

Abstract
In the post-genomic era, increasingly sophisticated genetic tools are being developed with the long-term goal of understanding how the coordinated activity of genes gives rise to a complex organism. With the advent of the next generation sequencing associated with effective computational approaches, wide variety of plant species have been fully sequenced giving a wealth of data sequence information on structure and organization of plant genomes. Since thousands of gene sequences are already known, recently developed functional genomics approaches provide powerful tools to analyze plant gene functions through various gene manipulation technologies. Integration of different omics platforms along with gene annotation and computational analysis may elucidate a complete view in a system biology level. Extensive investigations on reverse genetics methodologies were deployed for assigning biological function to a specific gene or gene product. We provide here an updated overview of these high throughout strategies highlighting recent advances in the knowledge of functional genomics in plants.
Keywords: Functional genomics, Omics platforms, Reverse genetics, Large-scale sequencing
INTRODUCTION
In recent decades, technological advances in gene sequencing and computational tools have generated massive genomic resources, which made possible the annotation and the identification of thousand genes. Although nucleotide sequences of several plant genes were released, the function of their encoding proteins and their involvement in specific networks remain unclear. In the post-genomic era, the current challenge of plant research is focusing increasingly on gene function analysis. To raise this challenge, various reverse genetics approaches including gene silencing strategies, transgene-induced ectopic expression, gene targeting, T-DNA/transposons insertional mutagenesis and target-induced local mutations are available to investigate the biological function of such genes [1-5]. In many cases these approaches have been applied to plant model systems as well as to agronomically important species to gain powerful insights into gene function [6-9].
Conversely to the static aspect of the genomic information (DNA sequence), functional genomics based on the dynamic of biological system, aims to assign a function (s) to genes and determines how genes and their products (RNA, proteins, metabolites) interact together [10]. Gain and loss of function approaches represent the most adapted tools to investigate gene function. Since generation of transgenic plants is not always recommended due to laborious work, time-consuming procedures and variable transformation efficiency among species, genome studies has driven an increasing interest in rapid assay systems as alternatives to stable transgenics for establishing gene function [10-12]. Here we will overview the main reverse genetics systems and we will further discuss the challenges associated with characterizing gene function in the post-genome era.
1. Advances in sequencing platforms
1.1. Next Generation Sequencing
The most obvious technological advance in recent years is undoubtedly the development of the Next Generation Sequencing (NGS) that had significantly reduced the cost of whole genome and transcriptome sequencing [13]. NGS is producing a vast array of genomic information and has led to the identification and annotation of high number of plant genes. However, determining the function of these genes and the networks they are involved in remains a major challenge for modern plant research. Many sequencing platforms were developed [14-16] opening new avenues of research in plant improvement [17]. Following, most relevant advances of high throughput sequencing strategies for gene discovery are presented including (i) the development of sequencing platforms and (ii) their wide range of applications in combination with network integration. This review provides an updated view of how advanced omics-based high throughput methodologies can effectively enhance the discovery of new candidate genes for crop improvement.
Publication of the first complete genome sequence of Arabidopsis thaliana in 2000 [18] and first monocotyledonous plant Oryza sativa in 2005 [19] revolutionized research in plant genomics. These genomes used the traditional Sanger sequencing based on clone-by-clone strategy which involved sequencing of overlapped bacterial artificial chromosomes (BAC) clones selected from a physical map. However, Sanger sequencing technology remains costly and low throughput. The demand for rapid and cost-effective sequencing technologies led to the development of several alternative approaches in the use of genomic template libraries, number of reads, read length and genome coverage. Thus the whole genome shotgun (WGS) approach was introduced to produce the draft sequences of many plants like Sorghum bicolor [20] or Vitis vinifera [21]. Beginning in 2005, the traditional Sanger-based approach to DNA sequencing has experienced revolutionary changes and new sequencing platforms have emerged, having the high-throughput and cost-efficient capabilities, called ‘Next Generation Sequencing (NGS)’ [22, 23].
A variety of NGS technologies include the 454 FLX (Roche) [14], the Genome Analyzer/Hiseq (Illumina Solexa) [24] and the SOLiD (Life Technologies), as well as newer platforms such as Heliscope (Helicos) [25], PacBio RS (Pacific Biosciences) [26] for single molecular sequencing, and Ion Torrent (Life Technologies), based on a semi-conductor chip [27], were also available (Table 1).
Table 1.
Evolution of sequencing platforms.
| Platforms | Companies | Detection Methods | References | Advantages + / Disadvantages - | |
|---|---|---|---|---|---|
| Conventional sequencing | Sanger | Sanger biochemistry | Capillary electrophoresis/ clone by clone strategy | [34] | Low throughput-, In vivo cloning,- Long reads+/ High costs- |
| Next Generation Sequencing | GS FLX | Roche 454 Life Sciences, | Pyrosequencing detection of pyrophosphate |
[14] | No in vivo cloning+ Better sequence quality+, High throughput+, Low cost+, wide range of uses+, Very short reads-. Difficulty for genome sequencing assemblies- |
| George Church’s Laboratory at Harvard University | Polony sequencing | Multiplex polony sequencing | [35] | ||
| Genome analyzer | Solexa, Illumina Inc | Fluorophore labeled reversible terminator nucleotides | [36] | ||
| Intelligent Bio-Systems | Qiagen | sequencing by synthesis | [37] | ||
| SOLiD (Supported oligo ligation detection) | Applied Biosystems | Fluorophore labeled oligonucleotide probes |
[38] | ||
| HeliScope | Helicos Bioscience | Signe molecule detection system, sequencing by synthesis | [15] | ||
| Ion PGM System | Life Technology Personal genome machine |
Post-light Ion semiconductor sequencing | [39] | ||
| Ion PGM System | Life Technology Personal genome machine |
Non-optical DNA sequencing | [40] | ||
| Third Generation Sequencing | PacBio RS | Pacific biosciences | Phospholinked fluorophore labeled nucleotides | [16] | Sequencing in real time+ Long reads length+ novo genome assemblies+ |
| PacBio RS | Pacific biosciences | specific single-molecule sequencing of 5-hydroxy methylcytosine | [41] |
NGS is rapidly becoming an established tool in translational research as well. Several plant genomes have recently been sequenced using NGS technology (Table 2), as a result, the number of sequenced genomes increased exponentially. More than 100 plant genomes were sequenced [see for review 28, 29] (List of sequenced plant genomes; URL: https://genomevolution.org/wiki/index.php/Sequenced-plant-genomes). Newly sequenced plant systems can be interpreted by a homology search against other plant model systems. Based on the detection of orthologeous genes in other organisms, many genes can be characterized only with reference to their homology. The high amount of data generated displays the enormous challenge for bioinformatics in genome
Table 2.
Major agronomically important plant genomes sequenced over the last years. (https://genomevolution.org/wiki/index.php/ Sequenced_plant_genomes). (For entire list 2015, see [29]).
| Plant Species | Genome Size | Number of Predicted Genes | Sequencing and Assembly Status | References |
|---|---|---|---|---|
|
Zea mays (maize) |
2.30 Gb | 39 656 | Contig N50/Scaffold N50/ Sanger | [42] |
|
Sorghum bicolor (sorghum) |
0.73 Gb | 34 496 | Contig N50/Scaffold N50/ Sanger/ whole-genome shotgun | [20] |
|
Triticum aestivum (bread wheat) |
3.92 Gb | Roche 454/ Illumina whole-genome shotgun | [43, 44] | |
|
Hordeum vulgare (barley) |
5.1 Gb | whole-genome shotgun / RNA seq |
[45] | |
|
Glycine max (soybean) |
1.115 Gb | 46 430 | Contig N50/Scaffold N50/ Sanger/ whole-genome shotgun | [46] |
| Solanum tuberosum (potato) | 0.856 Gb | 39 031 | Sanger/Roche 454/ Illumina/ Solid | [47] |
| Solanum lycopersicum (tomato) | 0.9 Gb | 34 727 | Sanger/Roche 454/ Illumina/ Solid | [48] |
|
Capsicum annuum (pepper) |
3.48 Gb | 34 476 | whole-genome shotgun / Illumina-sequencing | [49] |
|
Beta vulgaris (sugarbeet) |
0.758 Gb | 27 421 | Roche 454, Illumina and Sanger sequencing platforms | [50] |
|
Cicer arietinum (chickpea) |
0.738 Gb | 28 269 | Illumina sequencing | [51] |
| Phaseolus vulgaris (common bean) | 0.587 Gb | 43 627 | whole-genome shotgun/ Sanger /Roche454/Illumina-sequencing/RNA seq | [52] |
|
Vitis vinifera (grapevine) |
0.5 Gb | 54 216 | whole-genome shotgun | [21] |
|
Citrus sinensis (sweet orange) |
0.319 Gb | 25 376 | Sanger/454sequencing technology | [53] |
|
Prunus persica (peach) |
0.265 Gb | 27 852 | Sanger whole-genome shotgun methods/sanger sequencing | [54] |
| Musa acuminata (banana) | 0.523 Gb | 36 542 | Contig N50/Scaffold N50 | [55] |
| Solanum melongena (egg plants) | 1.126 Gb | 85 446 | HisSeq2000/454 GS FLX | [56] |
| Brassica oleracea (cabbage) | 0.630 Gb | 45 758 | Illumina, Roche 454 and Sanger sequencing | [57] |
sequences analysis [30]. An increasing number of alignment and de novo assembler tools have been developed specifically for rapid alignment of large sets of reads [31, 32].
The identification of genes affecting agronomically relevant traits and supports molecular breeding (Table 2) from economically important plants would be an important application. However, NGS has some limit especially in their short read lengths (~60 and 400bp for Illumina ABI-SOLiD and 454/Roche respectively) comparing to the fist Sanger sequencing (~1Kb)(454. Roche 454 GSFLX.http:// www.454.com/; SOLiD.http://www3.appliedbiosystems.com/AB_Home/applicationstechnologies/ SOLiDSystemSequencing/index.htm.; Illumina.http://www.illumina.com/;Helicos. http://www.helicos bio.com/, leading to sequence errors and difficulty for sequence assemblies [33]. Therefore, the development of the third generation sequencing with long read length and fast sequencing enhanced the genome assemblies [29].
1.2. Next Generation-based RNA-seq Technologies
Global analyses have become possible with the development of high throughput genomic technologies which facilitated the identification of putative gene function. In this context, methods have been developed for quantitative data acquisition such as suppressive substractive hybridization libraries and microarrays [58] in case of grapevines. However, the recent advent of high-throughput-based sequencing technologies became revolutionizing the analysis of transcriptomes [59]. In fact, RNA sequencing (RNA-Seq) involves direct sequencing of complementary DNAs (cDNAs) and followed by the mapping of the sequencing reads to the genome. It allows for the precise quantification of exon expression, generating absolute rather than relative gene expression measurements, providing greater insight and accuracy than microarrays [60, 61] since it can detect and measure rare transcripts even those with abundances of 1 to 10 RNA molecules per cell [61].
NGS technologies enable the completion and the characterization of the entire genome of several agronomically important plant species including grapevine [21] where Illumina RNA-seq method have been successfully applied to analyze the total transcriptome during berry development [62]. This transcriptomic approach will open up large application perspectives in terms of plant improvement and development of plants with a better tolerance to environmental stresses. Therefore, understanding the function of genes remains a major challenge of the post-genomic research. It should be noted that a successful NGS project requires expertise in lab work as well as in bioinformatics in order to warrant high quality data and data interpretation.
2. Genome annotation, Omics and System biology
2.1. Network Integration and Gene Function
It is known that metabolism is highly dynamic and cannot be directly derived from the genotype. Consequently, this static genomic information needs to be complemented with genome-wide molecular data to reveal the dynamic genotype–phenotype relationship. Assigning a function requires a phenotype and this is addressed by the phenomics platform [63]. Also, NGS can provide information about the methylation state of the entire genome (methylome), thereby enabling us to determine the epigenetic control of different genes within the genome [64]. Data integration from multiple Omics platforms is of considerable interest to provide a complete view of the biological system as illustrated in (Fig. 1). Transcriptomics, proteomics and metabolomics data should be used to predict and understand the function of each gene and also to study the gene network as a whole system [65-67]. Hence, the databases of the metabolic reconstruction of plant species are continuously growing and will provide the basis for comparative plant genomics (http://www.plantcyc.org/). Despite major progress in the development of proteomics and metabolomics tools, these sub-platforms are still costly compared to NGS [64].
Fig. (1).
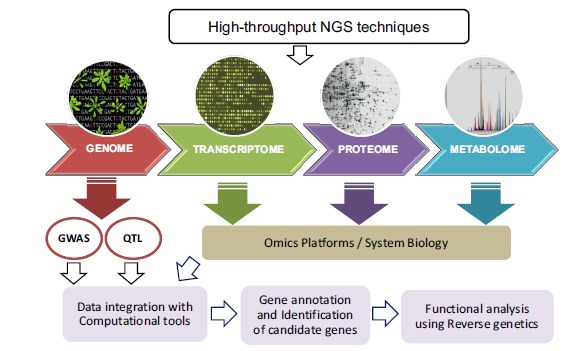
Data integration from multiple Omics platforms (genomics, transcriptomics, proteomics and metabolomics), computational annotation and functional characterization of candidate genes.
