

Abstract
Age-related Macular Degeneration (AMD) is the leading cause of blindness among the elderly in western societies. While antioxidant micronutrient treatment is available for intermediate non-neovascular disease, and effective anti-vascular endothelial growth factor treatment is available for neovascular disease, treatment for early AMD is lacking due to an incomplete understanding of the early molecular events. The role of lipids, which accumulate in the macula, and their oxidation, has emerged as an important factor in disease development. These oxidized lipids can either directly contribute to tissue injury or react with amine on proteins to form oxidation-specific epitopes, which can induce an innate immune response. If inadequately neutralized, the inflammatory response from these epitopes can incite tissue injury during disease development. This review explores how the accumulation of lipids, their oxidation, and the ensuing inflammatory response might contribute to the pathogenesis of AMD.
Keywords: Age-related macular degeneration, Bruch’s membrane, basal deposits, drusen, low-density lipoprotein (LDL), oxidation-specific epitopes, retinal pigmented epithelium (RPE)
Introduction
Age-related macular degeneration (AMD) is the leading cause of vision loss among the elderly in western societies[1]. Today, over 1.75 million people in the United States have advanced AMD, and as the “baby boomers” age, it is estimated that 3 million people will be affected by 2020[2]. While generally afflicting people over 60 years old[3], 7 million people are currently at risk of developing advanced AMD, and 1 in 3 people over 70 years old with early AMD will develop advanced disease over the next decade[3, 4]. AMD is a major public health problem that costs $30 billion annually in the United States[5], and is predicted to rise to $59 billion over the next 20 years[6]. The impact of AMD on an individual’s quality of life is unexpectedly high. The decrease in quality of life from early AMD is similar to a person with symptomatic HIV, and with advanced AMD, to one with metastatic prostate cancer having uncontrollable pain[7]. With vision loss, a person with AMD is less active[8] and is at higher risk of depression[9, 10] and anxiety[11] than unaffected elderly people. Because there is no therapy that restores vision, individuals with early AMD are as anxious over losing vision as patients who are actually blind[11].
AMD is a multifactorial, chronic, age-related disease that is influenced by both environmental and genetic factors. Oxidative stress has long been considered a major influence on AMD pathophysiology. A decade ago, the discovery of polymorphisms in complement factor H (CFH) with AMD risk introduced innate immunity as a pathophysiologic factor. Since this discovery, multiple other components of innate immunity have been implicated in AMD pathophysiology. One consequence of oxidative stress is the formation of oxidized lipids, which can elicit an immune response. Inadequate neutralization of the oxidized lipids can transform this initially protective response into a pathologic reaction that induces tissue injury. In this review, we will describe how oxidized lipids and innate immune dysfunction contribute to AMD pathophysiology.
What is the Macula and what is AMD?
The macula is the central retina that is designed for high acuity and color vision. It is composed of the neurosensory retina, the retinal pigmented epithelium (RPE), Bruch’s membrane, and choroid. While the clinician defines the macula as the central 6 mm diameter area located between the major retinal vascular arcades (Figure 1A), the anatomist defines the macula as the retina that contains more than one layer of ganglion cell nuclei (Figure 1B)[12]. The neurosensory retina is composed of the internal limiting membrane, nerve fiber layer, ganglion cell layer, inner plexiform layer, inner nuclear layer, outer plexiform layer, and photoreceptors. In communication with the photoreceptors, the RPE is a highly specialized, polarized cell that maintains the health of the photoreceptors. The RPE has multiple essential functions including the daily phagocytosis of photoreceptor outer segments, light absorption, heat exchange, vitamin A metabolism, outer blood retinal barrier, and maintenance of the choriocapillaris[13]. The RPE is adherent to Bruch’s membrane (BrM), a specialized, pentalaminar extracellular matrix composed of the RPE basement membrane, inner collagenous layer, elastic layer, outer collagenous layer, and the choriocapillaris basement membrane[14].
Figure 1.
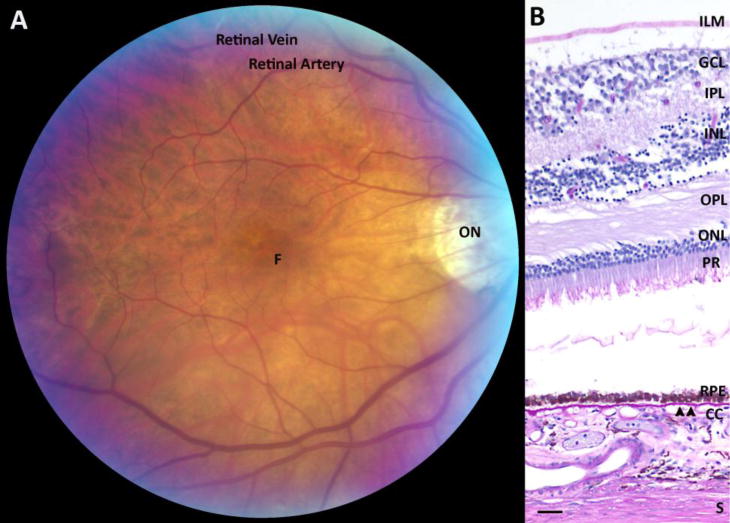
The clinician and anatomist’s view of the macula. A. Fundus photograph of the macula. ON, optic nerve; F, foveola. B. Histological section of the macula with the internal limiting membrane (ILM), multi-layered ganglion cell layer (GCL), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL), photoreceptor layer (PR), retinal pigmented epithelium (RPE), Bruch’s membrane (arrowheads), choriocapillaris (CC), and sclera (S). Bar=25μm.
AMD is categorized into two forms, a non-neovascular, “dry” form and a neovascular, “wet” form. In wet AMD, neovascular tufts grow in the subretinal space, within Bruch’s membrane, and/or the sub-RPE space. These vessels can hemorrhage or leak fluid, or develop into fibrovascular complexes, and cause rapid decrease in vision. With the dry form, vision loss is typically gradual. To the clinician, the hallmark signs are drusen, or yellowish subRPE deposits, and RPE hyper- or hypopigmentary abnormalities (Figure 2). The pigmentary abnormalities are the clinical manifestation of RPE degeneration, which ultimately culminates in cell death[15–17]. Confluent patches of RPE cell loss results in areas of “geographic atrophy”, a late form of dry AMD. When the geographic atrophy involves the foveola, vision loss is severe.
Figure 2.
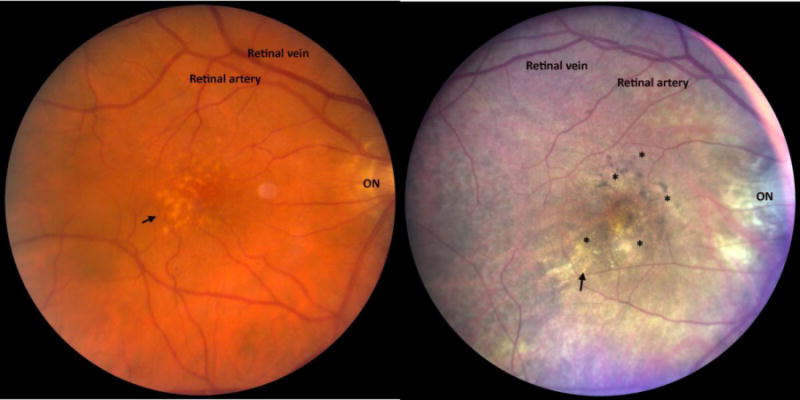
A. Fundus image of a patient with large drusen (arrow). B. Fundus image of a patient with drusen (arrow) and RPE hypo- and hyper-pigmentary changes (*). ON, optic nerve.
Besides drusen, deposits that form in the subretinal space between the photoreceptors and RPE are an integral component of the AMD pathology. Identified with little recognition by Mimoun et al. in 1990[18], reticular pseudodrusen were first clinically observed in AMD. Zweifel et al. showed that these deposits, which Mimoun et al. had hypothesized were derived from the choroid, were instead located in the subretinal space using optical coherence tomography (OCT)[19]. This clinical observation was confirmed by histopathological analysis showing subretinal deposits that contained lipoprotein particles and debris in autopsy eyes[20, 21]. These deposits have long been ignored, in part, because they can be challenging to visualize on clinical exam. These lesions are now within the mainstream assessment of clinicians due to improvements in imaging modalities such as OCT and fundus autofluorescence, and heightened awareness that these lesions are a marker for disease advancement[19, 22–25].
Histopathologically, the cardinal changes of AMD are RPE degeneration and extracellular deposits on either side of the RPE. The normal cuboidal RPE morphology becomes irregularly shaped, then flattened or atrophic, and finally dies[26–28]. These changes are reminiscent of epithelial-mesenchymal transition (EMT) where a cell dedifferentiates to survive a stressful microenvironment at the expense of losing function. In AMD, the RPE displays key features of “type 2” EMT, which is associated with tissue regeneration or repair[29], and includes: i) initiation by tissue injury or inflammation; ii) loss of cell polarity; iii) loss of cell-cell adhesion; and iv) a gain of cell migration. In particular, the RPE degenerates from a single nucleated hexagonal cell to a multinucleated, larger hexagonal cell, and then to a flattened cell that either “subducts into the sub-RPE or migrates to the subretinal space[17, 30, 31].
