

Abstract
OBJECTIVE
Improvement in treatment outcome for patients with glioblastoma multiforme (GBM) requires a multifaceted approach due to dysregulation of numerous signaling pathways. The murine double minute 2 (MDM2) protein may fulfill this requirement because it is involved in the regulation of growth, survival, and invasion. The objective of this study was to investigate the impact of modulating MDM2 function in combination with front-line temozolomide (TMZ) therapy in GBM.
METHODS
The combination of TMZ with the MDM2 protein–protein interaction inhibitor nutlin3a was evaluated for effects on cell growth, p53 pathway activation, expression of DNA repair proteins, and invasive properties. In vivo efficacy was assessed in xenograft models of human GBM.
RESULTS
In combination, TMZ/nutlin3a was additive to synergistic in decreasing growth of wild-type p53 GBM cells. Pharmacodynamic studies demonstrated that inhibition of cell growth following exposure to TMZ/nutlin3a correlated with: 1) activation of the p53 pathway, 2) downregulation of DNA repair proteins, 3) persistence of DNA damage, and 4) decreased invasion. Pharmacokinetic studies indicated that nutlin3a was detected in human intracranial tumor xenografts. To assess therapeutic potential, efficacy studies were conducted in a xenograft model of intracranial GBM by using GBM cells derived from a recurrent wild-type p53 GBM that is highly TMZ resistant (GBM10). Three 5-day cycles of TMZ/nutlin3a resulted in a significant increase in the survival of mice with GBM10 intracranial tumors compared with single-agent therapy.
CONCLUSIONS
Modulation of MDM2/p53-associated signaling pathways is a novel approach for decreasing TMZ resistance in GBM. To the authors’ knowledge, this is the first study in a humanized intracranial patient-derived xenograft model to demonstrate the efficacy of combining front-line TMZ therapy and an inhibitor of MDM2 protein–protein interactions. http://thejns.org/doi/abs/10.3171/2016.1.JNS152513
Keywords: glioblastoma, temozolomide, DNA repair, p53, MDM2, combination therapy, patient-derived xenograft, MDM2 inhibitor, oncology
Glioblastoma (GBM) is the most common brain cancer in adults and prognosis is dismal; the median progression-free period is less than 7 months and overall survival is generally less than 15 months.39 An elusive goal in brain cancer therapy is the development of treatment strategies that selectively target pathways that effectively lead to a long-term therapeutic response.32,39,48 In difficult-to-treat-malignancies such as GBM, the lack of a durable response is largely attributed to acquisition of chemoresistance due to activation of pathways that enhance cell survival, angiogenesis, and invasion.32,39,48 Successful treatment of GBM is further hindered by its genetic instability, which gives rise to heterogeneous populations of tumor cells, and the lack of therapeutic agents that effectively penetrate the blood-brain barrier.48 Surgery followed by temozolomide (TMZ) and radiation is the front-line therapeutic regimen to treat GBM, but current therapeutic success is limited in part by molecular heterogeneity20 and multiple resistance mechanisms.32 For example, in approximately 60% of patients with GBM, tumors are resistant to TMZ due to high levels of the direct reversal repair protein O6-methylguanine DNA methyltransferase (MGMT).42 Therefore, new treatments that can prevent or overcome resistance mechanisms in GBM are clearly needed.
Emerging evidence suggests that MDM2 may be a rational, multipronged therapeutic target for GBM. The MDM2 gene is amplified in approximately 7%–13% of GBM tumors, and Verreault et al. demonstrated that GBM tumors with MDM2 amplification are highly responsive to MDM2 inhibition.46 However, MDM2 protein is elevated in 45%–75% of GBM biopsy samples, indicating that additional mechanisms can regulate MDM2 expression.29 MDM2 is an E3 ubiquitin ligase that targets p53 for proteasomal-mediated degradation, blocks the transactivation domain of p53, and is responsible for the export of p53 from the nucleus.26 Additionally, p53 and MDM2 form an autoregulatory network in which MDM2 is a downstream transcriptional target of p53. Moreover, in the context of DNA damage, MDM2 localizes to the nucleus and modulates DNA repair in a p53-independent manner by binding to and inactivating Nbs1 in the MRE11/Rad50/NBS1 complex.1,8,27
The MDM2 protein–protein interaction (PPI) inhibitor nutlin3a binds to the N-terminal hydrophobic pocket of MDM2 and blocks interaction of MDM2 with not only p53 but also a number of other key signal transducers, including p73α, E2F1, Rb, and HIF-1α, thereby modulating their downstream effector function.2,21,22,34,35,45 Although it has been shown that nutlin3a and other MDM2 PPI inhibitors have single-agent activity in inhibiting the growth of a variety of tumor types including GBM,2,13,14,17,21,22,35,40,44–46 information on dosing regimens that build upon front-line chemotherapy for GBM is lacking.19,21,22,34,43,45
In the present study, we used nutlin3a to investigate MDM2 as a therapeutic target in GBM, both alone and in combination with TMZ. Our central hypothesis was that inhibition of MDM2 by nutlin3a would potentiate TMZ-mediated death of GBM cells in vivo, resulting in increased survival of mice with intracranial human GBM. We demonstrated that nutlin3a-mediated modulation of MDM2 function leads to an impairment in DNA repair that correlates with potentiation of TMZ-mediated cell death both in vitro and in vivo. We developed a TMZ/nutlin3a dosing regimen in an intracranial xenograft model using GBM10 cells, which were derived from a recurrent wild-type p53 GBM tumor that expresses high levels of the base excision repair (BER) protein apurinic/apyrimidinic endonuclease I (APE1) and MGMT. Both of these DNA repair proteins play an integral role in the repair of TMZ-induced DNA lesions, and increased levels can lead to resistance mechanisms in GBM. A combination regimen consisting of three 5-day cycles resulted in a significant increase in survival compared with mice treated with TMZ alone, with minimal bone marrow toxicity.
Methods
Nutlin3a Synthesis
Racemic nutlin3a,b was synthesized, and nutlin3a was purified as previously reported.33 Separation of nutlin enantiomers, nutlin3a and nutlin3b, was accomplished using a ChiralPak IB column (250 × 4.6 mm, Daicel Chemical Industries) at 40°C eluted with 70:30 hexane:ethanol at a flow rate of 0.8 ml/min. Dose-response curves confirmed that nutlin3a was the more potent enantiomer (data not shown).
Established and Primary Patient-Derived GBM Cell Lines
The U87-MG and SK28 cell lines were obtained from the American Type Culture Collection in March 2009 and stocks were cryopreserved at passage < 3. The patient cell lines GBM10 and GBM43 have been described previously. 9,31 A primary GBM cell line was established using a fresh tumor specimen obtained in a patient with pathologically diagnosed GBM (Aaron Cohen-Gadol IRB#0909–05, Dept. of Neurosurgery, Indiana University School of Medicine [IUSM]). The focused molecular profile of the MHBT91 cell line, determined via the qBiomarker Somatic Mutation PCR array (SABiosciences, Qiagen) for the p53/RB1 pathway, is wild type for p53, ataxia-telangiectasia mutated (ATM), Rb1, and CDKN2A/p16.
Cell Line Authentication and Characterization
Cell line identity was confirmed by DNA fingerprint analysis (IDEXX BioResearch) for species and baseline short-tandem repeat analysis testing. All cell lines were 100% human, and a 9-marker short-tandem repeat analysis is on file. The primary patient-derived GBM10 and GBM43 cells were initially expanded as flank tumors, harvested, and maintained in 2.5% fetal bovine serum (FBS) for 4 days on Matrigel-coated plates (BD Biosciences) to remove murine fibroblasts. Cells were expanded in DMEM/F12 (4.5 g/L D-glucose and L-glutamine) with 10% FBS for less than 2 weeks for intracranial implantation. All GBM cells were routinely checked for mycoplasma and were negative.
