary. In an attempt to define the breadth of the therapeutic window, we here developed transgenic mice that expressed FVIII at 23.4 mU per 108 platelets, which is an approximately 30-fold higher platelet-FVIII level than therapeutically required. We subjected these mice to a variety of methods for the assessment of a prothrombotic state including nWB-TGA, ex vivo whole blood clot formation analysis using ROTEM, in vivo clot formation upon ferric chloride induced carotid artery injury, and tissue fibrin deposition. We also evaluated plasma parameters associated with increased thrombosis risk, including D-Dimer [36–39], TAT [40], and fibrinogen [25]. None of these assays and parameters with the exception of TAT levels was elevated over WT levels in LV17/18tg mice in steady state.
Levels of TAT were about double in LV17/18tg compared to WT mice. We also detected elevated TAT levels in FVIIInull mice, which was similar to the levels obtained in LV17/18tg mice and therefore attributed the TAT increase to the FVIII-deficient background. Other studies did not report elevated TAT levels in hemophilia A or B mice compared with WT mice [41;42]. The different outcomes compared to our studies might be due to mouse strain differences and/or differences in plasma preparation. Plasma for TAT measurement was obtained by tail clipping in the aforementioned studies, which might have impacted sample activation. We obtained plasma by IVC draw, which is considered the blood drawing method in mice that results in least activated blood due to minimal tissue factor contamination and platelet activation [43]. In our studies, samples drawn from the IVC showed little activation, which was confirmed by a low VWF propeptide/VWF antigen ratio (<0.5, data not shown). The VWF propeptide/VWF antigen ratio is an indicator for sample activation mainly through platelet activation with a ratio below 0.5 indicating very little to no sample activation [44]. Low level activation in other studies might mask the differences in TAT levels that we found in our study.
To determine if 2bF8 exaggerates the thrombotic risk in the face of prothrombotic conditions, we applied two different approaches. Both inflammation and the FVL mutation have been shown to induce a prothrombotic state in humans and preclinical models [23;28;30;45]. We challenged mice with LPS to induce inflammation and in a second approach we backcrossed 2bF8 onto the prothrombotic FVL background. As expected, we detected elevated levels of fibrinogen, D-Dimer and TAT, and tissue fibrin deposition after LPS challenge in WT mice. The FVL mutation, a R506Q mutation in the FV gene, expectedly resulted in elevated levels of D-Dimer and TAT on an otherwise WT background. Importantly, these levels were not further elevated by 2bF8; not even in FVL mice that additionally expressed endogenous FVIII. These data demonstrate that 2bF8 does not appear thrombogenic even under prothrombotic conditions.
Hyper-activation and hyper-activatability of platelets increase the risk for thrombosis [46;47]. We found that platelet activation determined by P-selectin exposure and αIIbβ3 integrin activation in steady state (without stimulation) was similar whether platelets expressed FVIII or not. PAR4 agonist and ADP stimulation of whole blood dose-dependently increased platelet activation. Although we observed higher platelet activation in LV17/18tg than in mixed 129S/C57BL6 background WT mice, platelet activation in LV17/18tg mice was lower or similar as in WT mice on a pure C57BL6 background. Mouse strain-specific differences in platelet responsiveness have been reported [48]. Platelets from 129S mice appeared less sensitive to agonist stimulation compared with platelets from C57BL6 mice. It is possible that in our 129S/C57BL6 mixed background LV17tg and LV18tg transgenic founder colonies, which have been maintained in our facility for several generations, genetic traits affecting platelet activation, specifically PAR4 and ADP signaling, were preferentially propagated from the C57BL6 background. Whether signaling pathways resulting in platelet activation triggered by other agonists than PAR4-agonist or ADP show similar differences is unclear. The exact mechanism and potential differentially expressed molecules involved in platelet signaling and activation in different mouse strains have yet to be determined.