Significant changes are also seen in Bruch’s membrane. Sarks categorized AMD severity after thorough clinical and pathological examination on eyes over a wide age range[28]. The analysis focused on basal deposit formation, or the accumulation of heterogeneous debris within Bruch’s membrane. The composition and location define the extent that basal deposits are associated with aging or AMD. With aging, Bruch’s membrane thickens, mainly due to accumulations in the outer collagenous layer, which begin early in life[32]. Basal laminar deposits (BlamD) accumulate between the RPE cell and its basement membrane while basal linear deposits (BlinD) are heterogeneous deposits within the inner collagenous layer (Figure 3). BlamD accumulate with aging, and are not associated with AMD when they are thin and homogeneous in composition. They look identical to the RPE basement membrane on electron microscopy, and consist in part, of normal RPE basement membrane molecules such as collagen IV, laminin, and heparan sulfate proteoglycans[33]. In early AMD, BlamDs are thin and continuous, and associated with RPE pigmentary disturbance. With AMD, BlamD thicken and contain heterogeneous debris including long spacing collagen, inflammatory debris, and lipids. In AMD, Bruch’s membrane also accumulates BlinD and large drusen (>125microns in diameter)[34–36], which constitute different morphologic forms of the same lesion (Figure 3)[34, 36–38].
Figure 3.
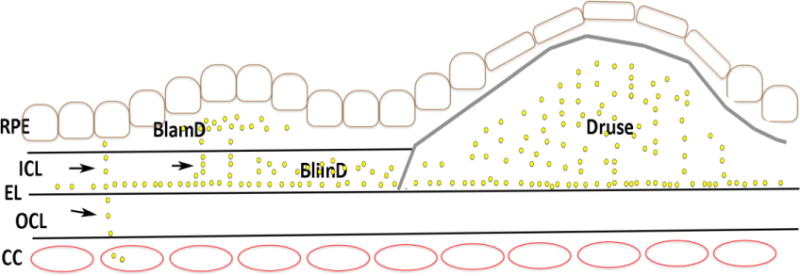
Cartoon of Bruch’s membrane deposits with AMD. Basal laminar deposits (BLamD) develop between the RPE basal lamina (RPE BL) and the RPE cell while basal linear deposits (BLinD) develop in the inner collagenous layer (ICL). A druse occupies the subRPE space and is denoted by the gray line. The yellow dots represent lipoprotein deposits, which can be released from the RPE and transit in linear streaks (arrows) toward the choriocapillaris (CC). With age-related changes to the elastic layer (EL), lipoproteins collect at the inner surface of the elastic layer (EL) and accumulate toward the RPE.
Genetic risk factors in AMD
Genetic variants in a number of genes are associated with AMD risk, and as a result, they have revolutionized how we think about AMD. The most compelling linkage has been identified at 1q25–32 and 10q26[39–41]. Patients who are homozygous for single nucleotide polymorphisms (SNP) in CFH on 1q32 have a 6.32 odds ratio risk of AMD[42–45]. Genome-wide association studies (GWAS) have also identified the chromosome 10q26 susceptibility locus that contains ARMS2 (age-related maculopathy susceptibility 2) and HTRA1, a serine protease, which carry similar risk as the CFH variant[46]. Just as the discovery of CFH polymorphisms has provided an unexpected role for complement activation into its pathophysiology, genetic links of lipid related genes may provide further insights into how lipid biology influences AMD. GWAS studies have identified risk variants in genes involved in lipid metabolism and the transfer of lipids among lipoproteins, such as ABCA1, LIPC, CETP, and LPL, and apolipoprotein E4 (APOE4), which has a protective effect[47, 48].
Epidemiologic Risk Factors in AMD
The incidence of AMD is approximately 3% per year for patients over 65 years old[49, 50]. A variety of non-genetic or environmental factors contribute to AMD risk[51]. Some risk factors are not modifiable, such as advancing age or gender[51, 52].Chronological age is the strongest risk factor for AMD while cigarette smoking is the second strongest, but modifiable risk factor[53]. Epidemiologic data from several large studies indicate that smoking influences both the onset and progression of dry AMD[53–56]. The smoking “pack-years” strongly correlates with AMD risk while smoking cessation reduces the risk[54]. In the Blue Mountains Eye study, the RPE was found to be a specific target of cigarette smoke as RPE abnormalities were a prominent change in smokers[57]. In the Age-related Eye Disease Study (AREDS), smoking was correlated with geographic atrophy[58]. A number of other factors have been found to influence AMD, including diet, high body mass index, cardiovascular disease, hypertension, nutrition, and sunlight exposure [51, 52, 56].
Epidemiologic studies have also evaluated the role of serum lipids in AMD. Thus far, the data supporting a role for serum lipids with AMD risk have been mixed. Prior studies have shown either no or an inverse association of HDL cholesterol and total cholesterol with AMD[50, 53, 59–69]. Alternatively, the Age, Gender/Environment Susceptibility Study (AGES) and Rotterdam Study found that elevated plasma HDL cholesterol, but not total cholesterol, low-density lipoprotein cholesterol, and triglycerides were associated with incidence of AMD[49, 62], and the Beaver Dam Eye Study found that HDL cholesterol was associated with geographic atrophy[50]. The ambiguous association of serum cholesterol with AMD risk might be explained by the local production of lipoproteins in the eye, which accumulate in Bruch’s membrane rather than a systemic origin, as discussed the next section, “Role for Lipids in AMD”.
As an alternative to serum cholesterol, omega-3 long-chain polyunsaturated fatty acids (n-3 LC-PUFAs), including eicosapentaenoic acid (EPA) and docohexanoic acid (DHA), have been investigated as biomarkers of AMD. DHA in particular, has important structural and protective functions in the retina, especially in photoreceptors[70]. Since photoreceptor cell death is a fundamental change in AMD, to determine what is released during retinal injury and identify molecular biomarkers for AMD, Orban et al. induced photoreceptor and RPE degeneration using light exposure in ABCA4−/−RD8−/− and wild type mice[71]. Serum DHA levels were significantly decreased after light exposure in both experimental groups. This finding prompted an evaluation in patients where serum DHA was also found to be decreased in AMD (n=24) relative to control patients (n=21). The suggestion of omega 3 fatty acids as a biomarker from this small cohort is supported by Merle et al. who showed that reduced red blood cell membrane EPA and serum EPA-DHA, as long-term biomarkers of n-3 dietary PUFA status were associated strongly with neovascular AMD[72]. The role that decreased PUFAs have in AMD pathophysiology requires further study because in two prospective trials, the Nutritional AMD Treatment 2 Study and the Age-related Eye Disease Study 2, dietary PUFAs had no effect on the progression of early to late stage AMD[73, 74].
Oxidized lipids offer a potential biomarker for AMD. In particular, carboxyethylpyrrole (CEP) is an oxidative protein modification generated from DHA-containing phospholipids, which are highly abundant in photoreceptors. As a result, CEP is a unique oxidized lipid originating from photoreceptors that could serve as an AMD biomarker. Indeed, CEP has been identified in photoreceptors, drusen, and choroid from patients with AMD, and is elevated in serum of AMD patients compared to age-matched control patients[75, 76]. The odds ratio for elevated CEP was more than 3-fold greater in AMD than in control patients[77]. When genotype is included in the model, the AMD risk for patients with elevated CEP and risk genotypes, especially ARMS2 and HTRA1, was 2–3-fold greater than the risk based on genotype alone.
Role for Lipids in AMD
Lipid particles, predominantly composed of cholesteryl esters, cholesterol, and phosphatidylcholine[78] in apolipoprotein B containing lipoproteins, accumulate in Bruch’s membrane prior to the development and in the same location as BlamD, BlinD, and drusen. The most current theory, as described by Christine Curcio, PhD[79], suggests that the RPE synthesizes and secretes very low density-like lipoproteins, which has also been observed by our laboratory[80, 81], rather than originating from the systemic circulation. These lipoproteins transit from the RPE through Bruch’s membrane in linear tracks[82] (Figure 3). Due to aging related changes to the elastic layer in Bruch’s membrane, lipoproteins first accumulate in the inner collagenous layer adjacent to the elastic layer, and then toward the RPE, eventually forming a “lipid wall”. The lipid wall prevents the normal passage of essential nutrients and oxygen as well as potentially toxic metabolites from passing between the RPE and choriocapillaris. In addition, heterogeneous material and inflammatory debris accumulates to thicken the BlamD, and with this accumulation and thickening, BlamD become histopathologically associated with AMD[34].
In addition to lipoprotein secretion, impaired reverse cholesterol transport in macrophages can impact drusen formation. In AMD, macrophages accumulate around drusen[83]. While it unclear whether these macrophages are protective or involved in AMD pathology, the Apte lab has elegantly shown that the cholesterol efflux regulators ABCA1 and ABCG1 are downregulated in macrophages with aging in both mice and humans, and in neovascular AMD[84]. Since macrophages likely transport cholesterol from drusen to high density lipoproteins (HDL) for removal into the systemic circulation as a protective response, the decline in cholesterol efflux regulators could result in accumulation of cholesterol either in macrophages or drusen. When cholesterol accumulates in macrophages, the macrophages switch from a predominantly pro-inflammatory M1 to pro-angiogenic M2 phenotype, which might help promote the conversion from dry to neovascular AMD. In support of these mechanistic data, ABCA1 variations are related with risk of intermediate AMD, which is characterized by lipid rich soft drusen, and advanced AMD[85]. The combination of apoB100 lipoprotein accumulation in Bruch’s membrane along with impaired reverse cholesterol transport from a decline in cholesterol efflux transporters in macrophages results in a net accumulation of drusen.
Although less well characterized, reticular pseudodrusen or subretinal drusenoid deposits (SDD) have been found to contain unesterified cholesterol, apolipoprotein E, vitronectin, and complement[20, 86, 87]. The presence of apoE and cholesterol suggests that HDLs are involved in the SDD process, and that they are distinct from and evolve by a different process than basal deposits and drusen. Recently, Greferath et al. found that SDD extend from the subretinal space through the outer nuclear layer, and even beyond the outer limiting membrane, and contained photoreceptor associated proteins and IBA-1 labeled immune cells. They also found through cellular analysis that SDDs were associated with photoreceptor disruption and loss, localized gliosis, and dysmorphic RPE[88].