The integration of protein and metabolite profiling significantly improves the identification of plant biomarkers under different environmental conditions [67, 68]. For instance, such integrated analysis was applied to wheat and rice in response to anoxia [69] and to A.thaliana for characterization of starch and raffinose metabolisms under different temperatures [70]. There are also a number of reports on the elucidation of gene function by combining the transcriptomics/proteomics approach to study grapevine stress tolerance [67], nitrogen assimilation in maize [71], growth to dormancy transition in white spruce stems [72], phytohormone crosstalk [73] and flour quality in wheat [74]. In the same context, integrated transcriptome-metabolome analysis was lately applied in rice grown under high-temperature conditions [75], and changing metabolic systems in rice mutants [76] and transgenic barley plants [77] growing in field conditions. Furthermore, since quantitative traits are regulated by differential expression of candidate genes, the combined approach of QTL mapping and transcriptome profiling was used in tomato [78], rice [79] and wheat [80]. The exploitation of these Omics tools in plant biotechnology and QTL-based marker-assisted breeding approaches is an obvious development.
Network integration has given rise to bioinformatics challenges of modeling the complexity and behavior of biological systems as a ‘whole’ computational means [65, 67, 81]. Recently, the development of bioinformatics tools that will integrate data from multiple Omics platforms will give not only a complete view of the cellular systems and networks [64], but also can predict the system behavior under new unexplored perturbations [82]. In this context, a network for heat transcriptome of three model plants (Arabidopsis, Populus and soybean) has been characterized [83] and a conceptual model that links traditional network analysis to whole plant physiology was later proposed [84]. These approaches have opened up many application perspectives in terms of plant improvement in non-favorable environmental conditions.
2.2. Plant Genome Annotations and Tools
NGS technology is revolutionizing genomics and many plant genomes are already available. The increasing in demands for large-scale genome annotation has resulted in placing several genomics databases for annotation. Currently these genomes are being collected and annotated by various resources and databases including Phytozome [85], Plant Metabolic Networks (PMN) [86], Plants [87], Plaza [88], and MetNet Online [89]. The newly web-based system for integrated plant genome annotation MEGANTE (https://megante.dna.affrc.go.jp/.) recently published [90] could target 24 genome plant species from the Brassicaceae, Fabaceae, Musaceae, Poaceae, Salicaceae, Solanaceae, Rosaceae and Vitaceae families. Annotation of plant genes with their correct functions is of considerable interest and can be limited by the “missing annotations,” in which a function is known to exist, but the corresponding gene remains unidentified. PlantSEED (http://plantseed.theseed.org/) appeared as a new resource to support functional identification, annotation, comparative analysis, and modeling of plant genomes allowing downstream data analyses [91].
3. Forward versus Reverse genetics
Progress in plant genomics have been accelerating when the genome of Arabidopsis thaliana was published in late 2000 [18]. Since then many genome sequencing projects have been undertaken including economically and biologically important species for biological research [28, 29, 92]. After a genome is sequenced and annotated, functional characterization of genes that are important for development, cellular processes, or stress response becomes a major effort in the scientific community [93]. In this way, bioinformatics tries to identify candidate genes with the aim of better understanding the genetic basis of disease, unique adaptations to pathogen and severe conditions, desirable properties in agricultural species, or differences between populations. Classically, gene function is determined using two opposite but complementary approaches: forward and reverse genetics.
Forward genetic screens (mutant phenotype -► gene) aim to determine the genetic basis responsible for a phenotype through screening of mutant phenotypes. Typically, there is a wide use of chemical mutagenesis for forward-genetic screens in many organisms including bacteria, C.elegans, Drosophila and plants [94]. The basis is to treat organism with a mutagen, then screen offspring for particular phenotypes of interest and seek to identify genes involved in a biological pathway or process. Using large-scale genetic screens, several genes involved plant stress or other developmental or biochemical processes have been identified [95-98]. Analysis of segregating populations and subsequent locus mapping within the genome reveals genes that are associated with the observed biological process [99]. The goal is to find all of the genes involved in a trait; this approach is known as a “genetic screen”. With the advent of whole genome sequencing, researchers have now access to all of the gene sequence information within a given organism to investigate their function. Instead of going from phenotype to sequence as in forward genetics, reverse genetics works in the opposite direction – from known sequence towards assigned gene function [1, 10]. In reverse genetics (gene -► mutant phenotype), the target gene was subjected to gene manipulation, modification or disruption of its activity followed by analyzing phenotypic effects of up- and down- regulations (Fig. 2).
Fig. (2).
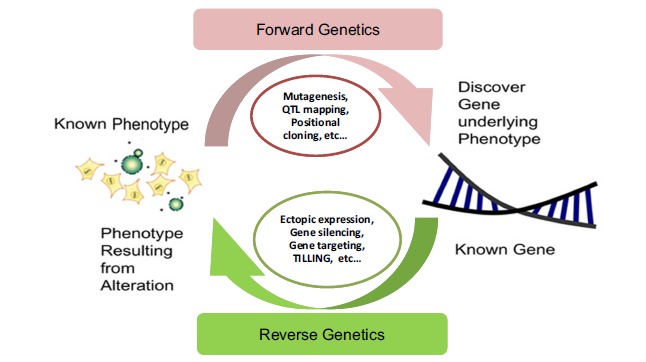
Forwards vs. reverse genetics tools for the identification and characterization of candidate genes.
4. Reverse genetics: Engineering loss and gain of function
Several plant genomes have been already sequenced [29] and in the few next years, we will be in front of many others fully-sequenced genomes (which are now in progress). The functions of a small number of genes were assigned, but those of many of them remain unclear. The current and future challenge is to investigate the biological function of identified and annotated gene sequences. Reverse genetics attempts to connect a given genetic sequence with specific effects on the organism and thus appears as an essential set of approaches for functional studies. A number of techniques have been developed, over the last 15 years, to enable researchers to identify plants with mutations in genes of known sequence. These reverse genetic approaches include silencing strategies (RNA interference: RNAi and virus-induced gene silencing: VIGS), screening of populations mutagenised by insertion (Insertional mutagenesis), deletion (Knock-out) and point mutation (TILLING: Target Induced Local Lesions in Genome), as well as gene targeting and ectopic expression using transgenics. Such tools are listed in (Table 3), each with its own strengths and weaknesses, try to explore gene function by analyzing the phenotypic effects of specific engineered gene sequences and seek to find what phenotypes arise as a result of particular genetic sequences.
Table 3.
Reverse genetics methods used for functional genomics in plants.
| Method | Advantages | Disadvantages | References |
|---|---|---|---|
| RNA interference | ▪ High-throughput vectors ▪ Heritable, ▪ Ability to silence multiple target genes at once ▪ Single copy of the target transgene is frequently sufficient to induce silencing |
▪ Variability in silencing efficiency ▪ Require efficient transformation ▪ Not developed for all species ▪ Effect on non-target genes |
[100-102] |
| Virus-induced gene silencing (VIGS) | ▪ No limitation imposed by transformation efficiency ▪ Rapid, easy to use ▪ Can be adapted for high-throughput screens ▪ Generate partial loss-of-function |
▪ Host range limitations ▪ Not established on all plant species ▪ Allows only transient expression ▪ Silencing level is variable |
[8, 100, 103, 104] |
| Ectopic expression | ▪ Can be adapted for high-throughput screens ▪ Suitable for transgene expression and gain-of-function analysis |
▪ Limited to transformable plant species ▪ Possibility of generating misleading neomorphs ▪ Expression level can be changed by exogenous regulatory sequences |
[1, 4, 11, 12] |
| Target Induced Local Lesions in Genome (TILLING) | ▪ Stable mutations ▪ Suitable for non-transformable species ▪ Allows identification of allelic series of mutants with a range of modified functions for a particular gene |
▪ Low/medium-throughput ▪ Desired mutation might never be found ▪ Need a large mutant population ▪ Relatively expensive |
[1-3, 105, 106] |
| Insertional mutagenesis | ▪ High-throughput ▪ Results in stable mutations ▪ Suitable for non-transformable species ▪ Can be adapted for both loss-of-function and gain-of-function studies |
▪ Desired mutation might never be found ▪ Variable effects depending on site of integration |
[1, 2, 107, 108] |
| Gene targeting and Genome editing | ▪ Highly specific ▪ Most revolutionary method that can be used for gene replacement ▪ Can be adapted for both loss-of-function and gain-of-function studies |
▪ Very low efficiency of homologous recombination | [1, 5, 9, 109] |
Classically, genes are transcribed into mRNA and translated into proteins determining the phenotypic traits. Each disruption in the genome (DNA sequence) can affect indirectly physical traits via the alteration of protein synthesis or activity. With the advent of recombinant DNA technology, gene engineering by gain- or loss-of-function approaches that mutate or knock out the gene function will switch on or off protein synthesis/activity resulting in new phenotypes. For example, changes in regulatory gene sequences or site-directed mutations targeting the open reading frame of gene-coding protein could identify amino residues for protein function. Reverse genetics experiments usually start with a cDNA, often corresponding to a transcript with an interesting pattern of expression, and then attempt to ascribe a biological function associated with a phenotypic trait to the target gene. The function is generally investigated by using the cDNA sequence to create a mutation of the wild-type allele in a transgenic plant (Ectopic expression using transgenics strategy) [4]. This mutation may be also inserted in a specific site or induced lesion in the genome (Insertional mutagenesis and TILLING). The phenotypic alteration will be further investigated. The target gene can be down-regulated by the expression of an antisense mRNA (Gene silencing) which interferes with the gene transcript causing RNA degradation or inhibiting translation. Similar approach was also used to deactivate transcripts of unknown genes and then check the phenotypic changes [100, 101]. Another way to elucidate the gene function is to exchange wild-type allele with an inactive mutant allele by homologous recombination (Gene targeting). These methods provide a way for linking genotype to phenotype to analyze the function of unknown genes for both fundamental and practically oriented studies.
4.1. RNA Silencing Approaches for Functional Gene Analysis in Plants
4.1.1. RNA-mediated Interference
RNA interference (RNAi) is induced by double-stranded RNAs (dsRNAs) leading to specific post-transcriptional gene silencing (PTGS) mechanism. This may be caused commonly by the expression of sequence homologous to an endogenous gene or by the expression of transgene that include a segment of gene sequence in an inverted repeat orientation that will generate dsRNAs. The amplification process of RNAi involves RNA-dependent RNA polymerases (RdRPs) that are required for the RNA silencing pathways [110, 111]. Besides its antiviral functions as part of the host defense response evolved to control plant virus replication, RNAi process fulfils fundamental regulatory roles through the activities of microRNAs and small interfering RNAs [112]. Previous studies on gene silencing have revealed two RNA-mediated epigenetic processes, RNA-directed RNA degradation and RNA-directed DNA methylation, providing new avenues for gene suppression technology in plants [113]. There is increasing evidence that components of the RNAi machinery are associated also with the formation of heterochromatin and that RNA-mediated chromatin modifications play an important role in epigenetic transcriptional gene silencing [114]. Since RNAi can specifically suppress the function of the targeted gene, the technique has been very useful in functional genomics studies [102]. RNAi have been widely described in plants and this is currently one of the hottest areas of biological research in the new era of functional genomics since interfering RNA-mediated silencing has revolutionized the understanding of gene function for basic and applied studies.
Transgene-induced RNAi has been effective at silencing one or more genes in a wide range of plants across the plant kingdom. The principle is simple: a fragment of a gene is introduced into a cell as dsRNA or as DNA that will give rise to dsRNA that would trigger silencing of endogenous genes in a homology dependent fashion. The dsRNA activates the DICER/RISC process so that the properties of the affected cell reflect a loss of function in the corresponding gene [115]. In this way, several functional genomics projects attempted to generate lines that are deficient for the activity of a subset of genes, and check their phenotypes to characterize the function of the knocked down gene [101]. A particularly useful property of RNA silencing is that it does not require neither full sequence of the target gene, nor complete sequence identity in the dsRNA and the target RNA. Since RNAi is highly sequence-specific, it is possible to knock down simultaneously multiple closely related genes by targeting their conserved sequences [102, 116]. For that reason, transgene-induced RNA silencing was implemented since years as a versatile reverse genetics tool and is well suited to the systematic analysis of gene function. RNAi technology has recently become a highly effective and powerful tool of functional genomics to explore plant genome [100] and has been applied to elucidate gene function in various crop species like tomato [11], potato [117], maize [116], wheat [118, 119] and rice [120].