It has been reported that platelet-targeted gene therapy directed by the GPIb promoter induces apoptosis in megakaryocytes resulting in a 30% reduction in platelet counts after therapy [49]. However, the platelet number was fully recovered after bone marrow reconstitution in 2bF8 gene therapy in which FVIII expression is driven by the αIIb promoter [12]. Aside from demonstrating that 2bF8 does not hyper-activate platelets, our studies show that 2bF8 does not impair platelet function as assessed with whole blood flow cytometry, whether we used weak or strong platelet activation. Our data is in agreement with studies performed by Damon et al [50] who showed that expression of FVIII under the control of the platelet-specific PF4 promoter did not impair PS-exposure or P-selectin expression upon stimulation with strong platelet agonists (thrombin or ionophore).
In conclusion, in the current study, we demonstrated that supra-therapeutic levels of 2bF8, which are 30-fold in excess of 2bF8 levels required for restoring hemostasis in hemophilic mice, appear non-thrombogenic and do not induce platelet hyper-activation or hyper-responsiveness. We showed that even under prothrombotic conditions induced by inflammation or FVL, platelet-targeted FVIII expression is not associated with increased thrombogenicity in mice. Thus, in regard to thrombosis risk, platelet-targeted FVIII gene therapy does not appear to raise safety concerns and seems to have a relatively wide therapeutic window. Targeting FVIII expression to platelets represents a promising approach to permanent restoration of hemostasis in patients with hemophilia A, but this needs to be tested in clinical trials. Even patients with co-existing thrombophilic disorders need not be excluded from platelet-targeted gene therapy.
Essentials.
Platelet-Factor (F) VIII gene therapy is a promising treatment in hemophilia A.
This study aims to evaluate if platelet-FVIII expression would increase the risk for thrombosis.
Targeting FVIII expression to platelets does not induce or elevate thrombosis risk.
Platelets expressing FVIII are neither hyper-activated nor hyper-responsive.
Acknowledgments
We thank Rachel Bercovitz at our institution for help with establishing whole blood flow cytometry for the detection of platelet activation, and Brian Cooley at the University of North Carolina (Chapel Hill) for critical discussion on the ferric chloride induced carotid artery injury model. This work was supported by the Novo Nordisk Access-to-Insight initiative (C.K.B.), the BloodCenter Research Foundation (R.R.M. and Q.S.), the National Institutes of Health grants HL-5P01HL044612 (R.R.M.), HL-5P01HL081588 (R.R.M.), HL-7R01HL112641 (R.R.M.), HL-R01HL102035 (Q.S.), the MACC fund (Q.S.), the Children’s Hospital Foundation (Q.S.), and funding from the Medical College and the Children’s Hospital of Wisconsin to the Pediatric Cardiac Hemostasis and Thrombosis (PCH&T) Research-Center (R.R.M.).
Footnotes
Authorship contributions:
C. K. Baumgartner designed and performed experiments, analyzed and interpreted data, and wrote manuscript. J. Mattson performed experiments. H. Weiler facilitated transgenic mouse generation and helped designed experiments. Q. Shi helped design experiments, analyzed and interpreted data, and revised manuscript. R. R. Montgomery helped design experiments, analyzed and interpreted data, and made critical comments on manuscript.
Disclosure of Conflict of Interest:
The authors state that they have no conflict of interest.