Pikuleva and Curcio have proposed a two lesion, two compartment model to explain the formation of basal deposits/drusen and SDDs[82]. The theory relies on the differences in lipid composition and topographical location of cones and rods. While cones are concentrated in the central macula, rods occupy both the perifovea and the peripheral retina[89]. Relative to cones, the apex of rod outer segments are cholesterol poor. In the macula where BlinD develop, in addition to endogenous cholesterol synthesis by the RPE and delivery of lipoproteins from the plasma, the phagocytosis of cones provides an additional source of cholesterol that the RPE must process, and the cholesterol is secreted in very low density like lipoproteins into Bruch’s membrane to form the nidus around which BlamD, BlinD, and drusen form. On the other hand, SDDs form in rod-rich areas, such as the perifovea. Rod cell membranes contain a cholesterol gradient with the least cholesterol content in the distal outer segments. The photoreceptors and Muller cells have an active bidirectional transport with the RPE for cholesterol and polyunsaturated fatty acids[90, 91]. Since HDLs cycle between the RPE and photoreceptors in the interphotoreceptor matrix, it is plausible that HDLs accumulate the released cholesterol by rod outer segments in the subretinal space as part of this lipid transport system. With RPE dysfunction, lipid transport system impairment could lead to the accumulation of HDLs as the nidus for SDD formation in the subretinal space. In addition to apoE and unesterified cholesterol, a component of HDLs, SDDs contain CD59, CFH, and vitronectin, which suggests that complement activation plays a role in SDD formation[20, 86, 87]. Thus, it is possible that the materials in SDD are themselves toxic or incite an inflammatory response that appears to include complement activation. With accumulation of SDDs, the normal movement of nutrients including oxygen from the choriocapillaris to photoreceptor outer segments is compromised, ultimately contributing to outer retinal atrophy.
Role of oxidative stress in AMD
The macula lives in a high oxidative stress environment due to a number of sources of reactive oxygen species (ROS), as reviewed in[92]. Due to the high energy demands of vision, the macula has a high metabolic rate that generates high levels of ROS. To meet these high energy demands, the macula receives some of the highest blood flows in the body, and as a result, the RPE is exposed to high ambient (70–90 mm Hg) oxygen partial pressures[92]. Remarkably, the RPE routinely phagocytizes 30,000 photoreceptor outer segments per day[93]. During this process, intracellular H2O2 resulting from NADPH oxidase in the phagosome or β-oxidation of rod outer segment lipids in peroxisomes is generated at levels that are similar to macrophages undergoing phagocytosis[94, 95]. Photo-oxidative stress is a unique, constant source of exogenous oxidative stress. The Chesapeake Bay study on watermen provided epidemiologic evidence of sunlight exposure with AMD risk[96, 97] and the Alienor Study suggested that ultraviolet light exposure is a risk for early AMD [98]. Photo-oxidative stress is linked with oxidative damage to the retina, RPE, and choroid, and multiple other works have substantiated this observation as reviewed in[99].
Cigarette smoking adds to the oxidative stress burden for patients who choose to smoke. Conceptually, it is not difficult to understand why smoking raises oxidative stress because cigarette smoke contains nearly 5000 toxins, many of which are strong oxidants[100, 101], and each puff of cigarette smoke contains 1015 free radicals[102]. Which or how many of these oxidants actually reach the eye, or how much oxidative stress is generated from a smoke induced inflammatory response is unknown. However, it is clear that cigarette smoke depletes tissues of ascorbic acid and protein sulfhydryl groups, causing the oxidation of DNA, lipids and proteins[103–105]. Many of these molecular changes such as malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and advanced glycation endproducts (AGE), have been identified in AMD, and suggest that oxidative damage is an important factor in the mechanism of disease development.
The AREDS enrolled nearly 5000 patients to evaluate the effectiveness of antioxidant micronutrients including β-carotene, vitamin C, vitamin E, and zinc[58]. Patients with intermediate, dry AMD who took this formulation had a 25% reduction in progression to advanced AMD. Moriarty-Craige et al., using the AREDS cohort, showed that patients on the AREDS formulation had improved plasma redox potential with increased cysteine/cystine despite no change in the glutathione/glutathione disulfide [106]. Since cysteine helps to regulate apoptosis and immune function, the benefit of the antioxidant treatment on progression could be partially explained by improved cysteine availability. Collectively, the AREDS data suggest that neutralizing oxidative stress with antioxidant micronutrients have a role in treating AMD.
The impaired antioxidant response in AMD
The accumulation of oxidatively damaged tissue indicates that the antioxidant response is insufficient Cells have evolved multiple strategies to neutralize harmful levels of ROS, and at the same time, allow for physiological ROS signaling. These strategies include restricting oxidant producing enzymes to a specific locale near the intended target or the entry of oxidants, such as H2O2, through aquaporin channels[107]. The short half-life of ROS also confines any potential spread of oxidative damage to a specific location.
A number of antioxidant systems are regulated through various transcription factors including Nf-κB, AP1, the FoxO family, or PGC-1α. However, the most comprehensive transcription system is mediated through Nuclear factor erythroid-2 related factor 2 (Nrf2), a basic leucine zipper transcription factor[108]. Nrf2 regulates a coordinated transcriptional program that maintains cellular redox homeostasis and protects the cell from oxidative injury[101, 109, 110]. Nrf2 is normally sequestered in the cytosol by Kelch-like ECH-associated protein 1 (Keap1)[111–114]. In the absence of acute stress, Keap1 constitutively suppresses Nrf2 signaling by both degrading Nrf2 through the ubiquitin-proteasomal degradation, which results in a baseline, low antioxidant gene expression[109]. However, upon an acute rise in ROS, Keap1 undergoes a conformational change after multiple cysteine residues interact with ROS, releasing Nrf2 for translocation to the nucleus where it dimerizes with Maf proteins, and binds to the antioxidant response element (ARE) in the promoters of its target genes to initiate the transcription[115–117]. Nrf2 regulates both an early acute phase through induction of “direct” antioxidant enzymes, such as catalase or SOD, and through maintenance of cellular glutathione and thioredoxin, and xenobiotic metabolism enzymes that produce reducing equivalents[118]. When ROS depletes cellular glutathione, cells can die from oxidatively mediated apoptosis[119–121].
Aging can reduce Nrf2 mRNA or protein, which impairs Nrf2 signaling. Suzuki et al. showed an age-dependent decline in Nrf2 from cigarette smoking[122]. These results have been corroborated in mice, where aging suppressed the ability of Nrf2 and its target genes in alveolar macrophages, in response to cigarette smoking[122]. We previously showed that aging impairs the Nrf2 response to acute oxidative stress in mice[123]. While the RPE of young mice elicits a robust induction of Nrf2 mediated antioxidants after exposure to the chemical oxidant sodium iodate, the RPE of old mice had a blunted antioxidant response with accumulation of MDA. MDA damage could be reversed by genetic rescue of Nrf2 signaling by conditionally knocking down Keap1.
Nrf2 also declines with disease. Nrf2 mRNA and protein, and Nrf2-dependent antioxidants and glutathione levels are reduced, and oxidative stress is increased in human emphysematous compared to normal lungs[122, 124] as well as Nrf2 deficient mice, which develop emphysema after chronic cigarette smoke exposure[101]. We have provided evidence that Nrf2 is decreased in AMD[125]. In AMD maculas, Nrf2 immunolabeling was increased with nuclear labeling in morphologically normal RPE compared to the RPE of non-AMD maculas, which suggests that Nrf2 signaling is induced by the high oxidative stress microenvironment of the AMD macula. However, in dysmorphic RPE cells from AMD maculas, Nrf2 immunolabeling was decreased relative to morphologically normal RPE cells. This finding suggests that in AMD as in other tissues, when the antioxidant system fails, the delicate balance between physiological oxidative stress and the onset of pathological oxidative stress is upset, resulting in tissue damage, as diagramed in Figure 4.
Figure 4.
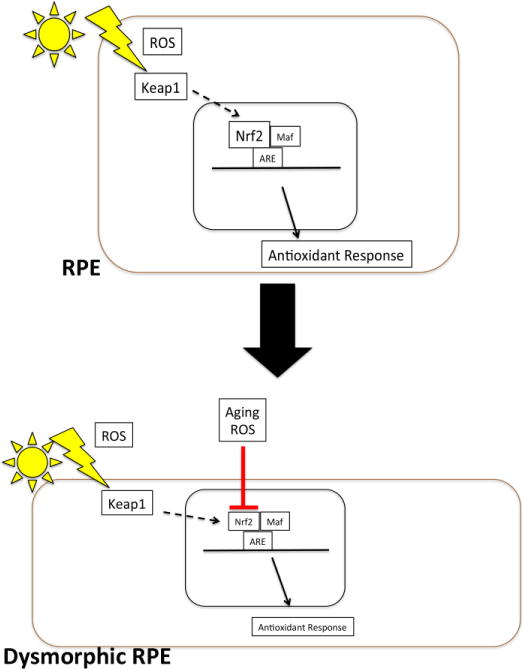
Impaired Nrf2 signaling in AMD. Oxidative stress with reactive oxygen species (ROS) induce Nrf2 production and release from Keap1 with transit to the nucleus. After binding to antioxidant response elements in the promoters of antioxidant and cytoprotective genes with Maf proteins, antioxidant genes are upregulated. With aging and chronic oxidative stress, Nrf2 production and activity is reduced, and the antioxidant response is impaired, which contributes to RPE dysfunction.