Preparation of Reagents
Temozolomide (LKT Laboratories, Inc.) was dissolved in phosphate-buffered saline (PBS) or cell culture media and used immediately. Nutlin3a was formulated in 100% dimethyl sulfoxide and diluted in media for in vitro studies, and in 0.5% methylcellulose (Sigma-Aldrich) and 0.05% Tween 80 (Fisher Scientific) for in vivo studies. Both compounds were given orally in vivo.
Cell Proliferation Assay
Cells were seeded in 96-well plates (U87-MG: 1400 cells/well; GBM10: 9000 cells/well; MHBT91: 10,000 cells/well) and treated with TMZ, nutlin3a, or combinations. After 5 days of incubation, cell growth was determined by methylene blue staining.4 Each experiment was conducted in triplicate and repeated 3 times. The final dimethyl sulfoxide concentration was ≤ 0.1%.
Western Blot Analysis
A RIPA extraction buffer (Roche) containing 1 complete Mini EDTA-free tablet (Roche) and 1% phosphatase inhibitor 3 (Sigma) was used for preparation of protein lysates, and protein quantification was determined by the DC Protein Assay (Bio-Rad) according to the manufacturer’s instructions. Proteins were resolved by SDS-PAGE on Criterion TGX Precast 4%–20% Gels (Bio-Rad) and transferred to nitrocellulose membranes. The transfer buffer consisted of 25 mM Tris, 192 mM glycine, pH 8.3 (Tris-glycine buffer), and 20% methanol. Membranes were blocked for 1 hour at room temperature in 5% dry milk in 137 mM NaCl, 20 mM Tris, and 0.05% Tween 20 (Tris-buffered saline [TBS] and Tween 20; TBST). Nitrocellulose membranes were blocked with 5% bovine serum albumin (BSA) and antibodies were diluted in 5% BSA and incubated with membranes overnight. Membranes were washed with 1× TBS containing 0.02% Tween 20 (TBST) for a total of three 12-minute washes. The correct molecular weight for each protein was confirmed by the PageRuler Prestained Protein Ladder (Thermo Scientific), and the following antibodies were used for detection: mouse anti-total p53 (53 kD, Santa Cruz Biotechnology); rabbit anti-p53 serine 15 (53 kD, Cell Signaling Technology); mouse anti-MDM2 antibodies (full-length form–90 kD, short form–76 kD: 2A9-Millipore, 2A10-Calbiochem, SMP14-Santa Cruz Biotechnology); mouse anti-APE1 (34 kD, clone 13B8E5C2, Novus Biologicals); mouse anti-p21(21 kD, Cell Signaling Technologies); mouse anti-MGMT (21.7 kD, Clone MT3.1, Millipore); mouse anti-α tubulin (50 kD, clone DM1A, Sigma-Aldrich); glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (37 kD, 14C10, Cell Signaling); mouse anti-retinoblastoma phosphoserine 780 (110 kD, p-Rb, BD Pharmingen); mouse total Rb (110 kD, Cell Signaling Technology); rabbit anti-topoisomerase IIα (170 kD, TopoIIα, Santa Cruz Biotechnology); and rabbit anti-phosphohistone H3 serine 10 (16 kD, pHH3, Millipore Upstate Biotechnology). All antibodies were diluted 1:1000 except for GAPDH and α-tubulin, which were diluted 1:10,000. Blots were incubated for 1 hour with appropriate secondary horseradish peroxidase–conjugated anti–mouse or anti–rabbit antibodies diluted at 1:5000 in 1× TBST. Membranes were washed with 1× TBS containing 0.02% Tween 20 (TBST) 3 times, and developed using the SuperSignal Western Chemiluminescent Substrate (Thermo Scientific); membranes were exposed to autoradiography film. Western blot densitometry was determined using ImageJ software (http://imagej.nih.gov/); proteins were normalized to loading control and expressed relative to vehicle-treated cells.
Analysis of γH2AX Foci
Glioblastoma cells were seeded on chamber slides (Lab-Teck Brand Products) 1 day before treatment. Cells were treated for 4 hours, fixed with 2% paraformaldehyde, and incubated in ice-cold 100% methanol for 10 minutes at −20°C. Fixed cells were rinsed in PBS for 5 minutes. For immunostaining, cells were blocked in 3% BSA, 0.3% TritonX-100 in PBS for 60 minutes and then incubated with a phospho-histone H2AX (Ser139) primary antibody (1:200 dilution, Cell Signaling Technologies) and incubated overnight at 4°C. Cells were then incubated with an Alexa Fluor 488 fluorochrome-conjugated secondary antibody (1:1000, Invitrogen) for 2 hours at room temperature in the dark. Coverslip slides with Prolong Gold Antifade reagent with DAPI were sealed with nail polish. Confocal images were acquired with a confocal/2-photon Olympus Fluoview FV-1000 multiphoton emission system (Olympus America), available at the Indiana Center for Biological Microscopy Imaging Facility, using the Olympus UPLSAPO 60×, N.A. 0.95 Water Immersion Objective. Images were collected in a sequential illumination mode using 405- and 488-nm laser lines. Fluorescent emission was collected in 2 spectral detectors with filter slit set up to 420–490 nm (DAPI) and 500–550 nm (fluorescein isothiocyanate [FITC]). All images were collected with constant scanning parameters. Fields were randomly selected on the basis of DAPI-counterstained nuclei. After acquisition of the DAPI image, the amplification of the FITC signal, z-step, and magnification parameters were kept constant during the study. The number of foci per nucleus was determined from 5 different fields per sample using ImageJ software. Data are representative of 2–3 independent experiments for each cell line.
Cell Cycle Analysis
The U87-MG cells were treated with nutlin3a, TMZ, or combination of TMZ and nutlin3a (T/N) for 72 hours in 3 independent experiments. Cells were collected and stained with propidium iodide solution containing 0.1% (volume/volume %) Triton X-100 (Sigma), 10 μg/ml propidium iodide (BD Biosciences), and 100 μg/ml DNase-free RNaseA (Invitrogen), and analyzed by flow cytometry. Cell cycle subpopulations were quantified using ModFit LT for Mac version 3.2 software (Verity Software House).
Alkaline Comet Assay
Comet assays were performed under alkaline conditions using the CometAssay kit according to the manufacturer’s instructions (Trevigen), and slides were analyzed with a fluorescence microscope. DNA damage (tail length) was analyzed by TriTek CometScore software. Three random fields of at least 50 cells per sample were scored to determine the average amount of DNA damage.
Apoptosis Assay
Treated GBM cells were collected and stained using Annexin V-FITC (BD Pharmingen) and 7-AAD (BD Pharmingen) and were analyzed by flow cytometry according to the manufacturer’s instructions.
Animal Studies and Xenograft Models
A breeding colony of NOD.Cg-PrkdcscidIL2rgtm1Wjl/Sz (NSG) mice was established at the IUSM Laboratory Animal Research Center (LARC) and maintained by the In Vivo Therapeutics Core (Indiana University Simon Cancer Center [IUSCC]/IUSM). The NSG mice were acclimated for at least 1 week on TD.06596 Irradiated Global 2018 feed (Uniprim 4100 ppm, Harlan Laboratories). All procedures and predeath monitoring criteria were approved by the Institutional Animal Care and Use Committee. The IUSM-LARC is accredited by the American Association for the Accreditation of Laboratory Animal Care.