Gene transfer-based techniques have also been used to induce RNAi in plants by delivering dsRNA or DNA
constructs that encode harpin-RNA into leaf epidermal cells of many crop plants [100]. Fast Agrobacterium-mediated transient gene expression/silencing methods have been applied for commercial plant species for successful transgene-induced PTGS via fruit agro-injection in tomato [11], agro-infiltration in potato [117], or through in planta agro-infiltration in grapevine [12]. Despite of its instability in plants, RNAi-mediated gene suppression approach open new avenues in the development of eco-friendly biotech approaches for crop improvement by knocking-out the specific genes for better stress tolerance and integrating novel traits in various plant species including insect/pest/pathogen resistance [121].
4.1.2. Virus-induced Gene Silencing Strategy
Another alternative to knock-down endogenous plant genes may be achieved via viral vectors through RNA-mediated post-transcriptional gene silencing mechanism. This powerful reverse genetics approach, known as Virus-induced gene silencing (VIGS), is a virus vector technology that exploits RNA-mediated anti-viral defense mechanism [122-124]. Production of dsRNA activates the RNA silencing pathway, resulting in down-regulation of the host gene transcript. VIGS offers an easy way to test the function of several genes in a short time, as it only requires a fragment (typically 300-800 bp) of the target plant gene inserted into a suitable viral vector to form a recombinant virus. Upon infection of a plant host usually via Agrobacterium tumefaciens, this recombinant viral vector induces PTGS targeting both the virus RNA and homologous endogenous plant RNA sequences for degradation [125-128]. Besides the lack of suppressors of gene silencing needed in VIGS strategy, successful RNA virus-based VIGS requires simultaneous infiltration of both viral clones including genomic RNA1 and RNA2. Nowadays, there are infectious clones of several plant viruses that have been used as VIGS vectors, most of which have RNA genomes [100]. Other source of VIGS vectors used recently for silencing is the small subviral RNA satellite along with helper virus-dependent replication [129]. While several plant viruses have been so far developed into VIGS vectors, the Tobacco Rattle Virus (TRV) established by David Baulcombe’s group [130] provides the most robust results in terms of efficiency, ease of application, and absence of disease symptoms [128]. VIGS has been broadly regarded as the tool of choice for transient induction of silencing that occurs for only few weeks with further decrease resulting in plant recovery [131]. However, recent evidences suggest that using some vectors under specific conditions, VIGS can persist for years or even transmitted to progeny [132, 133] behaving like stable transgenic plants. Besides many advantages of VIGS over other approaches including methodological simplicity, switching off genes specifically, rapid monitoring, robustness and speedy results, the system allows the study of genes whose functions are essential to plant viability [103, 127]. VIGS has been widely used in model plants as tool to assess function of candidate genes and to discover new genes required for diverse pathways. VIGS technology has been successfully used to validate and functionally analyze the contribution of candidate genes in many plant species [8, 134-137]. For instance, several stress-responsive-genes were studied using VIGS system in various model plants including tobacco [133], chili pepper [138], soybean [139], rose [140] and wheat [136, 141]. Such strategy can be also very helpful to assess gene function, especially in species recalcitrant to transformation making the VIGS an attractive alternative instrument for high-throughput functional genomics [103, 104].
4.2. Transgene-induced Ectopic Expression/Silencing
The development of powerful “omics” technologies has enabled researchers to identify many genes of interest for which comprehensive functional analyses are highly desirable. Large number of plant genes continues to be identified every day with no defined functions. Although powerful in silico techniques can predict the function of many gene-encoding proteins, evidence of gene function have to be constantly verified in vivo using transgenic approach allowing analysis of phenotypic changes occurred by transgene integration and expression. The production of transgenic lines which ectopically express recombinant genes or those in which endogenous genes are knocked down remains a major bottleneck. Gene transfer technology appears to be an essential tool implemented for functional studies through gain- or loss-of-function approaches. Overexpression of a wild-type gene product, however, can cause mutant phenotypes, providing geneticists with an alternative yet powerful tool to identify pathway components that might remain undetected using loss-of-function screens [142]. Gain-of-function is achieved by increasing gene expression level through the activation of endogenous genes by transcriptional enhancers or through ectopic expression of individual transgene via transformation [4]. Heterologous expression has also been exploited to study gene functions across species barriers and transcript abundance is increased by cloning the full open reading frame (ORF) downstream of a strong constitutive promoter such as 35S promoter of cauliflower mosaic virus (CaMV35S), or using chemical- or stress-inducible promoters and transcription factors to control ectopic expression level [10, 143, 144].
Several approaches were used for gene transfer to plants, depending on the fact of how the gene construct is expressed in a stable or in a transient manner. Over the last decades, hundreds of model and crop plant species have been genetically transformed essentially via Agrobacterium- or viral-mediated gene transfer technologies [145] as well as by direct gene delivery using bombardment of DNA-coated particles [146-148] or protoplast electroporation [149]. Methods for effective DNA transfer into regenerable competent cells were extensively developed and optimized for stable transformation and generation of transgenic plants. However, transgenic approaches for reverse genetic studies are not yet practical in several plants in which transformation methodology is so far not efficient or not available. A heterologous expression approach provides a solution for the high-throughput characterization of gene functions in these plant species [4].
To overcome these in vitro regeneration-dependent procedures which are laborious and time-consuming, rapid and versatile transient assays were widely investigated as an alternative to the transgenic approach. Agroinoculation became a reliable method for plant functional gene analysis after being firstly developed as a simple procedure for plant systemic infection with viruses by delivery of viral genomes into plant tissues using Agrobacterium binary vectors [150, 151]. A variety of agroinoculation methods were developed during the last decade in order to examine the effect of transient gene expression, with applications ranging from monitoring of plant-pathogen interactions [152], subcellular localization [153], ectopic up-/down-regulation [12], in vivo analysis of plant promoters and transcription factors [154, 155] to the production of recombinant proteins [156, 157]. Efforts of several laboratories have resulted in the development of various methods for Agrobacterium tumefaciens–mediated gene transfer. Among these, vacuum infiltration [158, 159] and floral dipping [160, 161] are efficient methods widely used for generating transgenic Arabidopsis plants. Similarly, Agrodrench was established as an effective agroinoculation method where soil adjacent to the plant root is drenched with an Agrobacterium suspension carrying virus-derived VIGS vectors [131]. Agrobacterium infiltration into leaf tissues remains a method of choice for transiently expressing foreign genes in many plant species. Localized introduction and expression of T-DNA constructs have been largely implemented over the last years, even in recalcitrant plants, for assigning gene function directly in other target tissues and organs by fruit agroinjection [11] or in situ agroinoculation of floral tissues [162]. Currently, in planta agroinfiltration-based procedures were substantially improved for either inducing transgene expression or gene silencing in rice [163] and grapevine [12]. Infiltrating leaves (using a needle-less syringe or vacuum) with Agrobacterium culture, in which the T-DNA plasmid contains a transgene that encodes an endogenous plant gene sequence, can trigger RNAi against the target endogenous gene and generates dsRNA. For instance, agroinfiltration of transgenic tobacco and grapevine plants expressing GFP resulted in GFP overexpression during the first days after infiltration, while this expression subsides to undetectable levels few days post-infiltration, concomitant with a reduction of endogenous GFP expression and induction of a silencing mechanism, firstly localized and subsequently spreads throughout the plant [12, 100].
In some cases where agroinfiltration is not appropriate, direct DNA uptake is useful for both stable transformation and transient gene expression. For instance, biolistic - as an alternative physical system that avoids the need for a pathogen-host interaction - could be easily performed for transient assays and have long been used for overexpression of foreign genes [146-148] as well as for silencing studies [162]. This particle delivery system allows use of simple multicopy vectors lacking T-DNA or virus sequences and even a minimal gene expression cassette carrying only the gene of interest with its regulatory sequences needed for its expression in plant cells, without the plasmid backbone [146, 161].
The advantages of gain-of-function approaches for the functional gene characterization include the abilities to characterize the function of genes from non-model plants using a heterologous expression system, and to identify genes that confer stress tolerance to plants as a result of ectopic transgene expression [4]. Overexpression of heterologous genes is widely used for the introduction of novel traits into transgenic plants. Overexpression can also be used in combination with down-regulation and controlled expression (e.g. induced, developmentally regulated, or tissue specific) studies as a tool for basic plant research and for functional analysis of native genes in various model plants [6]. One of the major progress in transgenic research relied to the technical advance in cloning technologies making possible the elaboration of genetic constructs. While traditional binary vectors have proven helpful in gene transfer assays, these plasmids rarely permit the cloning and transfer of more than a single target gene. The Gateway cloning system based on site-specific recombination has recently provided a significant improvement; in particular, the system enables assembly of multiple DNA fragments in a predefined order, orientation and frame register in a flexible manner and allows delivery of multiple transgenes to plant cells [6, 164]. Many of the principles described here arise from the numerous overexpression studies that have been reported in model species, where technical advantages facilitate overexpression screens and subsequent analysis, but examples are provided from other organisms that support or expand the concepts and demonstrate their validity in other plant systems.
4.3. Insertional Mutagenesis and TILLING
4.3.1. Insertional Mutagenesis
Insertional mutagenesis is a mutation caused by insertion of new genetic material (foreign DNA) into the target gene. Both T-DNA and transposon insertional mutants are being produced recently as an extremely valuable research tools for model plant systems to study gene function. Insertional mutagenesis has been developed within the last decade to generate specific mutations in organisms in which homologous recombination is a low frequency event [165]. The most effective method for insertional mutagenesis is targeted gene disruption. Large populations of T-DNA-tagged lines or mutants with transposon activation having an insertion at unique site in the genome have been generated in algae [165] and model plants [166, 167]. The approach relies on the generation of thousands of transformants followed by PCR-based screenings that allow for identification of lines harboring the introduced mutation within specific genes of interest [107]. Defining the insertion site for each transformant has allowed for the establishment of sequence-indexed libraries of mutant plants. T-DNA or transposon insertion has been exploited to create disruptions in target genes of interest, introduce new genes, or activate endogenous genes in the plant genome [2, 108, 167]. Transposons have several advantages over T-DNA including the ability to produce multiple independent insertion lines from individual starter lines, as well as producing revertants by remobilization [168].
4.3.2. Tilling
Targeting-induced local lesions in genomes (TILLING) is a strategy for the discovery and mapping of induced point mutations that was raised a decade ago as an alternative to insertional mutagenesis. This method was originally developed by the Henikoff laboratory to screen libraries of Ethyl methanesulfonate (EMS)-treated Arabidopsis for desired mutant alleles [169] and was subsequently adapted to others plant species. TILLING, which combines traditional mutagenesis with genome-wide high-throughput screening for point mutations in desired genes, is moving beyond functional genomics into crop improvement [94, 170, 171]. This technique was developed as a high-throughput and low cost reverse genetic method that has been successfully applied to many plant species making the TILLING process broadly applicable [172, 173]. Besides model plants, the feasibility of TILLING has already been demonstrated for a large number of agronomically important crops, including rice, barley, wheat, maize, sorghum, soybean, rapeseed and tomato plants. Furthermore, this method does not require transformation procedures and thus it is suitable for recalcitrant species and recommended as non-GMO technology to avoid controversies [3]. Recently, large-scale TILLING platforms for identifying mutants in plants offer great potential as a tool for functional genomics [3, 105, 106, 171].
4.4. Gene Targeting for Genome Engineering in Plants
4.4.1. Homologous Recombination-based Approaches for Genome Modification
Homologous recombination (HR) is one of the most universal events during the course of life evolution since it plays a central role in creating genetic diversity. It is most widely used by cells to accurately repair harmful breaks that occur on both strands of DNA known as double-strand breaks and thus safeguarding genomic integrity. HR was applied to several species to induce directed-site mutations and gene-disruption activity. HR-dependent targeted gene replacement (also called gene targeting) has been used extensively in yeast genome to elucidate the biological function of all predicted ORF. For plant cells, this method was firstly established on tobacco protoplasts [174] and then in moss Physcomitrella which exhibits high frequencies of gene targeting through HR [175, 176]. However, HR-based gene targeting has been for long term inappropriate and far from common practice for higher plants [177-179]. Despite of the low frequency of HR-based approaches that remains the major barrier, successful gene targeting in tobacco [174], Arabidopsis [180], and rice [181] using HR were reported.
In general, a targeting DNA construct contains typically part of the gene to be targeted along with a reporter gene or/and a selectable marker. The transformed organism carrying a loss-of-function mutation can then be analyzed for its phenotype. Gene targeting is thus a powerful tool for directed ‘knockout’ of genes [177]. Plant DNA-repair machinery predominantly uses non-homologous end-joining (NHEJ), making the homologous recombination (HR)-based methods, which have proved fruitful for gene targeting in non-plant systems, unsuitable for use in plants [109, 182].