The ocular “response to retention” hypothesis
The accumulation of lipoproteins in Bruch’s membrane is reminiscent of the “response to retention” hypothesis of atherosclerosis, which proposes that plasma lipoproteins accumulate in the arterial subendothelial space, to initiate a cascade of inflammatory events leading to plaque formation[126, 127]. The accumulation of cholesterol and lipids in Bruch’s membrane is a necessary step in basal deposit and drusen formation, but it is not sufficient. Our lab investigated the role of lipoprotein deposition in Bruch’s membrane in a mouse model. A single APOB gene encodes the apoB100 and apoB48 isoforms. In humans, apoB48 is synthesized in the small intestines and is present in chylomicrons while apoB100 is mainly produced by the liver. In contrast, apoB48 is the major apoB form expressed in all tissues of mice. To simulate apoB100 lipoprotein production in humans, “apoB100” mice that have a mutation in codon 2153 corresponding to the apo-B48 editing codon, which reduces the translation of apoB48 so that apoB100 is predominantly produced, have been used for study of atherosclerosis[128]. We showed that these mice accumulate apoB100 lipoproteins in Bruch’s membrane as early as two months of age[81]. However, when aged to 18 months, these mice do not develop basal deposits or drusen, suggesting that the accumulation of lipoproteins is not sufficient for basal deposit formation. Part of the ocular response to retention theory relies on an inciting agent that will generate an innate immune response with the accumulation of inflammatory debris to form BlinD. Oxidized lipids are an obvious choice given the high oxidative stress environment, the reduced antioxidant capability with aging, and that oxidized lipids induce a pro-inflammatory response. Polyunsaturated fatty acids are in particular, vulnerable to oxidation, which results in highly reactive degradative products such as MDA, 4-HNE, and core aldehyde, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC)[129]. Indeed, multiple oxidized lipids have been identified in various layers of the macula with AMD including MDA, 4-HNE, CEP, oxidized phospholipids (OxPL), and 7-ketocholesterol (7KCh). In the macula, CEP localizes to photoreceptor outer segments, the RPE, and Bruch’s membrane[76, 130]. OxPL has also been identified in photoreceptor outer segments[131]. The RPE contains MDA and 4-HNE[132, 133], while Bruch’s membrane, including drusen, contains MDA, CEP, 7KCh and OxPL[75, 130, 134–136].
Consequences of oxidized lipids, oxidation specific epitopes, and activation of innate immunity in AMD
As mentioned above, a trigger for the inflammatory component of the response to retention hypothesis is oxidized lipids. Oxidized lipids can generate oxidative stress themselves, or they can incite an inflammatory response. For example, the oxysterol, 7-ketocholesterol (7KCh), is an abundant oxidized lipid found in oxidized lipoproteins in both atherosclerosis and Bruch’s membrane[136–139]. 7KCh is toxic to a number of cell types including the RPE[140]. In atherosclerosis, oxysterols contribute to the conversion of macrophages into foam cells[141]. In the RPE, oxysterols are taken up at a high rate, and can induce inflammation and apoptosis[142]. The Rodriguez laboratory has shown that the majority of the pro-inflammatory signaling by 7KCh is mediated through its interaction with Toll-like receptor 4 (TLR-4) both in vitro and in vivo[143]. Interaction of 7KCh with TLR-4 activates NF-kB signaling to induce pro-inflammatory cytokine expression. Given the aging associated accumulation of cholesteryl esters in Bruch’s membrane and drusen, and their tendency to become oxidized into oxysterols, the toxic role of 7KCh and/or the failure to neutralize oxysterols may explain in part, how oxidized lipoproteins contribute to AMD pathology[144].
Picard et al. found that in RPE cells, CD36 participates in oxLDL uptake and clearance of subRPE deposits[145]. Furthermore, CD36 deficient mice accumulated oxLDL in Bruch’s membrane even when fed a normal chow diet. ApoE knockout mice given a high fat, high cholesterol diet, typically accumulate lipids in Bruch’s membrane, but when given a CD36 agonist, had diminished Bruch’s membrane thickening and preserved photoreceptor function. These results suggest that CD36, by neutralizing oxLDLs, can protect against basal deposit formation.
The Wang laboratory recently showed that retinal microglia have chemotropism for, and internalize 7KCh[146]. 7KCh was capable of inducing the migration retinal microglia, which were activated into an M1 pro-inflammatory phenotype, into the subretinal space. These activated microglia also expressed pro-angiogenic cytokines with reduced expression of neurotrophic factors. This profile favors retinal degeneration and initiation of choroidal neovascularization, or the advanced, neovascular form of AMD.
The recruitment of microglia to the subretinal space is significant because it may constitute an initiating step in the immune microenvironment during AMD progression. It is possible that resident choroidal macrophages or circulatory monocytes are also recruited with 7KCh accumulation via CCL2 and CXCR2 chemokine signaling[146–148]. In support of this notion, Sennlaub et al. have identified increased intraocular CCL2 and CCR2+circulating monocytes in atrophic AMD[149]. They showed that CX3CR1, which is constitutively expressed in the retina, represses CCL2 expression, and the recruitment of CCR2 pro-inflammatory monocytes. In Cx3Cr1 deficient mice, they found that aging or light exposure induced subretinal inflammation and photoreceptor degeneration, that subretinal monocytes overexpressed CCL2, and that impairment of CCL2 or CCR2 prevented inflammatory monocyte recruitment and photoreceptor degeneration. While these findings in mouse models are suggestive of a role for retinal microglia in the subretinal space, evidence of a role for retinal microglia in early AMD is lacking.
Oxidized lipid products can react from covalent bonds with primary amines of proteins and amino groups of lipids such as phosphatidylethanolamine (PE), and thereby form oxidation-specific epitopes (OSEs), which are recognized by innate immune receptors in a hapten-specific manner[150]. Unless neutralized in a timely manner, OSEs can generate unwanted inflammation and contribute to tissue injury. Whether as a component of oxLDLs or independently accumulating in the RPE/Bruch’s membrane/choroid, MDA is an abundant lipid peroxidation degradation product that accumulates in a number of oxidative stress related diseases including AMD. Innate immunity has placed substantial natural selection on MDA adducts because 15% of all IgM natural antibodies bind MDA adducts[151]. In a multi-laboratory collaboration headed by the Binder laboratory, our group found that CFH, previously recognized only for its major regulation of the alternative complement pathway, is a novel pattern recognition receptor that specifically binds MDA adducts, but not other oxidized epitopes such as oxidized phosphocholine, CEP, or 4-HNE[152]. This finding is significant because CFH SNPs are highly associated with AMD risk[42–45]. The short consensus repeats SCR7 and SCR20 of CFH that contain various disease-related mutations including the 402H variant in SCR7, bind to MDA. Importantly, plasma from patients who have the 402H CFH variant had impaired MDA binding[153]. CFH also bound to apoptotic debris that contained MDA epitopes, which increased the generation of iC3b inactivation fragments. In contrast, the 402H CFH variant showed impaired generation of iC3b inactivation fragments when incubated with MDA decorated apoptotic particles. Since RPE apoptosis is likely an early event in AMD[15] and iC3b opsonins promote the clearance of apoptotic cells without generating inflammation[154], the reduced iC3b could translate into unwanted inflammation during clearance of apoptotic debris. CFH was also found to block the uptake of MDA-modified proteins and their proinflammatory effects by macrophages and RPE cells in vitro. When mice were given intravitreal injections of MDA-BSA, Il-8 was induced by the RPE/choroid, which was neutralized by co-injection with CFH. Toomey et al. provided interesting insight into the role of CFH in AMD pathogenesis from studying CFH deficient mice[155]. While decreased CFH was associated with subRPE deposit formation, decreased vision was only seen in CFH+/− mice with increased complement activation while CFH−/− mice, which have impaired complement function due to secondary complement depletion, had intact ERG responses. This finding indicates that it is not subRPE deposits, but complement activation that induces RPE dysfunction sufficient to decrease vision. However, given the alternative function of CFH, of binding MDA adducts and neutralizing pro-inflammatory cytokine expression, CFH could regulate visual function by controlling non-complement inflammation, too. Future studies will be needed to determine the relative contribution of complement and complement independent inflammation regulated by CFH.
Another OSE that was recently recognized is OxPL, a prominent consequence of lipid peroxidation of the polyunsaturated fatty acids found in high abundance in photoreceptors. Oxidized phosphatidylcholine appears in the retina and RPE with aging, and increases in AMD[135]. C-reactive protein (CRP) and CD14 are pattern recognition receptors that recognize oxidized phosphatidylcholine, and have been identified in AMD tissue[156, 157]. A unique OxPL species is the oxidation of phosphocholine (PC) containing PL, which is detected with the IgM natural antibody E06 that binds to the PC moiety of OxPL, but not to the PC moiety of unoxidized phospholipids[158, 159]. This OxPL accounts for 85% or more of all phosphocholine (PC) containing OxPL found on lipoproteins in plasma and on apoptotic cells, as determined by the binding of murine monoclonal antibody E06[160, 161].
Lipoprotein (a) [Lp(a)] is a lipoprotein composed of apolipoprotein(a) [apo(a)] covalently bound to apolipoprotein B-100 (apoB) by a disulfide bond between Cys4326 of apoB and Cys4057 of apo(a) on kringle IV type 9 (KIV9)[162–164]. Apo(a) is highly homologous to the plasminogen gene, which contains 5 kringles (K) and a protease domain. However, apo(a) is distinct from plasminogen as it only contains KIV (10 subtypes of which KIV-2 is present in multiple and variable copies) and KV, and has an inactive protease domain due to a Ser561-Ile562 substitution for Arg561-Val562 that prevents plasminogen activators from converting apo(a) to plasmin[165]. Unlike plasminogen, apo(a) is present only in humans, non-human primates, and old world monkeys. In humans, Lp(a) has been shown to bind OxPL[160]. While likely a protective effect, it is theorized that Lp(a) can become atherogenic with enhanced binding to arterial intimal proteoglycans, which increases local concentrations of OxPL, inducing an inflammatory response by the tissue. Our laboratory recently found by immunohistochemistry that OxPL, apo(a), and apoB accumulate in maculas including drusen of AMD and age-matched controls[131] (Figure 5). Since immunohistochemistry is not quantitative, to elucidate the impact of Lp(a) in the fundus, we evaluated transgenic Lp(a) mice and mutant Lp(a) mice, which are unable to bind OxPL, that were stressed with a high fat diet. Lp(a) mice developed mild age-related changes in the fundus. In contrast, high fat diet-fed mutant Lp(a) mice that are unable to bind OxPL, developed RPE cell degeneration, basal deposits, and occasional drusen, which were visualized by light and electron microscopy. These morphologic changes were associated with increased oxidized phospholipids, decreased antioxidant proteins, increased complement, and decreased complement regulators. Our data suggest that in the eye, Lp(a), by binding OxPL, neutralizes its potential pro-inflammatory, tissue injuring effect, and may be protective against any contribution of OxPL on AMD pathology.