References
- 1.Ehrenforth S, Kreuz W, Scharrer I, Linde R, Funk M, Gungor T, Krackhardt B, Kornhuber B. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339:594–8. doi: 10.1016/0140-6736(92)90874-3. [DOI] [PubMed] [Google Scholar]
- 2.Kempton CL, Meeks SL. Toward optimal therapy for inhibitors in hemophilia. Blood. 2014;124:3365–72. doi: 10.1182/blood-2014-05-577643. [DOI] [PubMed] [Google Scholar]
- 3.Monahan PE, Gui T. Gene therapy for hemophilia: advancing beyond the first clinical success. Curr Opin Hematol. 2013;20:410–6. doi: 10.1097/MOH.0b013e328363c1a1. [DOI] [PubMed] [Google Scholar]
- 4.Roth DA, Tawa NE, Jr, O’Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344:1735–42. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 5.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, Moon TE, Cole V, Ramanathan-Girish S, Roehl H, Sajjadi N, Jolly DJ, Hurst D. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–45. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 6.Yarovoi HV, Kufrin D, Eslin DE, Thornton MA, Haberichter SL, Shi Q, Zhu H, Camire R, Fakharzadeh SS, Kowalska MA, Wilcox DA, Sachais BS, Montgomery RR, Poncz M. Factor VIII ectopically expressed in platelets: efficacy in hemophilia A treatment. Blood. 2003;102:4006–13. doi: 10.1182/blood-2003-05-1519. [DOI] [PubMed] [Google Scholar]
- 7.Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, Cooley BC, Desai D, Morateck PA, Gorski J, Montgomery RR. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116:1974–82. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Q, Wilcox DA, Fahs SA, Fang J, Johnson BD, DU LM, Desai D, Montgomery RR. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5:352–61. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- 9.Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, Weiler H, Montgomery RR. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112:2713–21. doi: 10.1182/blood-2008-02-138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuether EL, Schroeder JA, Fahs SA, Cooley BC, Chen Y, Montgomery RR, Wilcox DA, Shi Q. Lentivirus-mediated platelet gene therapy of murine hemophilia A with pre-existing anti-factor VIII immunity. J Thromb Haemost. 2012;10:1570–80. doi: 10.1111/j.1538-7836.2012.04791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Q, Kuether EL, Chen Y, Schroeder JA, Fahs SA, Montgomery RR. Platelet gene therapy corrects the hemophilic phenotype in immunocompromised hemophilia A mice transplanted with genetically manipulated human cord blood stem cells. Blood. 2014;123:395–403. doi: 10.1182/blood-2013-08-520478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroeder JA, Chen Y, Fang J, Wilcox DA, Shi Q. In vivo enrichment of genetically manipulated platelets corrects the murine hemophilic phenotype and induces immune tolerance even using a low multiplicity of infection. J Thromb Haemost. 2014;12:1283–93. doi: 10.1111/jth.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greene TK, Wang C, Hirsch JD, Zhai L, Gewirtz J, Thornton MA, Miao HZ, Pipe SW, Kaufman RJ, Camire RM, Arruda VR, Kowalska MA, Poncz M. In vivo efficacy of platelet-delivered, high specific activity factor VIII variants. Blood. 2010;116:6114–22. doi: 10.1182/blood-2010-06-293308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gewirtz J, Thornton MA, Rauova L, Poncz M. Platelet-delivered factor VIII provides limited resistance to anti-factor VIII inhibitors. J Thromb Haemost. 2008;6:1160–6. doi: 10.1111/j.1538-7836.2008.02992.x. [DOI] [PubMed] [Google Scholar]
- 15.DU LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, Haberichter SL, Merricks E, Raymer RA, Fang J, Koukouritaki SB, Jacobi PM, Hawkins TB, Cornetta K, Shi Q, Wilcox DA. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the predominant and possibly exclusive source of plasma factor VIII. Blood. 2014 doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003;23:2131–7. doi: 10.1161/01.ATV.0000095974.95122.EC. [DOI] [PubMed] [Google Scholar]