Precise genome modification is an attractive method for understanding gene function. Gene targeting is known as one of the best method currently available to induce specific change of endogenous gene via HR [183]. This strategy can be used to delete (knock-out) or substitute a gene (knock-in), remove exons, and introduce point mutations. Gene targeting can be applied to the whole organism or may be limited to a particular developmental stage and definite plant tissue. It can be used also for any gene, regardless of transcriptional activity or gene size.
4.4.2. Artificial Endonucleases-mediated Genome Editing
Targeted genome engineering (also known as genome editing) has emerged as an option to classical plant breeding and transgenic methods for crop improvement [184] and it is currently the most attractive topic in plant molecular biology and genomics research. A key step in genome editing is the generation of a double-stranded DNA break that is specific to the target gene. This is achieved by engineered endonucleases, which enable site-directed mutagenesis via a NHEJ repair pathway and/or gene targeting via HR to occur efficiently at specific sites in the genome [185, 186].
Some technical advances described targeted mutagenesis and gene targeting by either NHEJ machinery using site-specific induction of double-strand breaks (DSBs), or by activation of a HR pathway through overexpression of a yeast DNA recombination gene in transgenic plants [109]. Although gene targeting efficiency has been restricted to some plant species, its frequency was significantly enhanced by the recent use of programmable nucleases [5, 187, 188]. Since that, many plant genes have been knocked out by this method. Over the past 15 years, tremendous efforts have been made and a variety of technologies have been developed to target mutations to a specific location in the genome using artificial endonucleases.
One of the earliest nuclease technologies involved mega-nucleases that were very difficult to engineer and requiring a long and costly process [189, 190]. The DNA-binding domains of Zinc finger transcription factors (ZFNs) were the first to be used as genome editing tools [191] and have proven easier to manipulate and have been used in tobacco [187], Arabidopsis [188] and maize [192]. More recently, transcription activator-like effector nucleases (TALENs) [193], as well as the clustered regularly interspaced short palindromic repeats/Cas (CRISPR/Cas9) system using an RNA-guided bacterial immune system complex were reported [194]. First studies confirmed that CRISPR provides protection against invading viruses when combined with Cas9 genes involving an RNA-mediated DNA targeting [195]. Thereafter, CRISPR/Cas9 system is revolutionizing the field of genomic editing and have proved very useful and functional [9] providing scientists with a powerful tool able to change any gene, in any cell in a highly targeted manner and without introducing foreign DNA.
So far, several reports demonstrated the immense versatility of the technology in the field of plant biology by using a range of transformation platforms (PEG-protoplast transfection [196, 197], agroinfiltration [198, 199], virus transfection [200] and generation of stable transgenic plants [185, 201, 202]. This CRISPR/Cas9 allows for specific genome disruption and replacement in a flexible, robust and simple system resulting in high specificity and low cell toxicity, by targeting both endogenous genes and transgenes and by exploiting NHEJ and HR to generate small deletions, targeted insertions and multiplex genome modifications [9, 185]. This method has just been applied to a number of species including Arabidopsis [184, 201], tobacco [196, 198], tomato [201], sweet orange [199], wheat [197] and rice [203, 204]. For all instances, genome editing was shown to work in both model and crop plants, as well as in a variety of other organisms using engineered nucleases as powerful tools to target specific DNA sequences to edit genes precisely in the plant genomes.
5. Challenges and future outlooks
With the advent of high-throughput sequencing technologies, functional genomics have become a key platform in determining the biological function of individual gene. Today, many approaches are available for reverse genetics studies in plants that offer powerful tools to identify target genes and explore their functions in different manner. Functional analysis of the plant genome relies through loss or gain of function approaches and could be carried out using stable transgenics or alternative tools of transient expression assays depending on the target plant species and the gene studied.
It is not clear however how the availability in the near future of many hundreds of fully sequenced plant genomes would be integrated; and new levels of data organization may need to be developed. The complexity of plant genome and gene expression, regulation, function and interaction in a single cell as well as within a whole organism should be deeply investigated in the future via multiple omics platforms. In addition, new platforms for analyzing others biomolecules require to be implemented including hormones (hormonome), signaling and transduction pathways (signalome), as well as methodological advance in cell imaging, cell micro-manipulation and other tools for studying the dynamic at cytoplasm, organelle and membrane levels. It seems also that the largest part of research studies focus on the genomic information and subsequent putative function of target genes, while it is already known that cytoplasmic environment is the crucial factor that coordinate gene expression and cell differentiation during developmental stages or under adverse conditions. The direction of how the plant genome will react to any stimuli requires specific environment with proper cytoplasmic components that could be investigated in the near future. Integration of such tools will provide new insights for functional genomics/system biology in plant science fundamental and applied research towards food safety and crop improvement.
ACKNOWLEDGEMENTS
This work was supported in part by the International Centre for Genetic Engineering and Biotechnology (ICGEB -CRP/TUN06-01), the German Academic Exchange Service (DAAD) and the COST 858 “Abiotic stress in grapevine” to SD. Funding was also provided by the Tunisian Government (Ministry of Higher Education and Scientific Research) and a scientific mission grant from COST 858 “Grapevine Gene Transfer Technologies” to AB.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Alonso J.M., Ecker J.R. Moving forward in reverse: genetic technologies to enable genome-wide phenomic screens in Arabidopsis. Nat. Rev. Genet. 2006;7(7):524–536. doi: 10.1038/nrg1893. [DOI] [PubMed] [Google Scholar]
- 2.Gilchrist E., Haughn G. Reverse genetics techniques: engineering loss and gain of gene function in plants. Brief. Funct. Genomics. 2010;9(2):103–110. doi: 10.1093/bfgp/elp059. [DOI] [PubMed] [Google Scholar]
- 3.Kurowska M., Daszkowska-Golec A., Gruszka D., Marzec M., Szurman M., Szarejko I., Maluszynski M. TILLING - a shortcut in functional genomics. J. Appl. Genet. 2011;52:371–390. doi: 10.1007/s13353-011-0061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdeeva I., Abdeev R., Bruskin S., Piruzian E. Transgenic plants as a tool for plant functional genomics. In: Çiftçi Y.O., editor. Transgenic plants - Advances and Limitations. Croatia: InTech; 2012. [Google Scholar]
- 5.Kim H., Kim J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 6.Dafny-Yelin M., Tzfira T. Delivery of multiple transgenes to plant cells. Plant Physiol. 2007;145(4):1118–1128. doi: 10.1104/pp.107.106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shelden M.C., Roessner U. Advances in functional genomics for investigating salinity stress tolerance mechanisms in cereals. Front. Plant Sci. 2013;4(123) doi: 10.3389/fpls.2013.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramegowda V., Mysore K.S., Senthil-Kumar M. Virus-induced gene silencing is a versatile tool for unraveling the functional relevance of multiple abiotic-stress-responsive genes in crop plants. Front. Plant Sci. 2014;5(323) doi: 10.3389/fpls.2014.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belhaj K., Chaparro-Garcia A., Kamoun S., Patron N.J., Nekrasov V. Editing plant genomes with CRISPR/Cas9. Curr. Opin. Biotechnol. 2015;32:76–84. doi: 10.1016/j.copbio.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Vidal J.R., Gomez C., Cutanda M.C., Shrestha B.R., Bouquet A., Thomas M.R., Torregrosa L. Use of gene transfer technology for functional studies in grapevine. Aust. J. Grape Wine Res. 2010;16:138–151. [Google Scholar]
- 11.Orzaez D., Mirabel S., Wieland W.H., Granell A. Agroinjection of tomato fruits. A tool for rapid functional analysis of transgenes directly in fruit. Plant Physiol. 2006;140:3–11. doi: 10.1104/pp.105.068221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Amar A., Cobanov P., Buchholz G., Mliki A., Reustle G. In planta agroinfiltration system for transient gene expression in grapevine. Acta Physiol. Plant. 2013;35(11):3147–3156. [Google Scholar]
- 13.Edwards D., Batley J., Snowdon R.J. Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 2013;126:1–11. doi: 10.1007/s00122-012-1964-x. [DOI] [PubMed] [Google Scholar]
- 14.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Dewell S.B., Du L., Fierro J.M., Gomes X.V., Godwin B.C., He W., Helgesen S., Ho C.H., Irzyk G.P., Jando S.C., Alenquer M.L., Jarvie T.P., Jirage K.B., Kim J.B., Knight J.R., Lanza J.R., Leamon J.H., Lefkowitz S.M., Lei M., Li J., Lohman K.L., Lu H., Makhijani V.B., McDade K.E., McKenna M.P., Myers E.W., Nickerson E., Nobile J.R., Plant R., Puc B.P., Ronan M.T., Roth G.T., Sarkis G.J., Simons J.F., Simpson J.W., Srinivasan M., Tartaro K.R., Tomasz A., Vogt K.A., Volkmer G.A., Wang S.H., Wang Y., Weiner M.P., Yu P., Begley R.F., Rothberg J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pushkarev D., Neff N.F., Quake S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009;27(9):847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schadt E.E., Turner S., Kasarskis A. A window into third-generation sequencing. Hum. Mol. Genet. 2010;19(R2):R227–R240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 17.Pérez-de-Castro A.M., Vilanova S., Cañizares J., Pascual L., Blanca J.M., Díez M.J., Prohens J., Picó B. Application of genomic tools in plant breeding. Curr. Genomics. 2012;13:179–195. doi: 10.2174/138920212800543084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 19.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 20.Paterson A.H., Bowers J.E., Bruggmann R., Dubchak I., Grimwood J., Gundlach H., Haberer G., Hellsten U., Mitros T., Poliakov A., Schmutz J., Spannagl M., Tang H., Wang X., Wicker T., Bharti A.K., Chapman J., Feltus F.A., Gowik U., Grigoriev I.V., Lyons E., Maher C.A., Martis M., Narechania A., Otillar R.P., Penning B.W., Salamov A.A., Wang Y., Zhang L., Carpita N.C., Freeling M., Gingle A.R., Hash C.T., Keller B., Klein P., Kresovich S., McCann M.C., Ming R., Peterson D.G. Mehboob-ur-Rahman; Ware, D.; Westhoff, P.; Mayer, K.F.; Messing, J.; Rokhsar, D.S. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 21.Jaillon O., Aury J.M., Noel B., Policriti A., Clepet C., Casagrande A., Choisne N., Aubourg S., Vitulo N., Jubin C., Vezzi A., Legeai F., Hugueney P., Dasilva C., Horner D., Mica E., Jublot D., Poulain J., Bruyère C., Billault A., Segurens B., Gouyvenoux M., Ugarte E., Cattonaro F., Anthouard V., Vico V., Del Fabbro C., Alaux M., Di Gaspero G., Dumas V., Felice N., Paillard S., Juman I., Moroldo M., Scalabrin S., Canaguier A., Le Clainche I., Malacrida G., Durand E., Pesole G., Laucou V., Chatelet P., Merdinoglu D., Delledonne M., Pezzotti M., Lecharny A., Scarpelli C., Artiguenave F., Pè M.E., Valle G., Morgante M., Caboche M., Adam-Blondon A.F., Weissenbach J., Quétier F., Wincker P., French-Italian Public Consortium for Grapevine Genome Characterization The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 22.Mardis E.R. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 23.Mardis E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 2013;6:287–303. doi: 10.1146/annurev-anchem-062012-092628. [DOI] [PubMed] [Google Scholar]