Figure 5.
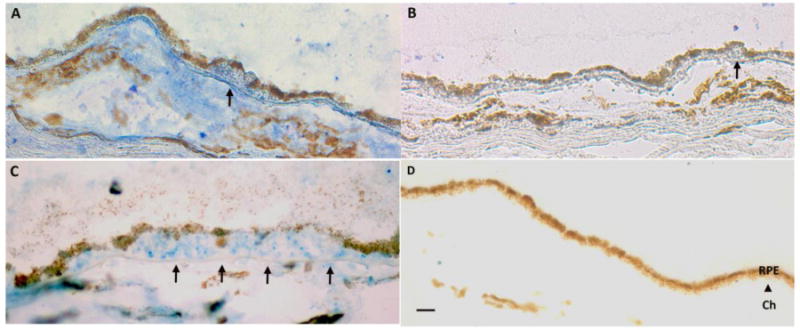
OxPL and Lp(a) in the macula. A. Immunostaining for OxPL identified with E06 antibody in Bruch’s membrane and choroid including a druse (arrow). B. IgM control. C. Immunostaining for apo(a) in Bruch’s membrane and choroid including a large druse (arrow). D. IgG control. Ch, choroid. Bar=25μm.
As mentioned previously, CEP is a distinct and perhaps signature OSE in the macula. In the retina, docosohexanoic acid (DHA) is the most abundant fatty acid in photoreceptor tips, and is also the most oxidizable fatty acid in the body because of its unsaturated structure. In the retina, DHA is oxidatively modified to CEP[76]. Matching the distribution of DHA, CEP adducts immunolocalize in photoreceptor outer segments, the RPE, and drusen in higher abundance than age-matched controls patients[75]. This distribution suggests that CEP is formed in photoreceptor outer segments and is phagocytized by the RPE as the routine regeneration of outer segment processing, and ultimately released by the RPE through Bruch’s membrane into the choriocapillaris for removal into the circulation. Indirect proof of this theory is that CEP autoantibodies are found in higher concentration in the plasma of AMD patients than age-matched controls. Hollyfield et al. immunized mice with CEP adducts and found elevated anti-CEP antibodies compared to controls[166]. Upon pathologic examination, photoreceptors were edematous overlying areas of RPE degeneration. RPE atrophy and loss were seen in patches, which was reminiscent of geographic atrophy. Bruch’s membrane developed areas of prominent basal laminar deposits. These ultrastructural changes were associated with complement deposition of C3d, a marker for C3 activation, in Bruch’s membrane. In this model, since the anti-CEP antibodies are both IgMs and IgGs, it is likely that both the innate (i.e. IgM) and adaptive (i.e. IgG) immune response is involved in the response to CEP adducts.
Final Thoughts
AMD is a highly prevalent, complex aging related disease that occurs within a 6 mm diameter area. While multifactorial in nature, the collision of oxidative stress, from the high ambient oxidative stress environment, the addition of exogenous oxidants, and failure of the anti-oxidant protective systems jeopardizes the integrity of the basic structural and functional molecules within cells. It is clear that oxidatively modified lipids form in all layers of the macula, and either directly contribute to tissue injury as a source of oxidative stress, or by adduct formation, develop oxidation-specific epitopes. The OSEs initially incite a protective immune response that is designed to neutralize or remove these potentially toxic molecules. However, if inadequately neutralized, the OSEs can convert the protective inflammatory response into a pathologic, chronic tissue injuring reaction. Currently, our understanding of how these OSEs participate in AMD pathogenesis has not been elucidated in part, because of our reliance on mouse models of AMD, which do not develop a complete AMD phenotype. While rejuvenating the antioxidant systems in the macula might reduce the formation of these potentially toxic oxidized lipids, fully categorizing the oxidized lipids that develop and the specific pattern recognition receptors that neutralize them, will offer new insights into disease etiology, and to develop alternative targets that prevent tissue injury in AMD.
Highlights.
Lipoprotein and lipid accumulation in the macula play an essential role in Age-related macular degeneration.
These lipids become oxidized and can induce tissue injuring inflammation unless neutralized by innate immunity.
Some oxidized lipids can form adducts to generate oxidation-specific epitopes which can also cause tissue injuring inflammation unless adequately neutralized.
Acknowledgments
National Eye Institute EY019044 (JTH) EY14005 (JTH), RPE Senior Scientist Award (JTH), Wilmer Core Grant EY001765, unrestricted grant from Research to Prevent Blindness to the Wilmer Eye Institute, Gifts from the Merlau family and Aleda Wright. Dr Handa is the Robert Bond Welch Professor of Ophthalmology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: Drs. Handa and Cano receive grant funding from Bayer Pharmaceuticals, Inc.
References
- 1.Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 2.Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 3.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukesh BN, Dimitrov PN, Leikin S, Wang JJ, Mitchell P, McCarty CA, Taylor HR. Five-year incidence of age-related maculopathy: the Visual Impairment Project. Ophthalmology. 2004;111:1176–1182. doi: 10.1016/j.ophtha.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 5.Brown GC, Brown MM, Sharma S, Stein JD, Roth Z, Campanella J, Beauchamp GR. The burden of age-related macular degeneration: a value-based medicine analysis. Trans Am Ophthalmol Soc. 2005;103:173–184. discussion 184–176. [PMC free article] [PubMed] [Google Scholar]
- 6.Centre for Eye Research Australia. Centrally Focused: The Impact of Age-Related Macular Degeneration. Access Australia. 2006 [Google Scholar]
- 7.Brown MM, Brown GC, Stein JD, Roth Z, Campanella J, Beauchamp GR. Age-related macular degeneration: economic burden and value-based medicine analysis. Can J Ophthalmol. 2005;40:277–287. doi: 10.1016/S0008-4182(05)80070-5. [DOI] [PubMed] [Google Scholar]
- 8.Wysong A, Lee PP, Sloan FA. Longitudinal incidence of adverse outcomes of age-related macular degeneration. Arch Ophthalmol. 2009;127:320–327. doi: 10.1001/archophthalmol.2008.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brody BL, Gamst AC, Williams RA, Smith AR, Lau PW, Dolnak D, Rapaport MH, Kaplan RM, Brown SI. Depression, visual acuity, comorbidity, and disability associated with age-related macular degeneration. Ophthalmology. 2001;108:1893–1900. doi: 10.1016/s0161-6420(01)00754-0. discussion 1900–1891. [DOI] [PubMed] [Google Scholar]
- 10.Augustin A, Sahel JA, Bandello F, Dardennes R, Maurel F, Negrini C, Hieke K, Berdeaux G. Anxiety and depression prevalence rates in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:1498–1503. doi: 10.1167/iovs.06-0761. [DOI] [PubMed] [Google Scholar]
- 11.Berman K, Brodaty H. Psychosocial effects of age-related macular degeneration. Int Psychogeriatr. 2006;18:415–428. doi: 10.1017/S1041610205002905. [DOI] [PubMed] [Google Scholar]
- 12.Orth DH, Fine BS, Fagman W, Quirk TC. Clarification of foveomacular nomenclature and grid for quantitation of macular disorders. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83:OP506–514. [PubMed] [Google Scholar]
- 13.Strauss O. The retinal pigment epithelium in visual function. Physiological reviews. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 14.Hogan MJ. Role of the retinal pigment epithelium in macular disease. Trans Am Acad Ophthalmol Otolaryngol. 1972;76:64–80. [PubMed] [Google Scholar]
- 15.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 16.Zanzottera EC, Messinger JD, Ach T, Smith RT, Freund KB, Curcio CA. The Project MACULA Retinal Pigment Epithelium Grading System for Histology and Optical Coherence Tomography in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2015;56:3253–3268. doi: 10.1167/iovs.15-16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ach T, Tolstik E, Messinger JD, Zarubina AV, Heintzmann R, Curcio CA. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56:3242–3252. doi: 10.1167/iovs.14-16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mimoun G, Soubrane G, Coscas G. J Fr Ophtalmol. 1990;13:511–530. [Macular drusen] [PubMed] [Google Scholar]
- 19.Zweifel SA, Spaide RF, Curcio CA, Malek G, Imamura Y. Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology. 2010;117:303–312 e301. doi: 10.1016/j.ophtha.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Rudolf M, Malek G, Messinger JD, Clark ME, Wang L, Curcio CA. Sub-retinal drusenoid deposits in human retina: organization and composition. Exp Eye Res. 2008;87:402–408. doi: 10.1016/j.exer.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarks J, Arnold J, Ho IV, Sarks S, Killingsworth M. Evolution of reticular pseudodrusen. Br J Ophthalmol. 2011;95:979–985. doi: 10.1136/bjo.2010.194977. [DOI] [PubMed] [Google Scholar]
- 22.Arnold JJ, Sarks SH, Killingsworth MC, Sarks JP. Reticular pseudodrusen. A risk factor in age-related maculopathy. Retina. 1995;15:183–191. [PubMed] [Google Scholar]
- 23.Cohen SY, Dubois L, Tadayoni R, Delahaye-Mazza C, Debibie C, Quentel G. Prevalence of reticular pseudodrusen in age-related macular degeneration with newly diagnosed choroidal neovascularisation. Br J Ophthalmol. 2007;91:354–359. doi: 10.1136/bjo.2006.101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee MY, Yoon J, Ham DI. Clinical characteristics of reticular pseudodrusen in Korean patients. Am J Ophthalmol. 2012;153:530–535. doi: 10.1016/j.ajo.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Hamel CP, Meunier I, Arndt C, Ben Salah S, Lopez S, Bazalgette C, Bazalgette C, Zanlonghi X, Arnaud B, Defoort-Dellhemmes S, Puech B. Extensive macular atrophy with pseudodrusen-like appearance: a new clinical entity. Am J Ophthalmol. 2009;147:609–620. doi: 10.1016/j.ajo.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 26.van der Schaft TL, Mooy CM, de Bruijn WC, Oron FG, Mulder PG, de Jong PT. Histologic features of the early stages of age-related macular degeneration. A statistical analysis. Ophthalmology. 1992;99:278–286. doi: 10.1016/s0161-6420(92)31982-7. [DOI] [PubMed] [Google Scholar]