Ectopic Flank Implantation of Tumor Cells in Mice
The right flank of NSG mice was implanted with 4 × 106 U87-MG or 5 × 106 GBM10 cells. When U87-MG tumors reached approximately 150 mm3, mice were randomized into cohorts of 6–8 animals. Tumor volumes were measured by caliper twice weekly and tumor volumes were calculated according to the formula (α2 × β)/2, where α was the shorter and β was the longer of the 2 dimensions. For pharmacodynamics studies with GBM10 flank tumors, mice were treated for 3 days once tumors reached approximately 200 mm3. Mice were euthanized and tumors were harvested for pharmacodynamic analyses as described in the text.
Orthotopic Surgical Implantation of GBM10 Tumor Cells in Mice
For intracranial implantation of tumor cells, a digitalized stereotactic delivery system was used (David Kopf Instruments, Model 5000 microinjection unit) and tumors were implanted as previously described.9,31 For stereotactic delivery of tumor cells, mice were placed under general anesthesia (intraperitoneal injection of 16 mg/kg xylazine and 120 mg/kg ketamine) and were positioned in the stereotaxic device. A digitalized drill assembly was used to bore a hole (0.3 mm in depth and 0.8 mm in diameter) in the cranium at a position 0.5-mm anterior and 1.2-mm lateral to the bregma anatomical landmark. Tumor cells (3 × 105) in 5 μl of PBS were introduced slowly using a 10-μl Hamilton syringe at a depth of 3.5 mm at a rate of 2 μl/min. Once injection was completed, the needle was kept in place for 5 minutes and then slowly removed, and the hole was sealed with bone wax. The incision was closed with 3M Vetbond Tissue Adhesive (3M Animal Care Products), and triple antibiotic cream was applied. Prior to initiation of efficacy studies, pilot studies were first performed in NSG mice to optimize cell dose, assess tumor take frequency, and track survival. Among the 125 mice used in this study for intracranial implants, 7 (6%) did not die of tumor growth by 105 days postimplantation and were censored from the study. For animals with intracranial tumors, the mice were observed at least twice daily and were euthanized based on predeath end point criteria.
Wound-Healing Assay
The GBM10 cells were grown to 80%–90% confluence in 35-mm culture dishes (Corning), and the cell layer was wounded with a sterile pipette tip to generate a cell-free gap. Cell debris was removed by washing twice with cell culture medium. A homogeneous wound area free of cell debris was marked. Cells were washed with serum-free DMEM and photographed to record the wound width at Hour 0. GBM10 cells were then cultured in DMEM with 10% FBS. Cells were periodically examined over 12 hours by microscopy; photographs were taken again at the marked wound location for migration measurement, and gaps were measured and analyzed by ImageJ software. Each experiment was repeated 3 times.
Matrigel Invasion Assay
Insert wells (Costar) were coated with fibronectin (Sigma) at 30 ng/μl for 1 hour at room temperature. The cells were starved overnight in serum-free medium. The cells were harvested and resuspended in serum-free medium. Each of 6 groups of cells (106 cells per group) was exposed to treatment in 1 ml of DMEM containing 0.1% BSA for 6 hours and then transferred (3 × 105 cells per group) to the coated insert wells. These cells were assayed in triplicate for invasion at 37°C in a humidified incubator (95% air, 5% CO2) for 8 hours using Costar chambers (Corning, Inc.). DMEM containing 10% FBS was used as the chemoattractant and added to the lower chamber. After 8 hours of incubation, the filters were removed and fixed with methanol for 15 minutes; cells on the upper side of the membrane were removed. Cells that invaded the 8-μm pores and grew on the lower side were stained with 1% crystal violet. Cells that had migrated to the lower side of the polyvinylidene difluoride membrane were counted under the inverted microscope in 5 fields selected randomly (100× magnification). Each experiment was repeated 3 times.
In Vivo Pharmacokinetics of Nutlin3
Plasma, normal brain, or tumor tissue was collected, flash-frozen, and stored at −80°C until analysis. As described in the Results section, racemic or nutlin3a was extracted by a liquid-liquid extraction procedure, and high-performance liquid chromatography–mass spectrometry (MS)/MS (API 4000; Applied Biosystems) was used for quantification. The lower limit of quantification using 10 μl of blood was 1 ng/ml.
Statistical Analysis
The IC50 values were calculated according to the method of Chou and Talalay12 and were used to construct isobolograms as previously described.41 Comet assay data were analyzed as recommended.25 Other data were analyzed by 1- or 2-way ANOVA, as appropriate, with repeated measures across varying time points using SigmaPlot 11.2 (Systat Software, Inc.). Differences among individual pairs of means were determined by the Holm-Šídák post hoc test. Kaplan-Meier survival plots were generated using Sigma-Plot, and changes in survival were analyzed by the logrank test. Data were considered significant at p < 0.05.
Results
Nutlin3a Decreases TMZ Resistance in GBM Cells and Leads to Persistence of DNA Double-Strand Breaks In Vitro
A panel of human wtp53 GBM cell lines (U87-MG, GBM10, and MHBT91) was selected that represents molecular profiles commonly found clinically in GBM (Fig. 1). The IC50 values for inhibition of proliferation were determined at multiple dose-ratios of TMZ to nutlin3a. The primary patient cell lines GBM10 and MHBT91 were resistant to TMZ, i.e., the IC50 concentrations for TMZ were 11- to 47-fold higher than in U87-MG cells. Isobologram analysis demonstrated that doses of TMZ plus nutlin3a required to inhibit cell proliferation were additive to synergistic. These data demonstrate that low micromolar concentrations of nutlin3a markedly decreased the concentration of TMZ required to inhibit cell proliferation of GBM lines with high levels of DNA repair proteins that are involved in the repair of TMZ-mediated methylation of DNA.
FIG. 1.

Combination TMZ and nutlin3a is additive to synergistic in blocking proliferation of wtp53 GBM cells in vitro. Effects of TMZ and nutlin3a (Nut3a), alone and in combination, were analyzed by isobologram in U87-MG, GBM10, and MHBT91 cells. Isobole points falling below the diagonal line of additivity indicate synergism; on the line indicate additivity; and above the line indicate antagonism. Each point represents the mean of 3 experiments conducted in triplicate. Vertical and horizontal lines indicate ± 1 SEM and are absent when less than the size of the point.
To determine if repair of DNA damage is affected in GBM cells exposed to T/N, the prevalence of γH2AX foci was investigated. There were significantly more γH2AX foci in T/N-treated U87-MG cells compared with all groups treated with single agents (Fig. 2A and B), and there were significantly more γH2AX foci in T/N-treated GBM10 cells than in nutlin3a or control groups (Fig. 2D and E). These data indicate that modulation of MDM2 function is linked to DNA damage and may potentially block DNA repair mechanisms. As expected, analysis of cell cycle–specific markers Rb phosphorylated at serine 780 (pRb [S780]) (decreased phosphorylation in early G1 arrest), histone H3 phosphorylated at serine 10 (pHH3) (increased phosphorylation in M phase), and TopoIIα (total protein increased in S phase), indicated that both the U87-MG and GBM10 cells arrested in G0/G1 following exposure to nutlin3a or T/N (Fig. 2C and F).
FIG. 2.

Increased numbers of γH2AX foci following combination TMZ and nutlin3a. A and B: U87-MG cells were treated for 4 hours with vehicle (Veh), 25 μM TMZ (25T), 5 μM nutlin3a (5N), or combination 25T/5N, and γH2AX foci were evaluated by confocal microscopy. For quantification of γH2AX foci, the number of foci per nucleus were compiled from 3 independent experiments. Three different regions with ≥ 50 cells per region were counted for each group. C: U87-MG cells were treated as described above for 72 hours and analyzed for cell cycle–specific markers by Western blot. D and E: GBM10 cells were treated for 4 hours with vehicle, 25 μM TMZ (25T), 10 μM nutlin3a (10N), or combination 25T/10N, and γH2AX foci were evaluated by confocal microscopy. F: GBM10 cells were treated as described above for 72 hours and analyzed for cell cycle–specific markers by Western blot. * = p < 0.05 versus control; † = p < 0.05 versus TMZ; # = p < 0.05 versus nutlin3a. Bar = 10 μm (A) and 20 μm (D).