- 19.Neyman M, Gewirtz J, Poncz M. Analysis of the spatial and temporal characteristics of platelet-delivered factor VIII-based clots. Blood. 2008;112:1101–8. doi: 10.1182/blood-2008-04-152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 21.Baumgartner CK, Zhang G, Kuether EL, Weiler H, Shi Q, Montgomery RR. Comparison of platelet-derived and plasma factor VIII efficacy using a novel native whole blood thrombin generation assay. J Thromb Haemost. 2015;13:2210–9. doi: 10.1111/jth.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Schroeder JA, Chen J, Luo X, Baumgartner CK, Montgomery RR, Hu J, Shi Q. The immunogenicity of platelet-derived FVIII in hemophilia A mice with or without preexisting anti-FVIII immunity. Blood. 2016;127:1346–54. doi: 10.1182/blood-2015-08-662916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui J, Eitzman DT, Westrick RJ, Christie PD, Xu ZJ, Yang AY, Purkayastha AA, Yang TL, Metz AL, Gallagher KP, Tyson JA, Rosenberg RD, Ginsburg D. Spontaneous thrombosis in mice carrying the factor V Leiden mutation. Blood. 2000;96:4222–6. [PubMed] [Google Scholar]
- 24.Weiler H, Lindner V, Kerlin B, Isermann BH, Hendrickson SB, Cooley BC, Meh DA, Mosesson MW, Shworak NW, Post MJ, Conway EM, Ulfman LH, von Andrian UH, Weitz JI. Characterization of a mouse model for thrombomodulin deficiency. Arterioscler Thromb Vasc Biol. 2001;21:1531–7. doi: 10.1161/hq0901.094496. [DOI] [PubMed] [Google Scholar]
- 25.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117:4953–63. doi: 10.1182/blood-2010-11-316885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida H, Russell J, Senchenkova EY, Almeida Paula LD, Granger DN. Interleukin-1beta mediates the extra-intestinal thrombosis associated with experimental colitis. Am J Pathol. 2010;177:2774–81. doi: 10.2353/ajpath.2010.100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida H, Yilmaz CE, Granger DN. Role of tumor necrosis factor-alpha in the extraintestinal thrombosis associated with colonic inflammation. Inflamm Bowel Dis. 2011;17:2217–23. doi: 10.1002/ibd.21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 29.Kerr R, Stirling D, Ludlam CA. Interleukin 6 and haemostasis. Br J Haematol. 2001;115:3–12. doi: 10.1046/j.1365-2141.2001.03061.x. [DOI] [PubMed] [Google Scholar]
- 30.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de RH, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–7. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 31.Eitzman DT, Westrick RJ, Bi X, Manning SL, Wilkinson JE, Broze GJ, Ginsburg D. Lethal perinatal thrombosis in mice resulting from the interaction of tissue factor pathway inhibitor deficiency and factor V Leiden. Circulation. 2002;105:2139–42. doi: 10.1161/01.cir.0000017361.39256.82. [DOI] [PubMed] [Google Scholar]
- 32.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. 2002;48:80–6. doi: 10.1002/cyto.10114. [DOI] [PubMed] [Google Scholar]
- 33.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood. 1991;78:1730–7. [PubMed] [Google Scholar]
- 34.Li N, Hu H, Lindqvist M, Wikstrom-Jonsson E, Goodall AH, Hjemdahl P. Platelet-leukocyte cross talk in whole blood. Arterioscler Thromb Vasc Biol. 2000;20:2702–8. doi: 10.1161/01.atv.20.12.2702. [DOI] [PubMed] [Google Scholar]
- 35.Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, Marchese P, Frelinger AL, III, Goldberg RJ, Michelson AD. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol. 2001;38:1002–6. doi: 10.1016/s0735-1097(01)01485-1. [DOI] [PubMed] [Google Scholar]