- 27.Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- 28.Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–341. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zanzottera EC, Messinger JD, Ach T, Smith RT, Curcio CA. Subducted and melanotic cells in advanced age-related macular degeneration are derived from retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2015;56:3269–3278. doi: 10.1167/iovs.15-16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ach T, Huisingh C, McGwin G, Jr, Messinger JD, Zhang T, Bentley MJ, Gutierrez DB, Ablonczy Z, Smith RT, Sloan KR, Curcio CA. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2014;55:4832–4841. doi: 10.1167/iovs.14-14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Schaft TL, de Bruijn WC, Mooy CM, Ketelaars DA, de Jong PT. Is basal laminar deposit unique for age-related macular degeneration? Arch Ophthalmol. 1991;109:420–425. doi: 10.1001/archopht.1991.01080030122052. [DOI] [PubMed] [Google Scholar]
- 33.van der Schaft TL, Mooy CM, de Bruijn WC, Bosman FT, de Jong PT. Immunohistochemical light and electron microscopy of basal laminar deposit. Graefes Arch Clin Exp Ophthalmol. 1994;232:40–46. doi: 10.1007/BF00176436. [DOI] [PubMed] [Google Scholar]
- 34.Curcio CA, Millican CL. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol. 1999;117:329–339. doi: 10.1001/archopht.117.3.329. [DOI] [PubMed] [Google Scholar]
- 35.Spraul CW, Lang GE, Grossniklaus HE. Morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:2724–2735. [PubMed] [Google Scholar]
- 36.Bressler NM, Silva JC, Bressler SB, Fine SL, Green WR. Clinicopathologic correlation of drusen and retinal pigment epithelial abnormalities in age-related macular degeneration. Retina. 1994;14:130–142. [PubMed] [Google Scholar]
- 37.Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 38.Abdelsalam A, Del Priore L, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999;44:1–29. doi: 10.1016/s0039-6257(99)00072-7. [DOI] [PubMed] [Google Scholar]
- 39.Weeks DE, Conley YP, Tsai HJ, Mah TS, Schmidt S, Postel EA, Agarwal A, Haines JL, Pericak-Vance MA, Rosenfeld PJ, Paul TO, Eller AW, Morse LS, Dailey JP, Ferrell RE, Gorin MB. Age-related maculopathy: a genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. American journal of human genetics. 2004;75:174–189. doi: 10.1086/422476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP, Jr, Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, A.M.D.G. Consortium Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45:433–439. 439e431–432. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YT, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GH, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Said S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanche H, Deleuze JF, Igo RP, Jr, Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA, Jr, Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NT, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CC, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JR, Allikmets R, Wang JJ, Schaumberg DA, Klein BE, Hagstrom SA, Chowers I, Lotery AJ, Leveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BH, Abecasis GR, Heid IM. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–143. doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement Factor H Variant Increases the Risk of Age-Related Macular Degeneration. Science. 2005 doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 43.Edwards AO, Ritter R, Iii, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement Factor H Polymorphism and Age-Related Macular Degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 44.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science. 2005 doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neale BM, Fagerness J, Reynolds R, Sobrin L, Parker M, Raychaudhuri S, Tan PL, Oh EC, Merriam JE, Souied E, Bernstein PS, Li B, Frederick JM, Zhang K, Brantley MA, Jr, Lee AY, Zack DJ, Campochiaro B, Campochiaro P, Ripke S, Smith RT, Barile GR, Katsanis N, Allikmets R, Daly MJ, Seddon JM. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC) Proc Natl Acad Sci U S A. 2010;107:7395–7400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J, Tosakulwong N, Pericak-Vance MA, Campochiaro PA, Klein ML, Tan PL, Conley YP, Kanda A, Kopplin L, Li Y, Augustaitis KJ, Karoukis AJ, Scott WK, Agarwal A, Kovach JL, Schwartz SG, Postel EA, Brooks M, Baratz KH, Brown WL, Brucker AJ, Orlin A, Brown G, Ho A, Regillo C, Donoso L, Tian L, Kaderli B, Hadley D, Hagstrom SA, Peachey NS, Klein R, Klein BE, Gotoh N, Yamashiro K, Ferris F, Iii, Fagerness JA, Reynolds R, Farrer LA, Kim IK, Miller JW, Corton M, Carracedo A, Sanchez-Salorio M, Pugh EW, Doheny KF, Brion M, Deangelis MM, Weeks DE, Zack DJ, Chew EY, Heckenlively JR, Yoshimura N, Iyengar SK, Francis PJ, Katsanis N, Seddon JM, Haines JL, Gorin MB, Abecasis GR, Swaroop A. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A. 2010;107:7401–7406. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conley YP, Thalamuthu A, Jakobsdottir J, Weeks DE, Mah T, Ferrell RE, Gorin MB. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum Mol Genet. 2005;14:1991–2002. doi: 10.1093/hmg/ddi204. [DOI] [PubMed] [Google Scholar]
- 49.Jonasson F, Fisher DE, Eiriksdottir G, Sigurdsson S, Klein R, Launer LJ, Harris T, Gudnason V, Cotch MF. Five-year incidence, progression, and risk factors for age-related macular degeneration: the age, gene/environment susceptibility study. Ophthalmology. 2014;121:1766–1772. doi: 10.1016/j.ophtha.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein R, Klein BE, Tomany SC, Cruickshanks KJ. The association of cardiovascular disease with the long-term incidence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 2003;110:1273–1280. doi: 10.1016/S0161-6420(03)00599-2. [DOI] [PubMed] [Google Scholar]
- 51.Heiba IM, Elston RC, Klein BE, Klein R. Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye Study. Genet Epidemiol. 1994;11:51–67. doi: 10.1002/gepi.1370110106. [DOI] [PubMed] [Google Scholar]
- 52.Vingerling JR, Klaver CC, Hofman A, de Jong PT. Epidemiology of age-related maculopathy. Epidemiol Rev. 1995;17:347–360. doi: 10.1093/oxfordjournals.epirev.a036198. [DOI] [PubMed] [Google Scholar]
- 53.Smith W, Assink J, Klein R, Mitchell P, Klaver CC, Klein BE, Hofman A, Jensen S, Wang JJ, de Jong PT. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology. 2001;108:697–704. doi: 10.1016/s0161-6420(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 54.Khan JC, Thurlby DA, Shahid H, Clayton DG, Yates JR, Bradley M, Moore AT, Bird AC. Smoking and age related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br J Ophthalmol. 2006;90:75–80. doi: 10.1136/bjo.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein R, Knudtson MD, Cruickshanks KJ, Klein BE. Further observations on the association between smoking and the long-term incidence and progression of age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol. 2008;126:115–121. doi: 10.1001/archopht.126.1.115. [DOI] [PubMed] [Google Scholar]
- 56.Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., 3rd Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112:533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell P, Wang JJ, Smith W, Leeder SR. Smoking and the 5-year incidence of age-related maculopathy: the Blue Mountains Eye Study. Arch Ophthalmol. 2002;120:1357–1363. doi: 10.1001/archopht.120.10.1357. [DOI] [PubMed] [Google Scholar]
- 58.Age-related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Risk factors for neovascular age-related macular degeneration. The Eye Disease Case-Control Study Group. Arch Ophthalmol. 1992;110:1701–1708. doi: 10.1001/archopht.1992.01080240041025. [DOI] [PubMed] [Google Scholar]
- 60.Klein R, Klein BE, Jensen SC, Meuer SM. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1997;104:7–21. doi: 10.1016/s0161-6420(97)30368-6. [DOI] [PubMed] [Google Scholar]
- 61.Hyman L, Schachat AP, He Q, Leske MC. Hypertension, cardiovascular disease, and age-related macular degeneration. Age-Related Macular Degeneration Risk Factors Study Group. Arch Ophthalmol. 2000;118:351–358. doi: 10.1001/archopht.118.3.351. [DOI] [PubMed] [Google Scholar]
- 62.Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, Vingerling JR, Klein BE, Smith W, De Jong PT. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 63.van Leeuwen R, Tomany SC, Wang JJ, Klein R, Mitchell P, Hofman A, Klein BE, Vingerling JR, Cumming RG, de Jong PT. Is medication use associated with the incidence of early age-related maculopathy? Pooled findings from 3 continents. Ophthalmology. 2004;111:1169–1175. doi: 10.1016/j.ophtha.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 64.Delcourt C, Michel F, Colvez A, Lacroux A, Delage M, Vernet MH. Associations of cardiovascular disease and its risk factors with age-related macular degeneration: the POLA study. Ophthalmic Epidemiol. 2001;8:237–249. doi: 10.1076/opep.8.4.237.1613. [DOI] [PubMed] [Google Scholar]
- 65.Nowak M, Swietochowska E, Marek B, Szapska B, Wielkoszynski T, Kos-Kudla B, Karpe J, Kajdaniuk D, Sieminska L, Glogowska-Szelag J, Nowak K. Changes in lipid metabolism in women with age-related macular degeneration. Clin Exp Med. 2005;4:183–187. doi: 10.1007/s10238-004-0054-z. [DOI] [PubMed] [Google Scholar]
- 66.Abalain JH, Carre JL, Leglise D, Robinet A, Legall F, Meskar A, Floch HH, Colin J. Is age-related macular degeneration associated with serum lipoprotein and lipoparticle levels? Clin Chim Acta. 2002;326:97–104. doi: 10.1016/s0009-8981(02)00288-7. [DOI] [PubMed] [Google Scholar]