Temozolomide- and Nutlin3a-Mediated Target Modulation and Efficacy in U87-MG Cells
The TMZ-sensitive U87-MG cell line was first used to investigate effects of nutlin3a on TMZ-mediated DNA damage. We evaluated 2 dose-ratios of T/N (20:1 and 5:1) to gain perspective on the ability of nutlin3a to decrease resistance to TMZ (Fig. 3A and B). At Day 5 posttreatment, there were significant increases in early and late apoptotic/necrotic cells treated with nutlin3a or T/N compared with control or TMZ alone. At the 20:1 dose-ratio (25 TMZ/1.25 nutlin3a), there was a significant increase in late apoptotic/necrotic cells in the T/N group compared with both single agents (Fig. 3B). This correlated with an increase in the percentage of cells arrested in G0/G1 and a decrease in TMZ-induced G2/M arrest (Fig. 3C). At 24 hours posttreatment (Fig. 3D), Western analyses indicated that levels of total p53, activated p53 (phosphorylation on serine 15-p53 [S15]), and MDM2 were increased by TMZ, nutlin3a, and T/N; p21 levels were increased following nutlin3a or T/N treatment. Moreover, total p53 levels were significantly increased by nutlin3a and T/N compared with TMZ alone, and T/N significantly increased total p53 and p53 (S15) compared with nutlin3a. Phosphorylation of p53 on serine 15 by ATM or DNA-dependent protein kinase is an indicator of DNA-strand breaks.26 Levels of p53 (S15) were elevated in the TMZ and T/N-treated groups at 72 hours (Fig. 3E and F), and the BER protein APE1 was decreased by TMZ, nutlin3a, and T/N, which would be expected to reduce capacity for repair of DNA adducts by the BER pathway.
FIG. 3.

In vitro target modulation and in vivo efficacy in ectopic U87-MG xenografts following combination TMZ and nutlin3a. A and B: U87-MG cells were treated with vehicle, 25 μM TMZ (25T), 1.25 or 5 μM nutlin3a (1.25N or 5N), or combination TMZ and nutlin3a (25T/1.25N or 25T/5N), and early apoptotic or late apoptotic/necrotic cells were evaluated by flow cytometry at Day 5 post-treatment. n.s. = nonsignificant; ** = p < 0.01; *** = p < 0.001 versus vehicle; †† = p < 0.01; ††† = p < 0.001 versus 25 TMZ. C: U87-MG cells were exposed to vehicle, nutlin3a (Nut), TMZ, or TMZ/nutlin3a (TMZ)/(Nut). The percent of cells in each phase of the cell cycle was determined using propidium iodide (PI) at 3 days posttreatment. These data are representative of 3 independent repeats and yielded similar results. D: U87 cells were treated with 25 μM TMZ and 5 μM nutlin3a for 24 hours. Relative levels of target proteins were determined by densitometry of Western blots in 6 independent experiments. * = p < 0.05; ** = p < 0.01; *** = p < 0.001 versus vehicle; †† = p < 0.01; ††† = p < 0.001 versus TMZ; # = p < 0.05 versus Nut3a. E and F: At 72 hours posttreatment, relative levels of target proteins were determined by densitometry. Results are from 2 independent experiments. G: Nutlin3 concentration was determined in mice with U87-MG tumor flank xenografts at 4 hours postadministration. As a background control, tumor was taken from 1 vehicle control mouse (V). Nutlin3 concentrations for 2 different tumor samples (N-1 and N-2) were determined by liquid-liquid extraction followed by high-performance liquid chromatography-MS/MS (API 4000) using caylin as the internal standard. H: NSG mice implanted with flank U87-MG tumors were treated with vehicle, 5 mg/kg TMZ, 100 mg/kg nutlin3, or TMZ/nutlin3 (TMZ/Nut3) for 2 treatment cycles. The horizontal line denotes the dosing period. Data points represent mean ± SEM. Inset: Effects of TMZ and TMZ/nutlin3 on tumor volume posttreatment. * = p < 0.05; *** = p < 0.001 versus vehicle on same day; + = p < 0.05; ‡ = p < 0.001 TMZ versus TMZ/Nut3 on the same day.
For the U87-MG efficacy study, results of pharmacokinetic (PK) analysis of racemic nutlin3 in NSG mice was in agreement with values previously reported in C57BL/6 mice (Table 1).51 In U87-MG flank xenografts, nutlin3 was detected by 4 hours postadministration (Fig. 3G). Mice with U87-MG flank xenografts received vehicle, TMZ, nutlin3, or T/N (Fig. 3H). We used a 5-day TMZ-dosing regimen, followed by a 2-day rest period, consistent with TMZ-dosing regimens used clinically.39 Following 2 treatment cycles, nutlin3 alone significantly slowed tumor growth, and TMZ alone produced a significant decrease in tumor volume compared with vehicle. Tumor volume was further decreased by approximately 50% in mice that received T/N compared with TMZ alone (Fig. 3H inset). Tumor growth for T/N versus TMZ alone was significantly inhibited for at least approximately 4 weeks beyond the dosing window, but tumor volume began to rebound thereafter. Body weights were unchanged (data not shown), and no apparent treatment-related toxicities were noted. These data indicate that nutlin3 can potentiate the antitumor activity of TMZ in GBM xenografts and that prolonged treatment is required to block tumor growth.
TABLE 1.
Pharmacokinetic analysis of nutlin3 in NSG mice
| PK Parameter | Nutlin3 |
|---|---|
| Cmax | 20,417 ng/ml (37 μM) |
| Tmax | 1 hr |
| T1/2 | 4.5 hrs |
| Cl/F | 0.29 ml/min |
| Vdss/F | 113 ml |
Cmax = maximum concentration; CI/F = clearance/bioavailability; Tmax = time to reach the maximum concentration; T1/2 = half-life; Vdss/F = apparent volume of distribution/bioavailability.
In Vitro and In Vivo Evaluation of TMZ- and Nutlin3a-Mediated Target Modulation in GBM10 Cells
The GBM10 cells were selected for further in vivo efficacy studies, because they are relevant to the clinical challenges of GBM therapy. The GBM10 cells were originally derived from a recurrent GBM, are highly resistant to TMZ and ionizing radiation, and express high levels of APE1 and MGMT in vivo.31 The mismatch repair proteins MSH6, PMS2, and MSH2 are expressed in GBM10 cells, which is consistent with mismatch repair proficiency (data not shown). To gain insight into the levels of TMZ-mediated DNA damage required for cell death, GBM10 cells were exposed to a clinically relevant dose of TMZ (200 μM)30 with increasing doses of nutlin3a (Fig. 4A and B). At 2 μM of nutlin3a, the combination of T/N significantly increased both early and late apoptosis/necrosis in GBM10 cells compared with single-agent treatment. Analysis of DNA-strand breaks by alkaline comet assays indicated that DNA-strand breaks persisted in GBM10 cells treated with the combination compared with single-agent treatment at both 4 and 24 hours (Fig. 4C).
FIG. 4.