- 24.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 25.Milos P. Helicos BioSciences. Pharmacogenomics. 2008;9:477–480. doi: 10.2217/14622416.9.4.477. [DOI] [PubMed] [Google Scholar]
- 26.Korlach J., Bjornson K.P., Chaudhuri B.P., Cicero R.L., Flusberg B.A., Gray J.J., Holden D., Saxena R., Wegener J., Turner S.W. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 2010;472:431–455. doi: 10.1016/S0076-6879(10)72001-2. [DOI] [PubMed] [Google Scholar]
- 27.Rothberg J.M., Hinz W., Rearick T.M., Schultz J., Mileski W., Davey M., Leamon J.H., Johnson K., Milgrew M.J., Edwards M., Hoon J., Simons J.F., Marran D., Myers J.W., Davidson J.F., Branting A., Nobile J.R., Puc B.P., Light D., Clark T.A., Huber M., Branciforte J.T., Stoner I.B., Cawley S.E., Lyons M., Fu Y., Homer N., Sedova M., Miao X., Reed B., Sabina J., Feierstein E., Schorn M., Alanjary M., Dimalanta E., Dressman D., Kasinskas R., Sokolsky T., Fidanza J., Namsaraev E., McKernan K.J., Williams A., Roth G.T., Bustillo J. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 28.Michael T.P., Jackson S. The first 50 plant genomes. Plant Genome. 2013:6. [Google Scholar]
- 29.Michael T.P., VanBuren R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 2015;24:71–81. doi: 10.1016/j.pbi.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Cantacessi C., Jex A.R., Hall R.S., Young N.D., Campbell B.E., Joachim A., Nolan M.J., Abubucker S., Sternberg P.W., Ranganathan S., Mitreva M., Gasser R.B. A practical, bioinformatic workflow system for large datasets generated by next-generation sequencing. Nucleic Acids Res. 2010;38(17):e171. doi: 10.1093/nar/gkq667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith A.D., Xuan Z., Zhang M.Q. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics. 2008;9(128) doi: 10.1186/1471-2105-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler J., MacCallum I., Kleber M., Shlyakhter I.A., Belmonte M.K., Lander E.S., Nusbaum C., Jaffe D.B. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grada A., Weinbrecht K. Next-Generation Sequencing: Methodology and application. J. Invest. Dermatol. 2013;133(e11) doi: 10.1038/jid.2013.248. [DOI] [PubMed] [Google Scholar]
- 34.Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shendure J., Porreca G.J., Reppas N.B., Lin X., McCutcheon J.P., Rosenbaum A.M., Wang M.D., Zhang K., Mitra R.D., Church G.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309(5741):1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 36.Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16(6):545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Ju J., Kim D.H., Bi L., Meng Q., Bai X., Li Z., Li X., Marma M.S., Shi S., Wu J., Edwards J.R., Romu A., Turro N.J. Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc. Natl. Acad. Sci. USA. 2006;103(52):19635–19640. doi: 10.1073/pnas.0609513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valouev A., Ichikawa J., Tonthat T., Stuart J., Ranade S., Peckham H., Zeng K., Malek J.A., Costa G., McKernan K., Sidow A., Fire A., Johnson S.M. A high resolution, nucleosome position map of C. elegans reveals a lack of universal sequence dictated positioning. Genome Res. 2008;18(7):1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metzker M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 40.Rothberg J.M., Hinz W., Rearick T.M., Schultz J., Mileski W., Davey M., Leamon J.H., Johnson K., Milgrew M.J., Edwards M., Hoon J., Simons J.F., Marran D., Myers J.W., Davidson J.F., Branting A., Nobile J.R., Puc B.P., Light D., Clark T.A., Huber M., Branciforte J.T., Stoner I.B., Cawley S.E., Lyons M., Fu Y., Homer N., Sedova M., Miao X., Reed B., Sabina J., Feierstein E., Schorn M., Alanjary M., Dimalanta E., Dressman D., Kasinskas R., Sokolsky T., Fidanza J.A., Namsaraev E., McKernan K.J., Williams A., Roth G.T., Bustillo J. An integrated semi-conductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 41.Song C.X., Clark T.A., Lu X.Y., Kislyuk A., Dai Q., Turner S.W., He C., Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnable P.S., Ware D., Fulton R.S., Stein J.C., Wei F., Pasternak S., Liang C., Zhang J., Fulton L., Graves T.A., Minx P., Reily A.D., Courtney L., Kruchowski S.S., Tomlinson C., Strong C., Delehaunty K., Fronick C., Courtney B., Rock S.M., Belter E., Du F., Kim K., Abbott R.M., Cotton M., Levy A., Marchetto P., Ochoa K., Jackson S.M., Gillam B., Chen W., Yan L., Higginbotham J., Cardenas M., Waligorski J., Applebaum E., Phelps L., Falcone J., Kanchi K., Thane T., Scimone A., Thane N., Henke J., Wang T., Ruppert J., Shah N., Rotter K., Hodges J., Ingenthron E., Cordes M., Kohlberg S., Sgro J., Delgado B., Mead K., Chinwalla A., Leonard S., Crouse K., Collura K., Kudrna D., Currie J., He R., Angelova A., Rajasekar S., Mueller T., Lomeli R., Scara G., Ko A., Delaney K., Wissotski M., Lopez G., Campos D., Braidotti M., Ashley E., Golser W., Kim H., Lee S., Lin J., Dujmic Z., Kim W., Talag J., Zuccolo A., Fan C., Sebastian A., Kramer M., Spiegel L., Nascimento L., Zutavern T., Miller B., Ambroise C., Muller S., Spooner W., Narechania A., Ren L., Wei S., Kumari S., Faga B., Levy M.J., McMahan L., Van Buren P., Vaughn M.W., Ying K., Yeh C.T., Emrich S.J., Jia Y., Kalyanaraman A., Hsia A.P., Barbazuk W.B., Baucom R.S., Brutnell T.P., Carpita N.C., Chaparro C., Chia J.M., Deragon J.M., Estill J.C., Fu Y., Jeddeloh J.A., Han Y., Lee H., Li P., Lisch D.R., Liu S., Liu Z., Nagel D.H., McCann M.C., SanMiguel P., Myers A.M., Nettleton D., Nguyen J., Penning B.W., Ponnala L., Schneider K.L., Schwartz D.C., Sharma A., Soderlund C., Springer N.M., Sun Q., Wang H., Waterman M., Westerman R., Wolfgruber T.K., Yang L., Yu Y., Zhang L., Zhou S., Zhu Q., Bennetzen J.L., Dawe R.K., Jiang J., Jiang N., Presting G.G., Wessler S.R., Aluru S., Martienssen R.A., Clifton S.W., McCombie W.R., Wing R.A., Wilson R.K. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 43.Brenchley R., Spannagl M., Pfeifer M., Barker G.L., D'Amore R., Allen A.M., McKenzie N., Kramer M., Kerhornou A., Bolser D., Kay S., Waite D., Trick M., Bancroft I., Gu Y., Huo N., Luo M.C., Sehgal S., Gill B., Kianian S., Anderson O., Kersey P., Dvorak J., McCombie W.R., Hall A., Mayer K.F., Edwards K.J., Bevan M.W., Hall N. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491(7426):705–710. doi: 10.1038/nature11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayer K.F., Rogers J., Doležel J., Pozniak C., Eversole K., Feuillet C., Gill B., Friebe B., Lukaszewski A.J., Sourdille P., Endo T.R., Kubaláková M., Cíhalíková J., Dubská Z., Vrána J., Sperková R., Simková H., Febrer M., Clissold L., McLay K., Singh K., Chhuneja P., Singh N.K., Khurana J., Akhunov E., Choulet F., Alberti A., Barbe V., Wincker P., Kanamori H., Kobayashi F., Itoh T., Matsumoto T., Sakai H., Tanaka T., Wu J., Ogihara Y., Handa H., Maclachlan P.R., Sharpe A., Klassen D., Edwards D., Batley J., Olsen O.A., Sandve S.R., Lien S., Steuernagel B., Wulff B., Caccamo M., Ayling S., Ramirez-Gonzalez R.H., Clavijo B.J., Wright J., Pfeifer M., Spannagl M., Martis M.M., Mascher M., Chapman J., Poland J.A., Scholz U., Barry K., Waugh R., Rokhsar D.S., Muehlbauer G.J., Stein N., Gundlach H., Zytnicki M., Jamilloux V., Quesneville H., Wicker T., Faccioli P., Colaiacovo M., Stanca A.M., Budak H., Cattivelli L., Glover N., Pingault L., Paux E., Sharma S., Appels R., Bellgard M., Chapman B., Nussbaumer T., Bader K.C., Rimbert H., Wang S., Knox R., Kilian A., Alaux M., Alfama F., Couderc L., Guilhot N., Viseux C., Loaec M., Keller B., Praud S. International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345(6194):1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 45.International Barley Genome Sequencing Consortium A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491(7426):711–716. doi: 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
- 46.Schmutz J., Cannon S.B., Schlueter J., Ma J., Mitros T., Nelson W., Hyten D.L., Song Q., Thelen J.J., Cheng J., Xu D., Hellsten U., May G.D., Yu Y., Sakurai T., Umezawa T., Bhattacharyya M.K., Sandhu D., Valliyodan B., Lindquist E., Peto M., Grant D., Shu S., Goodstein D., Barry K., Futrell-Griggs M., Abernathy B., Du J., Tian Z., Zhu L., Gill N., Joshi T., Libault M., Sethuraman A., Zhang X.C., Shinozaki K., Nguyen H.T., Wing R.A., Cregan P., Specht J., Grimwood J., Rokhsar D., Stacey G., Shoemaker R.C. Jackson; S.A. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 47.The Potato Genome Sequencing Consortium Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–195. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- 48.Tomato Genome Consortium The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim S., Park M., Yeom S.I., Kim Y.M., Lee J.M., Lee H.A., Seo E., Choi J., Cheong K., Kim K.T., Jung K., Lee G.W., Oh S., Bae C., Kim S.B., Lee H.Y., Kim S.Y., Kim M.S., Kang B.C., Jo Y.D., Yang H.B., Jeong H.J., Kang W.H., Kwon J.K., Shin C., Lim J.Y., Park J.H., Huh J.H., Kim J.S., Kim B.D., Cohen O., Paran I., Suh M.C., Lee S.B., Kim Y.K., Shin Y., Noh S.J., Park J., Seo Y.S., Kwon S.Y., Kim H.A., Park J.M., Kim H.J., Choi S.B., Bosland P.W., Reeves G., Jo S.H., Lee B.W., Cho H.T., Choi H.S., Lee M.S., Yu Y., Choi Y.D., Park B.S., van Deynze A., Ashrafi H., Hill T., Kim W.T., Pai H.S., Ahn H.K., Yeam I., Giovannoni J.J., Rose J.K., Sørensen I., Lee S.J., Kim R.W., Choi I.Y., Choi B.S., Lim J.S., Lee Y.H., Choi D. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014;46(3):270–278. doi: 10.1038/ng.2877. [DOI] [PubMed] [Google Scholar]
- 50.Dohm J.C., Minoche A.E., Holtgräwe D., Capella-Gutiérrez S., Zakrzewski F., Tafer H., Rupp O., Sörensen T.R., Stracke R., Reinhardt R., Goesmann A., Kraft T., Schulz B., Stadler P.F., Schmidt T., Gabaldón T., Lehrach H., Weisshaar B., Himmelbauer H. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature. 2014;505:546–549. doi: 10.1038/nature12817. [DOI] [PubMed] [Google Scholar]
- 51.Varshney R.K., Song C., Saxena R.K., Azam S., Yu S., Sharpe A.G., Cannon S., Baek J., Rosen B.D., Tar'an B., Millan T., Zhang X., Ramsay L.D., Iwata A., Wang Y., Nelson W., Farmer A.D., Gaur P.M., Soderlund C., Penmetsa R.V., Xu C., Bharti A.K., He W., Winter P., Zhao S., Hane J.K., Carrasquilla-Garcia N., Condie J.A., Upadhyaya H.D., Luo M.C., Thudi M., Gowda C.L., Singh N.P., Lichtenzveig J., Gali K.K., Rubio J., Nadarajan N., Dolezel J., Bansal K.C., Xu X., Edwards D., Zhang G., Kahl G., Gil J., Singh K.B., Datta S.K., Jackson S.A., Wang J., Cook D.R. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013;31:240–246. doi: 10.1038/nbt.2491. [DOI] [PubMed] [Google Scholar]
- 52.Schmutz J., McClean P.E., Mamidi S., Wu G.A., Cannon S.B., Grimwood J., Jenkins J., Shu S., Song Q., Chavarro C., Torres-Torres M., Geffroy V., Moghaddam S.M., Gao D., Abernathy B., Barry K., Blair M., Brick M.A., Chovatia M., Gepts P., Goodstein D.M., Gonzales M., Hellsten U., Hyten D.L., Jia G., Kelly J.D., Kudrna D., Lee R., Richard M.M., Miklas P.N., Osorno J.M., Rodrigues J., Thareau V., Urrea C.A., Wang M., Yu Y., Zhang M., Wing R.A., Cregan P.B., Rokhsar D.S., Jackson S.A. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014;46(7):707–713. doi: 10.1038/ng.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Q., Chen L.L., Ruan X., Chen D., Zhu A., Chen C., Bertrand D., Jiao W.B., Hao B.H., Lyon M.P., Chen J., Gao S., Xing F., Lan H., Chang J.W., Ge X., Lei Y., Hu Q., Miao Y., Wang L., Xiao S., Biswas M.K., Zeng W., Guo F., Cao H., Yang X., Xu X.W., Cheng Y.J., Xu J., Liu J.H., Luo O.J., Tang Z., Guo W.W., Kuang H., Zhang H.Y., Roose M.L., Nagarajan N., Deng X.X., Ruan Y. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013;45:59–66. doi: 10.1038/ng.2472. [DOI] [PubMed] [Google Scholar]