- 36.Wada H, Kobayashi T, Abe Y, Hatada T, Yamada N, Sudo A, Uchida A, Nobori T. Elevated levels of soluble fibrin or D-dimer indicate high risk of thrombosis. J Thromb Haemost. 2006;4:1253–8. doi: 10.1111/j.1538-7836.2006.01942.x. [DOI] [PubMed] [Google Scholar]
- 37.Eichinger S, Minar E, Bialonczyk C, Hirschl M, Quehenberger P, Schneider B, Weltermann A, Wagner O, Kyrle PA. D-dimer levels and risk of recurrent venous thromboembolism. JAMA. 2003;290:1071–4. doi: 10.1001/jama.290.8.1071. [DOI] [PubMed] [Google Scholar]
- 38.Wells PS, Anderson DR, Rodger M, Forgie M, Kearon C, Dreyer J, Kovacs G, Mitchell M, Lewandowski B, Kovacs MJ. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med. 2003;349:1227–35. doi: 10.1056/NEJMoa023153. [DOI] [PubMed] [Google Scholar]
- 39.Ridker PM, Hennekens CH, Cerskus A, Stampfer MJ. Plasma concentration of cross-linked fibrin degradation product (D-dimer) and the risk of future myocardial infarction among apparently healthy men. Circulation. 1994;90:2236–40. doi: 10.1161/01.cir.90.5.2236. [DOI] [PubMed] [Google Scholar]
- 40.Ginsberg JS, Brill-Edwards P, Panju A, Patel A, McGinnis J, Smith F, Dale I, Johnston M, Ofosu F. Pre-operative plasma levels of thrombin-antithrombin III complexes correlate with the development of venous thrombosis after major hip or knee surgery. Thromb Haemost. 1995;74:602–5. [PubMed] [Google Scholar]
- 41.Schlachterman A, Schuettrumpf J, Liu JH, Furlan FC, Toso R, Poncz M, Camire RM, Arruda VR. Factor V Leiden improves in vivo hemostasis in murine hemophilia models. J Thromb Haemost. 2005;3:2730–7. doi: 10.1111/j.1538-7836.2005.01639.x. [DOI] [PubMed] [Google Scholar]
- 42.Aljamali MN, Margaritis P, Schlachterman A, Tai SJ, Roy E, Bunte R, Camire RM, High KA. Long-term expression of murine activated factor VII is safe, but elevated levels cause premature mortality. J Clin Invest. 2008;118:1825–34. doi: 10.1172/JCI32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jirouskova M, Shet AS, Johnson GJ. A guide to murine platelet structure, function, assays, and genetic alterations. J Thromb Haemost. 2007;5:661–9. doi: 10.1111/j.1538-7836.2007.02407.x. [DOI] [PubMed] [Google Scholar]
- 44.Jacobi PM, Kanaji S, Gehrand A, Jakab DA, Haberichter SL. Von Willebrand factor propeptide (VWFpp): a marker useful for identifying adverse platelet activation in murine blood phlebotomy samples. J Thromb Haemost. 2013;11(Suppl 2):362. [Google Scholar]
- 45.Song D, Ye X, Xu H, Liu SF. Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood. 2009;114:2521–9. doi: 10.1182/blood-2009-02-205914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dayal S, Wilson KM, Motto DG, Miller FJ, Jr, Chauhan AK, Lentz SR. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127:1308–16. doi: 10.1161/CIRCULATIONAHA.112.000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arellano-Rodrigo E, Alvarez-Larran A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. 2006;91:169–75. [PubMed] [Google Scholar]
- 48.Zumbach A, Marbet GA, Tsakiris DA. Influence of the genetic background on platelet function, microparticle and thrombin generation in the common laboratory mouse. Platelets. 2001;12:496–502. doi: 10.1080/095371001317126392. [DOI] [PubMed] [Google Scholar]
- 49.Greene TK, Lyde RB, Bailey SC, Lambert MP, Zhai L, Sabatino DE, Camire RM, Arruda VR, Poncz M. Apoptotic effects of platelet factor VIII on megakaryopoiesis: implications for a modified human FVIII for platelet-based gene therapy. J Thromb Haemost. 2014;12:2102–12. doi: 10.1111/jth.12749. [DOI] [PubMed] [Google Scholar]
- 50.Damon AL, Scudder LE, Gnatenko DV, Sitaraman V, Hearing P, Jesty J, Bahou WF. Altered bioavailability of platelet-derived factor VIII during thrombocytosis reverses phenotypic efficacy in haemophilic mice. Thromb Haemost. 2008;100:1111–22. doi: 10.1160/th08-04-0242. [DOI] [PubMed] [Google Scholar]