In vitro and in vivo target modulation by TMZ and nutlin3a in GBM10 cells. A and B: GBM10 cells were treated with vehicle control, 200 μM TMZ (T) alone or in the presence of nutlin3a (Nut3a), and early apoptotic or late apoptotic/necrotic cells were evaluated by flow cytometry on Day 5 posttreatment. ‡ = p < 0.001 versus TMZ 200 + vehicle. C: GBM10 cells were treated for 4 or 24 hours with 200 μM TMZ and 2 μM nutlin3a. DNA-strand breaks were measured by alkaline comet assays. ** = p < 0.01 versus vehicle; # = p < 0.05; ## = p < 0.01 versus nut3a. D: GBM10 cells were treated with 200 μM TMZ and 2 μM nutlin3a for 24 hours as described above, and target proteins were analyzed by Western blot. Relative levels of target proteins were determined by densitometry of Western blots in 3 independent experiments. *** = p < 0.001 versus vehicle; ††† = p < 0.001 versus TMZ; # = p < 0.05 versus Nut3a. E: GBM10 cells were treated for 48 and 72 hours as described above and target proteins were analyzed by Western blot. Relative levels of target proteins at (F) 48 hours and (G) 72 hours were determined by densitometry of Western blots. Data are representative of 2 independent experiments.
Increases in total p53 and activated p53 (S15) were evident at 24 hours in all treatments compared with vehicle (Fig. 4D); activation of p53 (S15) was significantly increased by T/N compared with other treatments. Furthermore, MDM2 and p21 were significantly increased by nutlin3a and T/N. By 48 hours, total p53, p53 (S15), p21, and MDM2 levels remained elevated in TMZ- and T/N-treated cells (Fig. 4E and F). In contrast to T/N at 48 hours, MDM2 levels were lower in T/N-treated cells at 72 hours, which is consistent with reports of alkylator-mediated downregulation of MDM2 (Fig. 4E and G). The 2A9 MDM2 antibody was used to avoid possible epitope masking due to phosphorylation of MDM2.11 By 72 hours, downregulation of APE1 and increases in γH2AX were the highest in nutlin3a- and T/N-treated cells compared with other treatments; MGMT levels were modestly decreased by T/N treatment (Fig. 4E–G).
To assess whether the p53 pathway is activated and DNA repair proteins are modulated in vivo by T/N treatment, mice with GBM10 flank tumors received TMZ and/or nutlin3a for 3 consecutive days, and tumor lysates were analyzed by Western blot (Fig. 5). The MDM2 and p53 levels increased in tumors of mice treated with TMZ, nutlin3a, or T/N. Moreover, p53 (S15) levels were increased by TMZ and T/N. The MGMT and APE1 protein levels were greatly decreased in vivo by T/N compared with other treatments.
FIG. 5.
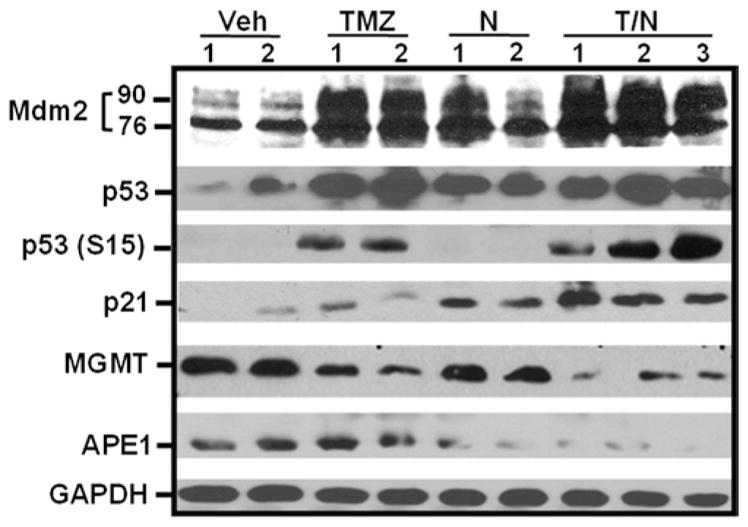
Temozolomide and nutlin3a activate the p53 pathway and downregulate DNA repair proteins MGMT and APE1 in GBM10 flank xenografts. NSG mice with flank GBM10 tumors were treated for 3 consecutive days with vehicle, 66 mg/kg TMZ, 100 mg/kg nutlin3a (N), or T/N. Tumors were harvested 2 hours after the last dose, and lysates were analyzed by Western blot. Both the 92- and 76-kD forms of MDM2 were detected.
Glioblastoma cells are highly invasive, and others have demonstrated that nutlin3a can inhibit migration and invasion via changes in the cytoskeleton in other cell types.25 The GBM10 cells were exposed to vehicle, TMZ, nutlin3a, or a combination (T/N). At the time points analyzed, cells remained viable and p53 and MDM2 levels increased (Fig. 6A). Both GBM10 migration (Fig. 6B) and invasion (Fig. 6C) were significantly inhibited by nutlin3a and the T/N combination.
FIG. 6.

Decreased migration and invasion of GBM10 cells exposed to TMZ and nutlin3a compared with single-agent exposure. A: GBM10 cells were treated for 8 hours with vehicle (V), 25 μM TMZ (T), 10 μM nutlin3a (N), or T/N; p53, MDM2, and GAPDH expression were analyzed by Western blot. B: A monolayer of GBM10 cells was wounded to generate a cell-free gap as described in the Methods. The cell-free gap was photographed to record the wound width (red arrows) at Hour 0. GBM10 cells were then treated for 12 hours with vehicle, 25 μM TMZ, 10 μM nutlin3a, or T/N. Cells were observed microscopically every 2 hours to monitor closing of the gap, and photographs were taken again at the marked wound location for measurement of migration. Representative photographs at 12 hours are shown. Each experiment was repeated 3 times and data are the mean ± SD of the 3 repetitions. Post hoc analyses demonstrated that all treatment pairwise comparisons were significant (*** = p < 0.001), except vehicle versus TMZ and nutlin3a versus T/N. C: GBM10 cells were incubated in fibronectin-coated insert wells and exposed to TMZ and nutlin3a as described above. After 8 hours of incubation, the filters were removed and fixed with methanol for 15 minutes; the cells on the upper side of the membrane were removed. Invasive cells, which were able to breach 8-μm pores and grow on the lower side, were stained with 1% crystal violet. Representative photographs at 8 hours posttreatment are shown. The cells that had migrated to the lower side of the membrane were counted under an inverted microscope in 5 fields selected randomly (100× magnification). Each experiment was repeated 3 times. Post hoc analyses demonstrated that all treatment pairwise comparisons were significant (** = p < 0.01, *** = p < 0.001), except vehicle versus TMZ.
In Vivo Efficacy of Combination T/N in an Orthotopic GBM10 Xenograft Model
The concentrations of nutlin3a in normal mouse brain and intracranial human tumors were determined to gain insight into concentrations of nutlin3a that can be obtained in vivo. Nutlin3a levels in intracranial GBM tumors and a melanoma (a tumor type that can metastasize to the brain) were determined. At 4 hours postadministration of nutlin3a, concentrations in tumor tissues ranged from 1 to 20 μM (Fig. 7A), which is within the range of concentrations required for efficacy in cell lines (Fig. 1). Brain tumor tissues had higher levels of nutlin3a than normal brain tissue, indicating that tumor tissues were relatively more permeable. As a single agent, however, nutlin3a did not increase survival of mice with intracranial GBM tumors (Fig. 7B).
FIG. 7.