- 54.The International Peach Genome Initiative The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013;45:487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 55.D'Hont A., Denoeud F., Aury J.M., Baurens F.C., Carreel F. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488(7410):213–217. doi: 10.1038/nature11241. [DOI] [PubMed] [Google Scholar]
- 56.Hirakawa H., Shirasawa K., Miyatake K., Nunome T., Negoro S., Ohyama A., Yamaguchi H., Sato S., Isobe S., Tabata S., Fukuoka H. Draft genome sequence of eggplant (Solanum melongena L.): the representative solanum species indigenous to the old world. DNA Res. 2014;6:649–660. doi: 10.1093/dnares/dsu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu S., Liu Y., Yang X., Tong C., Edwards D., Parkin I.A., Zhao M., Ma J., Yu J., Huang S., Wang X., Wang J., Lu K., Fang Z., Bancroft I., Yang T.J., Hu Q., Wang X., Yue Z., Li H., Yang L., Wu J., Zhou Q., Wang W., King G.J., Pires J.C., Lu C., Wu Z., Sampath P., Wang Z., Guo H., Pan S., Yang L., Min J., Zhang D., Jin D.L,i, W., Belcram H., Tu J., Guan M., Qi C., Du D., Li J., Jiang L., Batley J., Sharpe A.G., Park B.S., Ruperao P., Cheng F., Waminal N.E, Huang Y., Dong C., Wang L., Li J., Hu Z., Zhuang M., Huang Y., Huang J., Shi J., Mei D., Liu J., Lee T.H., Wang J., Jin H., Li Z., Li X., Zhang J., Xiao L., Zhou Y., Liu Z., Liu X., Qin R., Tang X., Liu W., Wang Y., Zhang Y., Lee J., Kim H.H., Denoeud F., Xu X., Liang X., Hua W., Wang X., Wang J., Chalhoub B., Paterson A.H. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014;5:3930. doi: 10.1038/ncomms4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daldoul S., Guillaumie S., Reustle G.M., Krczal G., Ghorbel A., Delrot S., Mliki A., Höfer M.U. Isolation and expression analysis of salt induced genes from contrasting grapevine (Vitis vinifera L.) cultivars. Plant Sci. 2010;179:489–498. doi: 10.1016/j.plantsci.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 59.Morozova O., Marra M.A. Applications of next generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 62.Zenoni S., Ferrarini A., Giacomelli E., Xumerle L., Fasoli M., Malerba G., Bellin D., Pezzotti M., Delledonne M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010;152:1787–1795. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furbank R.T., Tester M. Phenomics-technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011;16:635–644. doi: 10.1016/j.tplants.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 64.Mittler R., Shulaev V. Functional genomics, challenges and perspectives for the future. Physiol. Plant. 2013;148:317–321. doi: 10.1111/ppl.12060. [DOI] [PubMed] [Google Scholar]
- 65.Fukushima A., Kusano M., Redestig H., Arita M., Saito K. Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 2009;13(5-6):532–538. doi: 10.1016/j.cbpa.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 66.Daldoul S., Ben Amar A., Guillaumie S., Mliki A. Integration of omics and system biology approaches to study grapevine (Vitis vinifera L.) response to salt stress: a perspective for functional genomics - A review. J. Int. Sci. Vigne Vin. 2014;48:189–200. [Google Scholar]
- 67.Cramer G.R., Cushman J.C., Schooley D.A., Quilici D., Vincent D., Bohlman M.C., Ergul A., Tattersall E.A., Tillett R., Evans J., Delacruz R., Schlauch K., Mendes P. Progress in bioinformatics the challenge of integrating transcriptomic, proteomic and metabolomic information. Acta Hortic. 2005;689:417–425. [Google Scholar]
- 68.Gupta B., Saha J., Sengupta A., Gupta K. Plant abiotic stress: ‘Omics’ approach. J. Plant Biochem. Physiol. 2013;1(e108) [Google Scholar]
- 69.Shingaki-Wells R.N., Huang S., Taylor N.L., Carroll A.J., Zhou W., Millar A.H. Differential molecular responses of rice and wheat coleoptiles to anoxia reveal novel metabolic adaptations in amino acid metabolism for tissue tolerance. Plant Physiol. 2011;156(4):1706–1724. doi: 10.1104/pp.111.175570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mostafavi S., Ray D., Warde-Farley D., Grouios C., Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amiour N., Imbaud S., Clément G., Agier N., Zivy M., Valot B., Balliau T., Quilleré I., Tercé-Laforgue T., Dargel-Graffin C., Hirel B. An integrated “omics” approach to the characterization of maize (Zea mays L.) mutants deficient in the expression of two genes encoding cytosolic glutamine synthetase. BMC Genomics. 2014;15(1005) doi: 10.1186/1471-2164-15-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galindo-Gonzalez L.M., El Kayal W., Ju C.J., Allen C.C., King-Jones S., Cooke J.E. Integrated transcriptomic and proteomic profiling of white spruce stems during the transition from active growth to dormancy. Plant Cell Environ. 2012;35(4):682–701. doi: 10.1111/j.1365-3040.2011.02444.x. [DOI] [PubMed] [Google Scholar]
- 73.Proietti S., Bertini L., Timperio A.M., Zolla L., Caporale C., Caruso C. Crosstalk between salicylic acid and jasmonate in Arabidopsis investigated by an integrated proteomic and transcriptomic approach. Mol. Biol. Sys. 2013;9(6):1169–1187. doi: 10.1039/c3mb25569g. [DOI] [PubMed] [Google Scholar]
- 74.Altenbach S.B., Vensel W.H., DuPont F.M. Integration of transcriptomic and proteomic data from a single wheat cultivar provides new tools for understanding the roles of individual alpha gliadin proteins in flour quality and celiac disease. J. Cereal Sci. 2010;52:143–151. [Google Scholar]
- 75.Yamakawa H., Hakata M. Atlas of rice grain filling-related metabolism under high temperature: joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant Cell Physiol. 2010;51(5):795–809. doi: 10.1093/pcp/pcq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Izawa T., Mihara M., Suzuki Y., Gupta M., Itoh H., Nagano A.J., Motoyama R., Sawada Y., Yano M., Hirai M.Y., Makino A., Nagamura Y. Os-GIGANTEA confers robust diurnal rhythms on the global transcriptome of rice in the field. Plant Cell. 2011;23(5):1741–1755. doi: 10.1105/tpc.111.083238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kogel K.H., Voll L.M., Schafer P., Jansen C., Wu Y., Langen G., Imani J., Hofmann J., Schmiedl A., Sonnewald S., von Wettstein D., Cook R.J., Sonnewald U. Transcriptome and metabolome profiling of field-grown transgenic barley lack induced differences but show cultivar-specific variances. Proc. Natl. Acad. Sci. USA. 2010;107(14):6198–6203. doi: 10.1073/pnas.1001945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schauer N., Semel Y., Roessner U., Gur A., Balbo I., Carrari F., Pleban T., Perez-Melis A., Bruedigam C., Kopka J., Willmitzer L., Zamir D., Fernie A.R. Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 2006;24:447–454. doi: 10.1038/nbt1192. [DOI] [PubMed] [Google Scholar]
- 79.Sharma A., Deshmukh R.K., Jain N., Singh N.K. Combining QTL mapping and transcriptome profiling for an insight into genes for grain number in rice (Oryza sativa L.). Indian J. Genet. Plant Breed. 2011;71:115–119. [Google Scholar]
- 80.Kadam S., Singh K., Shukla S., Goel S., Vikram P., Pawar V., Gaikwad K., Khanna-Chopra R., Singh N. Genomic associations for drought tolerance on the short arm of wheat chromosome 4B. Funct. Integr. Genomics. 2012;12:447–464. doi: 10.1007/s10142-012-0276-1. [DOI] [PubMed] [Google Scholar]
- 81.Sheth B.P., Thaker V.S. Plant systems biology: insights, advances and challenges. Planta. 2014;240:33–54. doi: 10.1007/s00425-014-2059-5. [DOI] [PubMed] [Google Scholar]
- 82.Cramer G.R. Abiotic stress and plant responses from the whole vine to the genes. Aust. J. Grape Wine Res. 2010;16:86–93. [Google Scholar]
- 83.Weston D.J., Karve A.A., Gunter L.E., Jawdy S.S., Yang X., Allen S.M., Wullschleger S.D. Comparative physiology and transcriptional networks underlying the heat shock response in Populus trichocarpa, Arabidopsis thaliana and Glycine max. Plant Cell Environ. 2011;34:1488–1506. doi: 10.1111/j.1365-3040.2011.02347.x. [DOI] [PubMed] [Google Scholar]
- 84.Weston D.J., Hanson P.J., Norby R.J., Tuskan G.A., Wullschleger S.D. From systems biology to photosynthesis and whole plant physiology. Plant Signal. Behav. 2012;7(2):260–226. doi: 10.4161/psb.18802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang P., Dreher K., Karthikeyan A., Chi A., Pujar A., Caspi R., Karp P., Kirkup V., Latendresse M., Lee C., Mueller L.A., Muller R., Rhee S.Y. Creation of a genome-wide metabolic pathway database for Populus trichocarpa using a new approach for reconstruction and curation of metabolic pathways for plants. Plant Physiol. 2010;153(4):1479–1491. doi: 10.1104/pp.110.157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mueller L.A., Zhang P., Rhee S.Y. AraCyc: A biochemical pathway database for Arabidopsis. Plant Physiol. 2003;132(2):453–460. doi: 10.1104/pp.102.017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tatusova T., Smith-White B., Ostell J. A collection of plant-specific genomic data and resources at NCBI. Methods Mol. Biol. 2007;406:61–87. doi: 10.1007/978-1-59745-535-0_3. [DOI] [PubMed] [Google Scholar]
- 88.Van Bel M., Proost S., Wischnitzki E., Movahedi S., Scheerlinck C., Van de Peer Y., Vandepoele K. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 2012;158(2):590–600. doi: 10.1104/pp.111.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sucaet Y., Wang Y., Li J., Wurtele E.S. MetNet Online: A novel integrated resource for plant systems biology. BMC Bioinformatics. 2012;13:267. doi: 10.1186/1471-2105-13-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Numa H., Itoh T. MEGANTE: A Web-Based System for Integrated Plant Genome Annotation. 2013. [DOI] [PMC free article] [PubMed]
- 91.Seaver S.l., Gerdes S., Frelind O., Lerma-Ortize C., Bradburyd L.M., Zallot R., Hasnain G., Niehaus T.D., El Yacoubi B., Pasternak S., Olson R., Pusch G., Overbeek R., Stevens R., de Crécy-Lagard V., Ware D., Hanson A.D., Henry C.S. High throughput comparison, functional annotation, and metabolic modeling of plant genomes using the PlantSEED resource. Proc. Natl. Acad. Sci. USA. 2014;111(26):9645–9650. doi: 10.1073/pnas.1401329111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skuse G.R., Du C. Bioinformatics tools for plant genomics. Int. J. Plant Genomics. 2008 doi: 10.1155/2008/910474. [Editorial]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang C., Chen C., Huang Z., Liu R., Verdier J. ITIS, a bioinformatics tool for accurate identification of transposon insertion sites using next-generation sequencing data. BMC Bioinformatics. 2015;16:72. doi: 10.1186/s12859-015-0507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Henikoff S., Till B.J., Comai L. TILLING. Traditional mutagenesis meets functional genomics. Plant Physiol. 2004;135:630–636. doi: 10.1104/pp.104.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koiwa H., Bressan R.A., Hasegawa P.M. Identification of plant stress-responsive determinants in Arabidopsis by large-scale forward genetic screens. J. Exp. Bot. 2006;57(5):1119–1128. doi: 10.1093/jxb/erj093. [DOI] [PubMed] [Google Scholar]
- 96.Papdi C., Leung J., Joseph M.P., Salamó I.P., Szabados L. Genetic screens to identify plant stress genes. Methods Mol. Biol. 2010;639:121–139. doi: 10.1007/978-1-60761-702-0_7. [DOI] [PubMed] [Google Scholar]
- 97.Dobritsa A.A., Geanconteri A., Shrestha J., Carlson A., Kooyers N., Coerper D., Urbanczyk-Wochniak E., Bench B.J., Sumner L.W., Swanson R., Preuss D. A Large-scale genetic screen in Arabidopsis to identify genes involved in pollen exine production. Plant Physiol. 2011;157:2947–2970. doi: 10.1104/pp.111.179523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ding Y., Shaholli D., Mou Z. A large-scale genetic screen for mutants with altered salicylic acid accumulation in Arabidopsis. Front. Plant Sci. 2015;5:763. doi: 10.3389/fpls.2014.00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sikora P., Chawade A., Larsson M., Olsson J., Olsson O. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Waterhouse P.M., Helliwell C.A. Exploring plant genomes by RNA-induced gene silencing. Nat. Rev. Genet. 2003;4:29–38. doi: 10.1038/nrg982. [DOI] [PubMed] [Google Scholar]