- 67.Tan JS, Mitchell P, Smith W, Wang JJ. Cardiovascular risk factors and the long-term incidence of age-related macular degeneration: the Blue Mountains Eye Study. Ophthalmology. 2007;114:1143–1150. doi: 10.1016/j.ophtha.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 68.Klein R, Myers CE, Buitendijk GH, Rochtchina E, Gao X, de Jong PT, Sivakumaran TA, Burlutsky G, McKean-Cowdin R, Hofman A, Iyengar SK, Lee KE, Stricker BH, Vingerling JR, Mitchell P, Klein BE, Klaver CC, Wang JJ. Lipids, lipid genes, and incident age-related macular degeneration: the three continent age-related macular degeneration consortium. Am J Ophthalmol. 2014;158:513–524 e513. doi: 10.1016/j.ajo.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dashti N, McGwin G, Owsley C, Curcio CA. Plasma apolipoproteins and risk for age related maculopathy. Br J Ophthalmol. 2006;90:1028–1033. doi: 10.1136/bjo.2006.093856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.SanGiovanni JP, Chew EY. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog Retin Eye Res. 2005;24:87–138. doi: 10.1016/j.preteyeres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 71.Orban T, Johnson WM, Dong Z, Maeda T, Maeda A, Sakai T, Tsuneoka H, Mieyal JJ, Palczewski K. Serum levels of lipid metabolites in age-related macular degeneration. FASEB J. 2015;29:4579–4588. doi: 10.1096/fj.15-275289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merle BM, Benlian P, Puche N, Bassols A, Delcourt C, Souied EH, A.M.D.T.S.G. Nutritional Circulating omega-3 Fatty acids and neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2014;55:2010–2019. doi: 10.1167/iovs.14-13916. [DOI] [PubMed] [Google Scholar]
- 73.Souied EH, Delcourt C, Querques G, Bassols A, Merle B, Zourdani A, Smith T, Benlian P, A.M.D.T.S.G. Nutritional Oral docosahexaenoic acid in the prevention of exudative age-related macular degeneration: the Nutritional AMD Treatment 2 study. Ophthalmology. 2013;120:1619–1631. doi: 10.1016/j.ophtha.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 74.Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309:2005–2015. doi: 10.1001/jama.2013.4997. [DOI] [PubMed] [Google Scholar]
- 75.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Salomon RG. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–42035. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 77.Gu J, Pauer GJ, Yue X, Narendra U, Sturgill GM, Bena J, Gu X, Peachey NS, Salomon RG, Hagstrom SA, Crabb JW, Clinical G, A.M.D.S.G. Proteomic Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol Cell Proteomics. 2009;8:1338–1349. doi: 10.1074/mcp.M800453-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Curcio CA, Presley JB, Malek G, Medeiros NE, Avery DV, Kruth HS. Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp Eye Res. 2005;81:731–741. doi: 10.1016/j.exer.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 79.Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010;51:451–467. doi: 10.1194/jlr.R002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu T, Tian J, Cutler RG, Telljohann RS, Bernlohr DA, Mattson MP, Handa JT. Knockdown of FABP5 mRNA decreases cellular cholesterol levels and results in decreased apoB100 secretion and triglyceride accumulation in ARPE-19 cells. Lab Invest. 2010;90:963–965. doi: 10.1038/labinvest.2010.87. [DOI] [PubMed] [Google Scholar]
- 81.Fujihara M, Cano M, Handa JT. Mice that produce ApoB100 lipoproteins in the RPE do not develop drusen yet are still a valuable experimental system. Invest Ophthalmol Vis Sci. 2014;55:7285–7295. doi: 10.1167/iovs.14-15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res. 2014;41:64–89. doi: 10.1016/j.preteyeres.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cherepanoff S, McMenamin P, Gillies MC, Kettle E, Sarks SH. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol. 2010;94:918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- 84.Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, Chowers I, Edwards PA, Baldan A, Parks JS, Ory DS, Apte RS. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu Y, Reynolds R, Fagerness J, Rosner B, Daly MJ, Seddon JM. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:4663–4670. doi: 10.1167/iovs.10-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oak AS, Messinger JD, Curcio CA. Subretinal drusenoid deposits: further characterization by lipid histochemistry. Retina. 2014;34:825–826. doi: 10.1097/IAE.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 87.Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased Membrane Complement Regulators in the Retinal Pigmented Epithelium Contributes to Age-Related Macular Degeneration. The Journal of pathology. 2012 doi: 10.1002/path.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Greferath U, Guymer RH, Vessey KA, Brassington K, Fletcher EL. Correlation of Histologic Features with In Vivo Imaging of Reticular Pseudodrusen. Ophthalmology. 2016;123:1320–1331. doi: 10.1016/j.ophtha.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 89.Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. The Journal of comparative neurology. 1990;292:497–523. doi: 10.1002/cne.902920402. [DOI] [PubMed] [Google Scholar]
- 90.Tserentsoodol N, Gordiyenko NV, Pascual I, Lee JW, Fliesler SJ, Rodriguez IR. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol Vis. 2006;12:1319–1333. [PubMed] [Google Scholar]
- 91.Tserentsoodol N, Sztein J, Campos M, Gordiyenko NV, Fariss RN, Lee JW, Fliesler SJ, Rodriguez IR. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol Vis. 2006;12:1306–1318. [PubMed] [Google Scholar]
- 92.Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- 93.Ershov AV, Bazan NG. Photoreceptor phagocytosis selectively activates PPARgamma expression in retinal pigment epithelial cells. J Neurosci Res. 2000;60:328–337. doi: 10.1002/(SICI)1097-4547(20000501)60:3<328::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 94.Miceli MV, Liles MR, Newsome DA. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994;214:242–249. doi: 10.1006/excr.1994.1254. [DOI] [PubMed] [Google Scholar]
- 95.Tate DJ, Jr, Miceli MV, Newsome DA. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:1271–1279. [PubMed] [Google Scholar]
- 96.Taylor HR, Munoz B, West S, Bressler NM, Bressler SB, Rosenthal FS. Visible light and risk of age-related macular degeneration. Trans Am Ophthalmol Soc. 1990;88:163–173. discussion 173–168. [PMC free article] [PubMed] [Google Scholar]
- 97.Taylor HR, West S, Munoz B, Rosenthal FS, Bressler SB, Bressler NM. The long-term effects of visible light on the eye. Arch Ophthalmol. 1992;110:99–104. doi: 10.1001/archopht.1992.01080130101035. [DOI] [PubMed] [Google Scholar]
- 98.Delcourt C, Cougnard-Gregoire A, Boniol M, Carriere I, Dore JF, Delyfer MN, Rougier MB, Le Goff M, Dartigues JF, Barberger-Gateau P, Korobelnik JF. Lifetime exposure to ambient ultraviolet radiation and the risk for cataract extraction and age-related macular degeneration: the Alienor Study. Invest Ophthalmol Vis Sci. 2014;55:7619–7627. doi: 10.1167/iovs.14-14471. [DOI] [PubMed] [Google Scholar]
- 99.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 100.Smith CJ, Hansch C. The relative toxicity of compounds in mainstream cigarette smoke condensate. Food Chem Toxicol. 2000;38:637–646. doi: 10.1016/s0278-6915(00)00051-x. [DOI] [PubMed] [Google Scholar]
- 101.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996;21:669–681. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- 103.Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr. 2000;71:530–536. doi: 10.1093/ajcn/71.2.530. [DOI] [PubMed] [Google Scholar]
- 104.O’Neill CA, Halliwell B, van der Vliet A, Davis PA, Packer L, Tritschler H, Strohman WJ, Rieland T, Cross CE, Reznick AZ. Aldehyde-induced protein modifications in human plasma: protection by glutathione and dihydrolipoic acid. J Lab Clin Med. 1994;124:359–370. [PubMed] [Google Scholar]
- 105.Cross CE, O’Neill CA, Reznick AZ, Hu ML, Marcocci L, Packer L, Frei B. Cigarette smoke oxidation of human plasma constituents. Ann N Y Acad Sci. 1993;686:72–89. doi: 10.1111/j.1749-6632.1993.tb39157.x. discussion 89–90. [DOI] [PubMed] [Google Scholar]
- 106.Moriarty-Craige SE, Adkison J, Lynn M, Gensler G, Bressler S, Jones DP, Sternberg P., Jr Antioxidant supplements prevent oxidation of cysteine/cystine redox in patients with age-related macular degeneration. Am J Ophthalmol. 2005;140:1020–1026. doi: 10.1016/j.ajo.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 107.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 108.Biswas SK, Rahman I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Molecular aspects of medicine. 2008 doi: 10.1016/j.mam.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 110.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 111.Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res. 2005;591:93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 112.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and cellular biology. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Molecular and cellular biology. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adams J, Kelso R, Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends in cell biology. 2000;10:17–24. doi: 10.1016/s0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- 115.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 116.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 117.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 118.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, Taguchi K, Yamamoto M, Kensler TW. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicological sciences: an official journal of the Society of Toxicology. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal. 2005;7:42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- 120.Will O, Mahler HC, Arrigo AP, Epe B. Influence of glutathione levels and heat-shock on the steady-state levels of oxidative DNA base modifications in mammalian cells. Carcinogenesis. 1999;20:333–337. doi: 10.1093/carcin/20.2.333. [DOI] [PubMed] [Google Scholar]