Nutlin3a concentrations in the plasma, normal brain, and brain tumor tissue and lack of increased survival of mice with intracranial xenografts following a 2-week monotherapy. A: NSG mice were implanted with GBM10, GBM43, or SK28 melanoma cells in the right cerebral hemisphere and at 21 days postimplantation received 100 mg/kg nutlin3a. Normal mice also received 100 mg/kg nutlin3a. Four hours after nutlin3a administration, plasma and brains were collected, the brain tumor area was surgically excised (x axis: tumor types), and levels of nutlin3a were determined. Each point represents an individual mouse. B: Survival of NSG mice administered vehicle (control) or 100 mg/kg nutlin3a twice daily for 2 consecutive 5-day cycles. The numbers in parentheses represent the median survival time in days. C: Survival of NSG mice administered vehicle, twice-daily nutlin3a (Nut3a), once-daily TMZ alone, or TMZ with once-daily nutlin3a 6 hours after TMZ (TMZ+Nut3a), for 2 consecutive 5-day cycles. The numbers in parentheses represent the median survival time in days. *** = p < 0.001 versus vehicle. D: Body weights. E: Representative H & E–stained sections of orthotopic tumors at predeath end point in NSG mice. The black arrows indicate intracranial tumors in the mouse brain.
We next focused on development of a combination TMZ/nutlin3a regimen that would significantly increase the probability of survival of mice with intracranial GBM10 xenografts. An effect size of 20% increase in survival was considered to be biologically significant when comparing survival in TMZ versus combination T/N-treated mice.39 In initial studies, mice with GBM10 intracranial tumors received vehicle, TMZ (40 mg/kg once daily), nutlin3a (200 mg/kg twice daily), or T/N for two 5-day cycles. Although body weights were maintained, increases in survival of animals treated with T/N were not significant compared with other treatments (data not shown).
Based on our in vitro studies, a certain threshold of DNA damage is necessary to enhance tumor cell death. Therefore, the dose of TMZ was increased to 66 mg/kg, a dose that can significantly increase survival of NSG mice with GBM10 intracranial tumors. We also administered TMZ followed by nutlin3a 6 hours later to avoid potential drug-drug interactions between the compounds. Mice were treated for 2 cycles of vehicle, 66 mg/kg TMZ once daily, 100 mg/kg nutlin3a twice daily, or T/N. Survival was significantly increased by TMZ and T/N compared with vehicle or nutlin3a (Fig. 7C, p < 0.05). However, T/N did not significantly increase survival compared with TMZ alone. Body weights of all groups remained within normal ranges until the predeath end point (Fig. 7D), and mice at the predeath end point harbored intracranial tumors (Fig. 7E).
Following three 5-day cycles of therapy, survival of mice treated with T/N was significantly greater than that of mice treated with either TMZ or nutlin3a (Fig. 8A, p < 0.05) with no treatment-related changes in body weights (Fig. 8B). A major dose-limiting toxicity of TMZ is bone marrow suppression. Because this is one of the most sensitive indicators of normal tissue toxicity, we evaluated potential treatment effects on both peripheral blood and bone marrow.39,42 Peripheral blood counts were determined in mice on Day 30 (2 days after the end of the third treatment cycle) and on Day 43 (day on which the first vehicle-treated mouse approached the predeath end point) (Fig. 8C–E). Although there was a significant decrease in white and red blood cell counts in the TMZ- and T/N-treated mice compared with vehicle-treated mice, there was no difference in blood counts between the TMZ and T/N groups (Fig. 8C–E). Platelets were significantly decreased by TMZ and T/N on Day 30 only. Bone marrow cellularity was determined in each group at their predeath end point (Fig. 8F); no statistical differences in the number of bone marrow cells per femur were noted, suggesting that the bone marrow recovered following the dosing regimen. Stupp et al. originally reported that treatment of patients with GBM with TMZ and radiation resulted in an approximately 20% increase in progression-free survival compared with radiation therapy alone.39 In the present intracranial GBM10 model, TMZ treatment alone resulted in an approximately 13% increase in survival compared with vehicle-treated mice. Combination T/N treatment further improved survival, resulting in an approximately 29% increase in survival with minimal toxicity to the bone marrow.
FIG. 8.

Increased survival of mice with intracranial GBM10 xenografts treated with 3 cycles of combination TMZ and nutlin3a therapy. A: Survival of NSG mice with orthotopic GBM10 tumors after administration of three 5-day cycles of treatment. The numbers in parentheses represent the median survival time in days. * = p < 0.05; *** = p < 0.001 versus vehicle; † = p < 0.05 versus TMZ. B: Body weights. C–E: Complete blood counts in the peripheral blood were determined. C: White blood cells (WBC); D: Platelets; and E: Red blood cells (RBC). * = p < 0.05. F: Bone marrow cellularity was determined in 5–6 mice from each group when mice reached the predeath end point.
Discussion
A significant challenge in the development of efficacious treatments for GBM is the molecular heterogeneity and genetic instability of these tumors, even within the same patient.38 To overcome this complexity in GBM, a multipronged approach that targets multiple signaling pathways will be essential. MDM2 represents a rational therapeutic target in GBM. It is expressed at elevated levels in GBM following TMZ treatment and possesses both p53-dependent and p53-independent functions controlling aspects of cell survival, growth, invasion, and DNA repair.23,26 In the present study, the MDM2 PPI inhibitor nutlin3a potentiated TMZ-mediated GBM cell death and significantly increased survival of mice with orthotopic intracranial human GBM tumors that were highly resistant to TMZ.
The DNA damage sensors ATM, ATM- and RAD3-related (ATR), and the stress-activated tyrosine kinase c-Abl provide a well-documented link between chemotherapy-mediated DNA damage and regulation of the MDM2/mouse double-minute X (MDMX)/p53 complex.10 Following DNA damage, MDM2 is phosphorylated on serine 395 by ATM,24 on serine 407 by ATR,36 and on tyrosine 394 by c-abl,37,47 which along with other posttranslational modifications of MDMX and p53 neutralizes the inhibitory effect of MDM2/MDMX on p53 and results in sustained p53-mediated signaling.10 The present data are consistent with the interpretation that a central mechanism of action responsible for increased TMZ sensitivity via nutlin3a is sustained activation of p53-mediated signaling and downregulation of DNA repair. The TMZ-mediated DNA damage persisted in GBM cells exposed to TMZ/nutlin3a compared with either agent alone. The DNA repair proteins APE1 and MGMT can be expressed at high levels in GBM and are major predictors of TMZ resistance.49 APE1 and MGMT were both downregulated to a greater extent in GBM cells treated with TMZ/nutlin3a compared with either agent alone. Following DNA damage, p53 can act as a negative regulator of APE150 and MGMT5 by sequestering the Sp1 transcription factor required for promoter activation. Additionally, Busso et al. reported that overexpression of MDM2 increased monoubiquitination of APE1, and that nutlin3a, by increasing MDM2 levels, increased APE1 ubiquitination in wtp53 HCT116 cells.6 Furthermore, it is possible that DNA repair could be blocked in TMZ/nutlin3a-treated cells due to increased localization of MDM2 at DNA-strand breaks. Alt and colleagues demonstrated that MDM2 acts as a DNA repair inhibitor by binding to Nbs1 and blocking the function of the MRE11/Rad50/NBS1 complex, thereby masking the ability of the cells to sense DNA damage.1
The use of combination therapy is a centerpiece of cancer chemotherapy. Combination therapies that build upon standard-of-care therapy, and incorporate MDM2 PPI inhibitors, are being investigated in a variety of cancers. Costa et al. demonstrated that TMZ synergized with the MDM2 PPI inhibitor ISA27 to kill U87-MG cells in vitro, and administration of ISA27 alone inhibited growth of GBM flank xenografts.13
Our results provide new information on targeting the MDM2 signaling network in combination with TMZ in a clinically relevant humanized intracranial GBM xenograft model (GBM10). GBM10 cells represent a particularly challenging tumor type to treat because they are highly resistant to TMZ due to high levels of DNA repair proteins (MGMT and APE1) and deletion of the CDKN2A/p16 gene. In the present study, although nutlin3a could promote apoptosis as a single agent in vitro or modestly block growth of U87-MG flank tumors in vivo, this did not translate to the in vivo intracranial xenograft model.