- 101.McGinnis K.M. RNAi for functional genomics in plants. Brief. Funct. Genomics. 2010;9(2):111–117. doi: 10.1093/bfgp/elp052. [DOI] [PubMed] [Google Scholar]
- 102.Kumar S. RNAi (RNA interference) vectors for functional genomics study in plants. Natl. Acad. Sci. Lett. 2014;37(3):289–294. [Google Scholar]
- 103.Benedito V.A., Visser P.B., Angenent G.C., Krens F.A. The potential of virus-induced gene silencing for speeding up functional characterization of plant genes. Genet. Mol. Res. 2004;3(3):323–341. [PubMed] [Google Scholar]
- 104.Purkayastha A., Dasgupta I. Virus-induced gene silencing: a versatile tool for discovery of gene functions in plants. Plant Physiol. Biochem. 2009;47:967–976. doi: 10.1016/j.plaphy.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 105.Dalmais M., Antelme S., Ho-Yue-Kuang S., Wang Y., Darracq O., Bouvier d’Yvoire M., Cezard L., Legee F., Blondet E., Oria N., Troadec C., Brunaud V., Jouanin L., Hofte H., Bendahmane A., Lapierre C., Sibout R. A TILLING platform for functional genomics in Brachypodium distachyon. PLoS One. 2013;8(6):e65503. doi: 10.1371/journal.pone.0065503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang N., Shi L. Screening of mutations by TILLING in plants. Methods Mol. Biol. 2015;1245:193–203. doi: 10.1007/978-1-4939-1966-6_15. [DOI] [PubMed] [Google Scholar]
- 107.Stepanova A.N., Alonso J.M. PCR-based screening for insertional mutants. Methods Mol. Biol. 2006;323:163–172. doi: 10.1385/1-59745-003-0:163. [DOI] [PubMed] [Google Scholar]
- 108.Jung K.H., An G., Ronald P.C. Towards a better bowl of rice: assigning function to tens of thousands of rice genes. Nat. Rev. Genet. 2008;9:91–101. doi: 10.1038/nrg2286. [DOI] [PubMed] [Google Scholar]
- 109.Tzifira T., White C. Towards targeted mutagenesis and gene replacement in plants. Trends Biotechnol. 2005;23(12):567–569. doi: 10.1016/j.tibtech.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 110.Dalmay T., Hamilton A.J., Rudd S., Angell S., Baulcombe D. C. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell. 2000;101:543–553. doi: 10.1016/s0092-8674(00)80864-8. [DOI] [PubMed] [Google Scholar]
- 111.Wasseneger M., Krczal G. Nomenclature and functions of RNA-directed RNA polymerases. Trends Plant Sci. 2006;11(3):142–151. doi: 10.1016/j.tplants.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 112.Voinnet O. Induction and suppression of RNA silencing: insights from viral infections. Nat. Rev. Genet. 2005;6:206–221. doi: 10.1038/nrg1555. [DOI] [PubMed] [Google Scholar]
- 113.Wang M.B., Waterhouse P.M. Applications of gene silencing in plants. Curr. Opin. Plant Biol. 2001;5:146–150. doi: 10.1016/s1369-5266(02)00236-4. [DOI] [PubMed] [Google Scholar]
- 114.Wassenegger M. The role of the RNAi machinery in heterochromatin formation. Cell. 2005;122(1):13–16. doi: 10.1016/j.cell.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 115.Baulcombe D. RNA silencing. Curr. Biol. 2002;12(3):82–84. doi: 10.1016/s0960-9822(02)00665-6. [DOI] [PubMed] [Google Scholar]
- 116.McGinnis K., Murphy N., Carlson A.R., Akula A., Akula C., Basinger H., Carlson M., Hermanson P., Kovacevic N., McGill M.A., Seshadri V., Yoyokie J., Cone K., Kaeppler H.F., Kaeppler S.M., Springer N.M. Assessing the efficiency of RNA interference for maize functional genomics. Plant Physiol. 2007;143(4):1441–1451. doi: 10.1104/pp.106.094334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bhaskar P.B., Venkateshwaran M., Wu L., Ané J.M., Jiang J. Agrobacterium-mediated transient gene expression and silencing: a rapid tool for functional gene assay in potato. PLoS One. 2009;4(6):e5812. doi: 10.1371/journal.pone.0005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Travella S., Klimm T.E., Keller B. RNA interference-based gene silencing as an efficient tool for functional genomics in hexaploid bread wheat. Plant Physiol. 2006;142(1):6–20. doi: 10.1104/pp.106.084517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fu D., Uauy C., Blechl A., Dubcovsky J. RNA interference for wheat functional gene analysis. Transgenic Res. 2007;16(6):689–701. doi: 10.1007/s11248-007-9150-7. [DOI] [PubMed] [Google Scholar]
- 120.Li J., Jiang D., Zhou H., Li F., Yang J., Hong L., Fu X., Li Z., Liu Z., Li J., Zhuang C. Expression of RNA-interference/antisense transgenes by the cognate promoters of target genes is a better gene-silencing strategy to study gene functions in rice. PLoS One. 2011;6(7) doi: 10.1371/journal.pone.0017444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Younis A., Siddique M.I., Kim C.K., Lim K.B. RNA interference (RNAi) induced gene silencing: A promising approach of Hi-Tech plant breeding. Int. J. Biol. Sci. 2014;10(10):1150–1158. doi: 10.7150/ijbs.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Voinnet O. RNA silencing as a plant immune system against viruses. Trends Genet. 2001;17:449–459. doi: 10.1016/s0168-9525(01)02367-8. [DOI] [PubMed] [Google Scholar]
- 123.Waterhouse P.M. Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature. 2001;411:834–842. doi: 10.1038/35081168. [DOI] [PubMed] [Google Scholar]
- 124.Wang M.B., Metzlaff M. RNA silencing and antiviral defense in plants. Curr. Opin. Plant Biol. 2005;8(2):216–222. doi: 10.1016/j.pbi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 125.Vance V., Vaucheret H. RNA silencing in plants – defense and counter defense. Science. 2001;292:2277–2280. doi: 10.1126/science.1061334. [DOI] [PubMed] [Google Scholar]
- 126.Lu R., Martin-Hernandez A.M., Peart J.R., Malcuit I., Baulcombe D.C. Virus-induced gene silencing in plants. Methods. 2003;30:296–303. doi: 10.1016/s1046-2023(03)00037-9. [DOI] [PubMed] [Google Scholar]
- 127.Brigneti G., Martin-Hernandez A.M., Jin H., Chen J., Baulcombe D.C., Baker B.B., Jones J.D. Virus-induced gene silencing in Solanum species. Plant J. 2004;39:264–272. doi: 10.1111/j.1365-313X.2004.02122.x. [DOI] [PubMed] [Google Scholar]
- 128.Padmanabhan M., Dinesh-Kumar S.P. Virus-induced gene silencing as a tool for delivery of dsRNA into plants. Cold Spring Harb. Protoc. 2009;2(4):1–4. doi: 10.1101/pdb.prot5139. [DOI] [PubMed] [Google Scholar]
- 129.Cai X., Wang C., Xu Y., Xu Q., Zheng Z., Zhou X. Efficient gene silencing induction in tomato by a viral satellite DNA vector. Virus Res. 2007;125:169–175. doi: 10.1016/j.virusres.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 130.Ratcliff F., Martin-Hernandez A.M., Baulcombe D.C. Tobacco rattle virus as a vector for analysis of gene function by silencing. Plant J. 2001;25:237–245. doi: 10.1046/j.0960-7412.2000.00942.x. [DOI] [PubMed] [Google Scholar]
- 131.Ryu C.M., Anand A., Kang L., Mysore K.S. Agrodrench: a novel and effective agroinoculation method for virus-induced gene silencing in roots and diverse solanaceous species. Plant J. 2004;40(2):322–331. doi: 10.1111/j.1365-313X.2004.02211.x. [DOI] [PubMed] [Google Scholar]
- 132.Senthil-Kumar M., Mysore K.S. Virus-induced gene silencing can persist for more than 2 years and also be transmitted to progeny seedlings in Nicotiana benthamiana and tomato. Plant Biotechnol. J. 2011;9:797–806. doi: 10.1111/j.1467-7652.2011.00589.x. [DOI] [PubMed] [Google Scholar]
- 133.Senthil-Kumar M., Mysore K.S. Tobacco rattle virus-based virus-induced gene silencing in Nicotiana benthamiana. Nat. Protoc. 2014;9:1549–1562. doi: 10.1038/nprot.2014.092. [DOI] [PubMed] [Google Scholar]
- 134.Gould B., Kramer E.M. Virus-induced gene silencing as a tool for functional analyses in the emerging model plant Aquilegia (columbine, Ranunculaceae). Plant Methods. 2007;3(6) doi: 10.1186/1746-4811-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kirigia D., Runo S., Alakonya A. A virus-induced gene silencing (VIGS) system for functional genomics in the parasitic plant Striga hermonthica. Plant Methods. 2014;10(12) doi: 10.1186/1746-4811-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee W.S., Rudd J.J., Kanyuka K. Virus induced gene silencing (VIGS) for functional analysis of wheat genes involved in Zymoseptoria tritici susceptibility and resistance. Fungal Genet. Biol. 2015;79:84–88. doi: 10.1016/j.fgb.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Senthil-Kumar M. RameGowda, H.V.; Hema, R.; Mysore, K.S.; Udayakumar, M. Virus-induced gene silencing and its application in characterizing genes involved in water-deficit-stress tolerance. J. Plant Physiol. 2008;165:1404–1421. doi: 10.1016/j.jplph.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 138.Lim C.W., Lee S.C. Functional roles of the pepper MLO protein gene, CaMLO2, in abscisic acid signaling and drought sensitivity. Plant Mol. Biol. 2014;85:1–10. doi: 10.1007/s11103-013-0155-8. [DOI] [PubMed] [Google Scholar]
- 139.Kandoth P.K., Heinz R., Yeckel G., Gross N.W., Juvale P.S., Hill J., Whitham S.A., Baum T.J., Mitchum M. A virus-induced gene silencing method to study soybean cyst nematode parasitism in Glycine max. BMC Res. Notes. 2013;6(255) doi: 10.1186/1756-0500-6-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu D., Liu X., Meng Y., Sun C., Tang H., Jiang Y., Khan M.A., Xue J., Ma N., Gao J. An organ-specific role for ethylene in rose petal expansion during dehydration and rehydration. J. Exp. Bot. 2013;64:2333–2344. doi: 10.1093/jxb/ert092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ma M., Yan Y., Huang L., Chen M., Zaho H. Virus-induced gene-silencing in wheat spikes and grains and its application in functional analysis of HMW-GS-encoding genes. BMC Plant Biol. 2012;12(141) doi: 10.1186/1471-2229-12-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Prelich G. Gene overexpression: uses, mechanisms, and interpretation. Genetics. 2012;190:841–854. doi: 10.1534/genetics.111.136911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dou H., Xv K., Meng Q., Li G., Yang X. Potato plants ectopically expressing Arabidopsis thaliana CBF3 exhibit enhanced tolerance to high-temperature stress. Plant Cell Environ. 2014;38:61–72. doi: 10.1111/pce.12366. [DOI] [PubMed] [Google Scholar]
- 144.Khan Z.A., Abdin M.Z., Khan J.A. Functional characterization of a strong bi-directional constitutive plant promoter isolated from cotton leaf curl Burewala virus. PLoS One. 2015;10(3):e0121656. doi: 10.1371/journal.pone.0121656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chung S.M., Vaidya M., Tzfira T. Agrobacterium is not alone: gene transfer to plants by viruses and other bacteria. Trends Plant Sci. 2006;11(1):1–4. doi: 10.1016/j.tplants.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 146.Vidal J.R., Kikkert J.R., Donzelli B.D., Wallace P.G., Reisch B.I. Biolistic transformation of grapevine using minimal gene cassette technology. Plant Cell Rep. 2006;25:807–814. doi: 10.1007/s00299-006-0132-7. [DOI] [PubMed] [Google Scholar]
- 147.Ueki S., Lacroix B., Krichevsky A., Lazarowitz S.G., Citovsky V. Functional transient genetic transformation of Arabidopsis leaves by biolistic bombardment. Nat. Protoc. 2009;4:71–77. doi: 10.1038/nprot.2008.217. [DOI] [PubMed] [Google Scholar]
- 148.Ueki S., Magori S., Lacroix B., Citovsky V. Transient gene expression in epidermal cells of plant leaves by biolistic DNA delivery. Methods Mol. Biol. 2013;940:17–26. doi: 10.1007/978-1-62703-110-3_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yoo S.D., Cho Y.H., Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2007;2(7):1565–1572. doi: 10.1038/nprot.2007.199. [DOI] [PubMed] [Google Scholar]
- 150.Vaghchhipawala Z.E., Mysore K.S. Agroinoculation: a simple procedure for systemic infection of plants with viruses. Methods Mol. Biol. 2008;451:555–562. doi: 10.1007/978-1-59745-102-4_38. [DOI] [PubMed] [Google Scholar]
- 151.Vaghchipawala Z., Rojas C.M., Senthil-Kumar M., Mysore K.S. Agroinoculation and agroinfiltration: simple tools for complex gene function analyses. Methods Mol. Biol. 2011;678:65–76. doi: 10.1007/978-1-60761-682-5_6. [DOI] [PubMed] [Google Scholar]