- 121.Walsh AC, Michaud SG, Malossi JA, Lawrence DA. Glutathione depletion in human T lymphocytes: analysis of activation-associated gene expression and the stress response. Toxicology and applied pharmacology. 1995;133:249–261. doi: 10.1006/taap.1995.1149. [DOI] [PubMed] [Google Scholar]
- 122.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology. 2008;39:673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 123.Sachdeva MM, Cano M, Handa JT. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp Eye Res. 2014;119:111–114. doi: 10.1016/j.exer.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. American journal of respiratory and critical care medicine. 2008;178:592–604. doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 125.Wang L, Kondo N, Cano M, Ebrahimi K, Yoshida T, Barnett BP, Biswal S, Handa JT. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic Biol Med. 2014 doi: 10.1016/j.freeradbiomed.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009;28:393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Farese RV, Jr, Veniant MM, Cham CM, Flynn LM, Pierotti V, Loring JF, Traber M, Ruland S, Stokowski RS, Huszar D, Young SG. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc Natl Acad Sci U S A. 1996;93:6393–6398. doi: 10.1073/pnas.93.13.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta. 2012;1818:2465–2475. doi: 10.1016/j.bbamem.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun M, Finnemann SC, Febbraio M, Shan L, Annangudi SP, Podrez EA, Hoppe G, Darrow R, Organisciak DT, Salomon RG, Silverstein RL, Hazen SL. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J Biol Chem. 2005 doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Handa JT, Tagami M, Ebrahimi K, Leibundgut G, Janiak A, Witztum JL, Tsimikas S. Lipoprotein(A) with An Intact Lysine Binding Site Protects the Retina From an Age-Related Macular Degeneration Phenotype in Mice (An American Ophthalmological Society Thesis) Trans Am Ophthalmol Soc. 2015;113:T51–T522. [PMC free article] [PubMed] [Google Scholar]
- 132.Schutt F, Ueberle B, Schnolzer M, Holz FG, Kopitz J. Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS letters. 2002;528:217–221. doi: 10.1016/s0014-5793(02)03312-4. [DOI] [PubMed] [Google Scholar]
- 133.Schutt F, Bergmann M, Holz FG, Kopitz J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3663–3668. doi: 10.1167/iovs.03-0172. [DOI] [PubMed] [Google Scholar]
- 134.Farboud B, Aotaki-Keen A, Miyata T, Hjelmeland LM, Handa JT. Development of a polyclonal antibody with broad epitope specificity for advanced glycation endproducts and localization of these epitopes in Bruch’s membrane of the aging eye. Mol Vis. 1999;5:11. [PubMed] [Google Scholar]
- 135.Suzuki M, Kamei M, Itabe H, Yoneda K, Bando H, Kume N, Tano Y. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol Vis. 2007;13:772–778. [PMC free article] [PubMed] [Google Scholar]
- 136.Moreira EF, Larrayoz IM, Lee JW, Rodriguez IR. 7-Ketocholesterol is present in lipid deposits in the primate retina: potential implication in the induction of VEGF and CNV formation. Invest Ophthalmol Vis Sci. 2009;50:523–532. doi: 10.1167/iovs.08-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Garcia-Cruset S, Carpenter KL, Guardiola F, Stein BK, Mitchinson MJ. Oxysterol profiles of normal human arteries, fatty streaks and advanced lesions. Free Radic Res. 2001;35:31–41. doi: 10.1080/10715760100300571. [DOI] [PubMed] [Google Scholar]
- 138.Ohtsuka M, Miyashita Y, Shirai K. Lipids deposited in human atheromatous lesions induce apoptosis of human vascular smooth muscle cells. J Atheroscler Thromb. 2006;13:256–262. doi: 10.5551/jat.13.256. [DOI] [PubMed] [Google Scholar]
- 139.van Reyk DM, Brown AJ, Hult’en LM, Dean RT, Jessup W. Oxysterols in biological systems: sources, metabolism and pathophysiological relevance. Redox Rep. 2006;11:255–262. doi: 10.1179/135100006X155003. [DOI] [PubMed] [Google Scholar]
- 140.Rodriguez IR, Alam S, Lee JW. Cytotoxicity of oxidized low-density lipoprotein in cultured RPE cells is dependent on the formation of 7-ketocholesterol. Invest Ophthalmol Vis Sci. 2004;45:2830–2837. doi: 10.1167/iovs.04-0075. [DOI] [PubMed] [Google Scholar]
- 141.Larrayoz IM, Huang JD, Lee JW, Pascual I, Rodriguez IR. 7-ketocholesterol-induced inflammation: involvement of multiple kinase signaling pathways via NFkappaB but independently of reactive oxygen species formation. Invest Ophthalmol Vis Sci. 2010;51:4942–4955. doi: 10.1167/iovs.09-4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Joffre C, Leclere L, Buteau B, Martine L, Cabaret S, Malvitte L, Acar N, Lizard G, Bron A, Creuzot-Garcher C, Bretillon L. Oxysterols induced inflammation and oxidation in primary porcine retinal pigment epithelial cells. Curr Eye Res. 2007;32:271–280. doi: 10.1080/02713680601187951. [DOI] [PubMed] [Google Scholar]
- 143.Huang JD, Amaral J, Lee JW, Rodriguez IR. 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS One. 2014;9:e100985. doi: 10.1371/journal.pone.0100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Javitt NB, Javitt JC. The retinal oxysterol pathway: a unifying hypothesis for the cause of age-related macular degeneration. Curr Opin Ophthalmol. 2009;20:151–157. doi: 10.1097/ICU.0b013e32832af468. [DOI] [PubMed] [Google Scholar]
- 145.Picard E, Houssier M, Bujold K, Sapieha P, Lubell W, Dorfman A, Racine J, Hardy P, Febbraio M, Lachapelle P, Ong H, Sennlaub F, Chemtob S. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging (Albany NY) 2010 doi: 10.18632/aging.100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Indaram M, Ma W, Zhao L, Fariss RN, Rodriguez IR, Wong WT. 7-Ketocholesterol increases retinal microglial migration, activation, and angiogenicity: a potential pathogenic mechanism underlying age-related macular degeneration. Sci Rep. 2015;5:9144. doi: 10.1038/srep09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eibinger G, Fauler G, Bernhart E, Frank S, Hammer A, Wintersperger A, Eder H, Heinemann A, Mischel PS, Malle E, Sattler W. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Exp Cell Res. 2013;319:1828–1838. doi: 10.1016/j.yexcr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, Paniccia A, Musumeci A, Chiricozzi E, Trincavelli ML, Daniele S, Martini C, Gustafsson JA, Doglioni C, Feo SG, Leiva A, Ciampa MG, Mauri L, Sensi C, Prinetti A, Eberini I, Mora JR, Bordignon C, Steffensen KR, Sonnino S, Sozzani S, Traversari C, Russo V. The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med. 2013;210:1711–1728. doi: 10.1084/jem.20130440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E, Saederup N, Charo IF, Rooijen NV, Nandrot E, Bourges JL, Behar-Cohen F, Sahel JA, Guillonneau X, Raoul W, Combadiere C. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. 2013;5:1775–1793. doi: 10.1002/emmm.201302692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rosenfeld ME, Palinski W, Yla-Herttuala S, Butler S, Witztum JL. Distribution of oxidation specific lipid-protein adducts and apolipoprotein B in atherosclerotic lesions of varying severity from WHHL rabbits. Arteriosclerosis. 1990;10:336–349. doi: 10.1161/01.atv.10.3.336. [DOI] [PubMed] [Google Scholar]
- 151.Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Backhed F, Miller YI, Horkko S, Corr M, Witztum JL, Binder CJ. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jozsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol. 2008;29:380–387. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 154.Amarilyo G, Verbovetski I, Atallah M, Grau A, Wiser G, Gil O, Ben-Neriah Y, Mevorach D. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-kappaB-dependent blockade. Eur J Immunol. 2010;40:699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 155.Toomey CB, Kelly U, Saban DR, Bowes Rickman C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc Natl Acad Sci U S A. 2015;112:E3040–3049. doi: 10.1073/pnas.1424391112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Elner VM, Elner SG, Bian ZM, Kindezelskii AL, Yoshida A, Petty HR. RPE CD14 immunohistochemical, genetic, and functional expression. Exp Eye Res. 2003;76:321–331. doi: 10.1016/s0014-4835(02)00310-x. [DOI] [PubMed] [Google Scholar]
- 157.Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A. 2006;103:17456–17461. doi: 10.1073/pnas.0606234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, Witztum JL. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Friedman P, Horkko S, Steinberg D, Witztum JL, Dennis EA. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem. 2002;277:7010–7020. doi: 10.1074/jbc.M108860200. [DOI] [PubMed] [Google Scholar]
- 160.Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Horkko S, Krauss RM, Chapman MJ, Witztum JL, Tsimikas S. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- 161.Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, Witztum JL. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dube JB, Boffa MB, Hegele RA, Koschinsky ML. Lipoprotein(a): more interesting than ever after 50 years. Current opinion in lipidology. 2012;23:133–140. doi: 10.1097/MOL.0b013e32835111d8. [DOI] [PubMed] [Google Scholar]
- 163.Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 164.Hoover-Plow J, Huang M. Lipoprotein(a) metabolism: potential sites for therapeutic targets. Metabolism. 2013;62:479–491. doi: 10.1016/j.metabol.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 166.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]