Our data do suggest that nutlin3a levels obtained in the brain were sufficient to increase TMZ-mediated inhibition of tumor growth and to enhance survival of mice with GBM10 intracranial tumors. There was a significant increase in survival of mice treated with TMZ/nutlin3a in combination compared with TMZ alone, which could be achieved with three 5-day treatment cycles. We also showed that p53 reactivation by nutlin3a leads to a persistence of DNA damage and downregulation of DNA repair proteins in GBM cells treated with TMZ in combination with nutlin3a. MDM2 is an inducible target in the GBM cell lines described herein. Whereas MDM2 protein levels were low in nontreated cells, MDM2 protein accumulated following activation of the p53 pathway due to the p53-MDM2 autoregulatory loop.
It may be possible to target MDM2 not only in wild-type but also in mutant p53 cancers.21,22,34 To this end, we recently demonstrated in a mutant p53 breast-to-lung metastatic model of triple-negative breast cancer that combination carboplatin and nutlin3a results in a significant inhibition of cell growth at primary and secondary tumor sites in vivo. In addition, response to combination therapy was in part dependent on p73α levels.43 Moreover, others have shown that inhibition of MDM2 binding to p53 or p73α appears to be more effective at promoting p53- and p73α-mediated apoptosis in cancer cells compared with normal cells.19,45
In the GBM10 intracranial model, there was no difference in the bone marrow cellularity in mice treated with TMZ or TMZ/nutlin3a at the predeath end point. Toxicity data in mice should be interpreted with caution because differential sensitivity to oncolytics between murine and human bone marrow has been reported.7,15 In PK studies, Zhang et al. reported that low levels of nutlin3a were detected in murine bone marrow,51 suggesting that bone marrow is a potentially protected compartment. Iancu-Rubin et al. evaluated the impact of the nutlin3-derivative RG7112 on thrombopoiesis because this has been a dose-limiting toxicity in clinical trials.18 Administration of RG7112 for 10 days decreased platelet production in rats and monkeys, but the effects were reversible. These studies suggest that refinement of dosing regimens using MDM2 PPI inhibitors will be necessary to avoid normal tissue toxicity. To this end, Higgins et al. have proposed a modeling approach for strategic scheduling of MDM2 PPI inhibitors.16
The development of MDM2 PPI inhibitors with improved PK profiles is being tested in clinical trials for both hematological and solid tumors (www.clinicaltrials.gov). The clinical utility of MDM2 PPI inhibitors is still in the early phases of study. Initial clinical trials have yielded evidence of therapeutic responses in relapsed leukemia3 and liposarcoma, but indicate that molecular profiling could be essential in identifying responders versus nonresponders. 28 To effectively target MDM2 in GBM, delivery of an MDM2 PPI inhibitor to the tumor at concentrations effective in modulating MDM2 function for sufficient duration will be essential. We are currently evaluating clinically relevant MDM2 PPI inhibitors to determine if we can further improve survival in GBM xenograft models as well as to investigate effects of MDM2 PPI inhibitors on therapy-resistant GBM stem-like cells.
Conclusions
Modulation of MDM2/p53-associated signaling pathways is a novel approach for decreasing TMZ resistance in GBM. To our knowledge, this is the first study in a humanized intracranial patient-derived xenograft model to demonstrate the efficacy of combining front-line TMZ therapy and an inhibitor of MDM2 PPIs. We found that sustained p53 pathway activation following exposure to the combination of TMZ and an MDM2 PPI inhibitor correlated with downregulation of DNA repair proteins critical for the repair of TMZ-mediated DNA damage, leading to increased apoptosis of GBM cells and increased survival of mice with intracranial GBM. The potentiation of TMZ-mediated DNA damage by MDM2 PPI inhibitors offers a promising strategy that may ultimately improve clinical outcome in patients with GBM.
Acknowledgments
We thank Veronika Slivova and staff at the Methodist Research Institute Biorepository, Andi Masters (Clinical Pharmacology Analytical Core), Malgorzata M. Kamocka (Indiana Center for Biological Microscopy facility), Tony Sinn, and Tiaishia Spragins (In Vivo Therapeutics Core) for their expert assistance. We thank Drs. Melissa Fishel, Art Baluyut, and Mark Kelley for their critical reading of this manuscript. Research for this study was supported by the National Cancer Institute of the National Institutes of Health (award number R01CA138798 [H.W., B.B., E.T., L.M., and K.P.]), the DeVault Fellowship-IUSCC Cancer Biology Training Program (E.T.), the Indiana University Melvin and Bren Simon Cancer Center Translational Research Acceleration Collaboration program (K.P.), the Indiana Clinical and Translational Sciences Institute (K.P.), and the Indiana University-Purdue University Indianapolis Signature Center Initiative for the Cure of Glioblastoma. The In Vivo Therapeutics Core and the Clinical Pharmacology Analytical Core laboratory of the Indiana University Melvin and Bren Simon Cancer Center are both supported by the National Cancer Institute (P30 CA082709). We greatly appreciate the generous support of the Mary Ann and Gene Zink Family Glioblastoma Multiforme Research Fund, Team JOEY (a Heroes Foundation Program), the Riley Children’s Foundation, and the Jeff Gordon Foundation (H.W. and K.P.).
ABBREVIATIONS
- APE1
apurinic/apyrimidinic endonuclease I
- ATM
ataxia-telangiectasia mutated
- BER
base excision repair
- BSA
bovine serum albumin
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate-dehydrogenase
- GBM
glioblastoma
- IUSCC
IUSM, Indiana University Simon Cancer Center, Indiana University School of Medicine
- MDM2
murine double minute 2
- MGMT
O6-methylguanine DNA methyltransferase
- MS
mass spectroscopy
- NSG
NOD.Cg-PrkdcscidIL2rgtm1Wjl/Sz
- PBS
phosphate-buffered saline
- PK
pharmacokinetic
- PPI
protein–protein interaction
- S15
phosphorylation on serine 15-p53
- TBS
Tris-buffered saline
- TBST
TBS and Tween 20
- TMZ
temozolomide
- T/N
combination of TMZ and nutlin3a
Footnotes
Disclosures
Dr. Sarkaria received non–study-related clinical or research support from Novartis, Basilea, Genentech, Sanofi, Beigene, Lilly, GlaxoSmithKline, Peloton, and Glionova.
Author Contributions
Conception and design: Pollok, Wang, S Cai, Bailey, Saadatzadeh, Ding, Tonsing-Carter, Eitel, Waning, Gelbert, Jones, Mayo, Shannon. Acquisition of data: Wang, S Cai, Bailey, Saadatzadeh, Ding, Tonsing-Carter, W Cai, Eitel, Bringman, Gelbert, Jones, Shannon. Analysis and interpretation of data: Pollok, Saadatzadeh, Ding, Tonsing-Carter, Long, Waning, Murray, Gelbert, Jones, Cohen-Gadol, Mayo, Shannon. Drafting the article: all authors. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Pollok. Statistical analysis: Pollok, Ding, Tonsing-Carter, Shannon. Administrative/technical/material support: Pollok, Wang, S Cai, Bailey, Saadatzadeh, Tonsing-Carter, Georgiadis, Gunter, Long, Minto, Gordon, Sen, W Cai, Eitel, Bringman, Murray, Sarkaria, Gelbert, Cohen-Gadol. Study supervision: Pollok, Georgiadis, Long, Minto, Wells, Gelbert, Jones.