- 152.Pasin F., Kulasekaran S., Natale P., Simón-Mateo C., García J.A. Rapid fluorescent reporter quantification by leaf disc analysis and its application in plant-virus studies. Plant Methods. 2014;10(22) doi: 10.1186/1746-4811-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kokkirala V.R., Peng Y., Abbagani S., Zhu Z., Umate P. Subcellular localization of proteins of Oryza sativa L. in the model tobacco and tomato plants. Plant Signal. Behav. 2010;5(11):1336–1341. doi: 10.4161/psb.5.11.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang Y., Li R., Qi M. In vivo analysis of plant promoters and transcription factors by agroinfiltration of tobacco leaves. Plant J. 2000;22(6):543–551. doi: 10.1046/j.1365-313x.2000.00760.x. [DOI] [PubMed] [Google Scholar]
- 155.Liu W., Mazare M., Rudis M.R., Fethe M.H. Stewart, C.N. Rapid in vivo analysis of synthetic promoters for plant pathogen phytosensing. BMC Biotechnol. 2011;11(108) doi: 10.1186/1472-6750-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen Q., Lai H., Hurtado J., Stahnke J., Leuzinger K., Dent M. Agroinfiltration as an effective and scalable strategy of gene delivery for production of pharmaceutical proteins. . Adv. Tech. Biol. Med. 2013. [DOI] [PMC free article] [PubMed]
- 157.Dugdale B., Mortimer C.L., Kato M., James T.A., Harding R.M., Dale J.L. Design and construction of an in-plant activation cassette for transgene expression and recombinant protein production in plants. Nat. Protoc. 2014;9(5):1010–1027. doi: 10.1038/nprot.2014.068. [DOI] [PubMed] [Google Scholar]
- 158.Bechtold N., Ellis J., Pelletier G. In planta Agrobacterium mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. C.R. Acad. Sci. 1993;316:1194–1199. [Google Scholar]
- 159.Narusaka M., Shiraishi T., Iwabuchi M., Narusaka Y. The floral inoculating protocol: a simplified Arabidopsis thaliana transformation method modified from floral dipping. Plant Biotechnol. 2010;27:349–351. [Google Scholar]
- 160.Clough S.J., Bent A.F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16(6):735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 161.Zhang X., Henriques R., Lin S.S., Niu Q.W., Chua N.H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 2006;1:641–646. doi: 10.1038/nprot.2006.97. [DOI] [PubMed] [Google Scholar]
- 162.Shang Y., Schwinn K.E., Bennett M.J., Hunter D.A., Waugh T.A., Pathirana N.N., Brummell D.A., Jameson P.E., Davies K.M. Methods for transient assay of gene function in floral tissues. Plant Methods. 2007;3(1) doi: 10.1186/1746-4811-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Andrieu A., Breitler J.C., Meynard D., Gantet P., Guiderdoni E. An in planta, Agrobacterium-mediated transient gene expression method for inducing gene silencing in rice (Oryza sativa L.) leaves. Rice (N. Y.) 2012;5(23) doi: 10.1186/1939-8433-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Karimi M., Depicker A., Hilson P. Recombinational cloning with plant Gateway vectors. Plant Physiol. 2007;145(4):1144–1154. doi: 10.1104/pp.107.106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gonzalez-Ballester D., Pootakham W., Mus F., Yang W., Catalanotti C., Magneschi L., de Montaigu A., Higuera J.J., Prior M., Galván A., Fernandez E., Grossman A.R. Reverse genetics in Chlamydomonas: a platform for isolating insertional mutants. Plant Methods. 2011;7(24) doi: 10.1186/1746-4811-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Alonso J.M., Stepanova A.N., Leisse T.J., Kim C.J., Chen H., Shinn P., Stevenson D.K., Zimmerman J., Barajas P., Cheuk R., Gadrinab C., Heller C., Jeske A., Koesema E., Meyers C.C., Parker H., Prednis L., Ansari Y., Choy N., Deen H., Geralt M., Hazari N., Hom E., Karnes M., Mulholland C., Ndubaku R., Schmidt I., Guzman P., Aguilar-Henonin L., Schmid M., Weigel D., Carter D.E., Marchand T., Risseeuw E., Brogden D., Zeko A., Crosby W.L., Berry C.C., Ecker J.R. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301(5633):653–657. doi: 10.1126/science.1086391. [DOI] [PubMed] [Google Scholar]
- 167.Thole V., Peraldi A., Worland B., Nicholson P., Doonan J.H., Vain P. T-DNA mutagenesis in Brachypodium distachyon. J. Exp. Bot. 2012;63(2):567–576. doi: 10.1093/jxb/err333. [DOI] [PubMed] [Google Scholar]
- 168.Upadhyaya N.M., Zhu Q.H., Bhat R.S. Transposon insertional mutagenesis in rice. Methods Mol. Biol. 2010;678:147–177. doi: 10.1007/978-1-60761-682-5_12. [DOI] [PubMed] [Google Scholar]
- 169.McCallum C.M., Comai L., Greene E.A., Henikoff S. Targeting Induced Local Lesions IN Genomes (TILLING) for plant functional genomics. Plant Physiol. 2000;123:439–442. doi: 10.1104/pp.123.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Slade A.J., Knauf V.C. TILLING moves beyond functional genomics into crop improvement. Transgenic Res. 2005;14:109–115. doi: 10.1007/s11248-005-2770-x. [DOI] [PubMed] [Google Scholar]
- 171.Chen L., Hao L., Parry M.A., Phillips A.L., Hu Y.G. Progress in TILLING as a tool for functional genomics and improvement of crops. J. Integr. Plant Biol. 2014;56:425–443. doi: 10.1111/jipb.12192. [DOI] [PubMed] [Google Scholar]
- 172.Till B.J., Colbert T., Tompa R., Enns L.C., Codomo C.A., Johnson J.E., Reynolds S.H., Henikoff J.G., Greene E.A., Steine M.N., Comai L., Henikoff S. High-throughput TILLING for functional genomics. Methods Mol. Biol. 2003;236:205–220. doi: 10.1385/1-59259-413-1:205. [DOI] [PubMed] [Google Scholar]
- 173.Tadele Z., Chikelu M.B., Till B.J. TILLING for mutations in model plants and crops. In: Jain S.M., Brar D.S., editors. Molecular Techniques in Crop Improvement. Springer; 2010. pp. 307–332. [Google Scholar]
- 174.Paszkowski J., Baur M., Bogucki A., Potrykus I. Gene targeting in plants. EMBO J. 1988;7:4021–4026. doi: 10.1002/j.1460-2075.1988.tb03295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Reski R. Physcomitrella and Arabidopsis: the David and Goliath of reverse genetics. Trends Plant Sci. 1998;3:209–210. [Google Scholar]
- 176.Kamisugi Y., Schlink K., Rensing S.A., Schween G., von Stackelberg M., Cuming A.C., Reski R., Cove D.J. The mechanism of gene targeting in Physcomitrella patens: homologous recombination, concatenation and multiple integration. Nucleic Acids Res. 2006;34(21):6205–6214. doi: 10.1093/nar/gkl832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Puchta H. Gene replacement by homologous recombination in plants. Plant Mol. Biol. 2002;48:173–182. [PubMed] [Google Scholar]
- 178.Reiss B. Homologous recombination and gene targeting in plant cells. Int. Rev. Cytol. 2003;228:85–139. doi: 10.1016/s0074-7696(03)28003-7. [DOI] [PubMed] [Google Scholar]
- 179.Hanin M., Paszkowski J. Plant genome modification by homologous recombination. Curr. Opin. Plant Biol. 2003;6:157–162. doi: 10.1016/s1369-5266(03)00016-5. [DOI] [PubMed] [Google Scholar]
- 180.Hanin M., Volrath S., Bogucki A., Briker M., Ward E., Paszkowski J. Gene targeting in Arabidopsis. Plant J. 2001;28:671–677. doi: 10.1046/j.1365-313x.2001.01183.x. [DOI] [PubMed] [Google Scholar]
- 181.Terada R., Urawa H., Inagaki Y., Tsugane K., Iida S. Efficient gene targeting by homologous recombination in rice. Nat. Biotechnol. 2002;20:1030–1034. doi: 10.1038/nbt737. [DOI] [PubMed] [Google Scholar]
- 182.Weinthal D.M., Taylor R.A., Tzfira T. Nonhomologous end joining-mediated gene replacement in plant cells. Plant Physiol. 2013;162(1):390–400. doi: 10.1104/pp.112.212910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Endo M., Seiichi T. Toward establishing an efficient and versatile gene targeting system in higher plants. Trait introduction methods and innovation plateforms in plant biotechnology. Biocat. Agric. Biotechnol. 2014;3(1):2–6. [Google Scholar]
- 184.Belhaj K., Chaparro-Garcia A., Kamoun S., Nekrasov V. Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods. 2013;9(39) doi: 10.1186/1746-4811-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bortesi L., Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015;33(1):41–52. doi: 10.1016/j.biotechadv.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 186.Osakabe Y., Osakabe K. Genome editing with engineered nucleases in plants. Plant Cell Physiol. 2015;56(3):389–400. doi: 10.1093/pcp/pcu170. [DOI] [PubMed] [Google Scholar]
- 187.Townsend J.A., Wright D.A., Winfrey R.J., Fu F., Maeder M.L., Joung J.K., Voytas D.F. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature. 2009;459(7245):442–445. doi: 10.1038/nature07845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang F., Maeder M.L., Unger-Wallace E., Hoshaw J.P., Reyon D., Christian M., Li X., Pierick C.J., Dobbs D., Peterson T., Joung J.K., Voytas D.F. High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc. Natl. Acad. Sci. USA. 2010;107(26):12028–12033. doi: 10.1073/pnas.0914991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Puchta H., Dujon B., Hohn B. Homologous recombination in plant cells is enhanced by in vivo induction of double strand breaks into DNA by a site‐specific endonuclease. Nucleic Acids Res. 1993;21:5034–5040. doi: 10.1093/nar/21.22.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Daboussi F., Stoddard T.J., Zhang F. 2015. Engineering meganucleases for precise plant genome modification. [Google Scholar]
- 191.Porteus M.H., Caroll D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005;23(8):967–973. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 192.Shukla V.K., Doyon Y., Miller J.C., DeKelver R.C., Moehle E.A., Worden S.E., Mitchell J.C., Arnold N.L., Gopalan S., Meng X., Choi V.M., Rock J.M., Wu Y.Y., Katibah G.E., Zhifang G., McCaskill D., Simpson M.A., Blakeslee B., Greenwalt S.A., Butler H.J., Hinkley S.J., Zhang L., Rebar E.J., Gregory P.D., Urnov F.D. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature. 2009;459(7245):437–441. doi: 10.1038/nature07992. [DOI] [PubMed] [Google Scholar]
- 193.Bogdanove A.J., Voytas D.F. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 194.Nemudryi A.A., Valetdinova K.R., Medvedev S.P., Zakian S.M. TALEN and CRISPR/Cas Genome Editing Systems: Tools of Discovery. Acta Naturae. 2014;6(3):19–40. [PMC free article] [PubMed] [Google Scholar]
- 195.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li J.F., Norville J.E., Aach J., McCormack M., Zhang D., Bush J., Church G.M., Sheen J. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 2013;31:688–691. doi: 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shan Q., Wang Y., Li J., Gao C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014;9:2395–2410. doi: 10.1038/nprot.2014.157. [DOI] [PubMed] [Google Scholar]
- 198.Nekrasov V., Staskawicz B., Weigel D., Jones J.D., Kamoun S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:691–693. doi: 10.1038/nbt.2655. [DOI] [PubMed] [Google Scholar]
- 199.Jia H., Wang N. Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS One. 2014;9(4):e93806. doi: 10.1371/journal.pone.0093806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ali Z., Abul-faraj A., Li L., Ghosh N., Piatek M., Mahjoub A., Aouida M., Piatek A., Baltes N.J., Voytas D.F., Dinesh-Kumar S., Mahfouz M.M. Mol. Plant. doi: 10.1016/j.molp.2015.02.011. in press. [DOI] [PubMed] [Google Scholar]
- 201.Mao Y., Zhang H., Xu N., Zhang B., Gou F., Zhu J.K. Application of the CRISPR-Cas system for efficient genome engineering in plants. Mol. Plant. 2013;6:2008–2011. doi: 10.1093/mp/sst121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Brooks C., Nekrasov V., Lippman Z.B., Van Eck J. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/ CRISPR-associated9 system. Plant Physiol. 2014;166:1292–1297. doi: 10.1104/pp.114.247577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Miao J., Guo D., Zhang J., Huang Q., Qin G., Zhang X., Wan J., Gu H., Qu L.J. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 2013;23:1233–1236. doi: 10.1038/cr.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xu R., Li H., Qin R., Wang L., Li L., Wei P., Yang J. Gene targeting using the Agrobacterium tumefaciens-mediated CRISPR-Cas system in rice. Rice (N. Y.) 2014;7(5) doi: 10.1186/s12284-014-0005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]