References
- 1.Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J Biol Chem. 2005;280:18771–18781. doi: 10.1074/jbc.M413387200. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. 2007;26:3473–3481. doi: 10.1038/sj.onc.1210136. [DOI] [PubMed] [Google Scholar]
- 3.Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22:868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bijangi-Vishehsaraei K, Saadatzadeh MR, Huang S, Murphy MP, Safa AR. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol Cell Biochem. 2010;342:133–142. doi: 10.1007/s11010-010-0477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bocangel D, Sengupta S, Mitra S, Bhakat KK. p53-Mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009;29:3741–3750. [PMC free article] [PubMed] [Google Scholar]
- 6.Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009;28:1616–1625. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai S, Wang H, Bailey B, Ernstberger A, Juliar BE, Sinn AL, et al. Humanized bone marrow mouse model as a preclinical tool to assess therapy-mediated hematotoxicity. Clin Cancer Res. 2011;17:2195–2206. doi: 10.1158/1078-0432.CCR-10-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. 2011;Chapter 14(Unit 14.16) doi: 10.1002/0471141755.ph1416s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J. The roles of MDM2 and MDMX phosphorylation in stress signaling to p53. Genes Cancer. 2012;3:274–282. doi: 10.1177/1947601912454733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Q, Chen J. The phenotype of MDM2 auto-degradation after DNA damage is due to epitope masking by phosphorylation. Cell Cycle. 2011;10:1162–1166. doi: 10.4161/cc.10.7.15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 13.Costa B, Bendinelli S, Gabelloni P, Da Pozzo E, Daniele S, Scatena F, et al. Human glioblastoma multiforme: p53 reactivation by a novel MDM2 inhibitor. PLoS One. 2013;8:e72281. doi: 10.1371/journal.pone.0072281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 15.Erickson-Miller CL, May RD, Tomaszewski J, Osborn B, Murphy MJ, Page JG, et al. Differential toxicity of camptothecin, topotecan and 9-aminocamptothecin to human, canine, and murine myeloid progenitors (CFU-GM) in vitro. Cancer Chemother Pharmacol. 1997;39:467–472. doi: 10.1007/s002800050600. [DOI] [PubMed] [Google Scholar]
- 16.Higgins B, Glenn K, Walz A, Tovar C, Filipovic Z, Hussain S, et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin Cancer Res. 2014;20:3742–3752. doi: 10.1158/1078-0432.CCR-14-0460. [DOI] [PubMed] [Google Scholar]
- 17.Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res. 2009;7:1497–1509. doi: 10.1158/1541-7786.MCR-09-0144. [DOI] [PubMed] [Google Scholar]
- 18.Iancu-Rubin C, Mosoyan G, Glenn K, Gordon RE, Nichols GL, Hoffman R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp Hematol. 2014;42:137–145. 145.e1–145.e5. doi: 10.1016/j.exphem.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, et al. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem. 2007;282:2636–2645. doi: 10.1074/jbc.M606928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanu OO, Hughes B, Di C, Lin N, Fu J, Bigner DD, et al. Glioblastoma multiforme oncogenomics and signaling pathways. Clin Med Oncol. 2009;3:39–52. doi: 10.4137/cmo.s1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaRusch GA, Jackson MW, Dunbar JD, Warren RS, Donner DB, Mayo LD. Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1α and Hdm2. Cancer Res. 2007;67:450–454. doi: 10.1158/0008-5472.CAN-06-2710. [DOI] [PubMed] [Google Scholar]
- 22.Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27:997–1003. doi: 10.1038/sj.onc.1210707. [DOI] [PubMed] [Google Scholar]
- 23.Lehman JA, Mayo LD. Integration of DNA damage and repair with murine double-minute 2 (Mdm2) in tumorigenesis. Int J Mol Sci. 2012;13:16373–16386. doi: 10.3390/ijms131216373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran DM, Maki CG. Nutlin-3a induces cytoskeletal rearrangement and inhibits the migration and invasion capacity of p53 wild-type cancer cells. Mol Cancer Ther. 2010;9:895–905. doi: 10.1158/1535-7163.MCT-09-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27:254–271. doi: 10.7555/JBR.27.20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 29.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5:27–41. doi: 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- 30.Rudek MA, Donehower RC, Statkevich P, Batra VK, Cutler DL, Baker SD. Temozolomide in patients with advanced cancer: phase I and pharmacokinetic study. Pharmacotherapy. 2004;24:16–25. doi: 10.1592/phco.24.1.16.34800. [DOI] [PubMed] [Google Scholar]
- 31.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 32.Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. 2008;14:2900–2908. doi: 10.1158/1078-0432.CCR-07-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Secchiero P, Corallini F, Gonelli A, Dell’Eva R, Vitale M, Capitani S, et al. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res. 2007;100:61–69. doi: 10.1161/01.RES.0000253975.76198.ff. [DOI] [PubMed] [Google Scholar]
- 35.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinozaki T, Nota A, Taya Y, Okamoto K. Functional role of Mdm2 phosphorylation by ATR in attenuation of p53 nuclear export. Oncogene. 2003;22:8870–8880. doi: 10.1038/sj.onc.1207176. [DOI] [PubMed] [Google Scholar]
- 37.Sionov RV, Coen S, Goldberg Z, Berger M, Bercovich B, Ben-Neriah Y, et al. c-Abl regulates p53 levels under normal and stress conditions by preventing its nuclear export and ubiquitination. Mol Cell Biol. 2001;21:5869–5878. doi: 10.1128/MCB.21.17.5869-5878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 40.Supiot S, Hill RP, Bristow RG. Nutlin-3 radiosensitizes hypoxic prostate cancer cells independent of p53. Mol Cancer Ther. 2008;7:993–999. doi: 10.1158/1535-7163.MCT-07-0442. [DOI] [PubMed] [Google Scholar]
- 41.Tallarida RJ. Quantitative methods for assessing drug synergism. Genes Cancer. 2011;2:1003–1008. doi: 10.1177/1947601912440575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor JW, Schiff D. Treatment considerations for MGMT-unmethylated glioblastoma. Curr Neurol Neurosci Rep. 2015;15:507. doi: 10.1007/s11910-014-0507-z. [DOI] [PubMed] [Google Scholar]
- 43.Tonsing-Carter E, Bailey BJ, Saadatzadeh MR, Ding J, Wang H, Sinn AL, et al. Potentiation of carboplatin-mediated DNA damage by the Mdm2 modulator Nutlin-3a in a humanized orthotopic breast-to-lung metastatic model. Mol Cancer Ther. 2015;14:2850–2863. doi: 10.1158/1535-7163.MCT-15-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 45.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by smallmolecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 46.Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2 amplified and TP53 wild-type glioblastomas. Clin Cancer Res. 2016;22:1185–1196. doi: 10.1158/1078-0432.CCR-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waning DL, Lehman JA, Batuello CN, Mayo LD. c-Abl phosphorylation of Mdm2 facilitates Mdm2-Mdmx complex formation. J Biol Chem. 2011;286:216–222. doi: 10.1074/jbc.M110.183012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodworth GF, Dunn GP, Nance EA, Hanes J, Brem H. Emerging insights into barriers to effective brain tumor therapeutics. Front Oncol. 2014;4:126. doi: 10.3389/fonc.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshimoto K, Mizoguchi M, Hata N, Murata H, Hatae R, Amano T, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol. 2012;2:186. doi: 10.3389/fonc.2012.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S, et al. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008;36:1555–1566. doi: 10.1093/nar/gkm1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang F, Tagen M, Throm S, Mallari J, Miller L, Guy RK, et al. Whole-body physiologically based pharmacokinetic model for nutlin-3a in mice after intravenous and oral administration. Drug Metab Dispos. 2011;39:15–21. doi: 10.1124/dmd.110.035915. 4 hours. [DOI] [PMC free article] [PubMed] [Google Scholar]