

Abstract
Background:
In addition to neurons, all components of the neurovascular unit (NVU), such as glial, endothelial, and basal membranes, are destroyed during traumatic brain injury (TBI). Previous studies have shown that excessive stimulation of calpain is crucial for cerebral injury after traumatic insult. The objective of this study was to investigate whether calpain activation participated in NVU disruption and edema formation in a mouse model of controlled cortical impact (CCI).
Methods:
One hundred and eight mice were divided into three groups: the sham group, the control group, and the MDL28170 group. MDL28170 (20 mg/kg), an efficient calpain inhibitor, was administered intraperitoneally at 5 min, 3 h, and 6 h after experimental CCI. We then measured neurobehavioral deficits, calpain activity, inflammatory mediator levels, blood–brain barrier (BBB) disruption, and NVU deficits using electron microscopy and histopathological analysis at 6 h and 24 h after CCI.
Results:
The MDL28170 treatment significantly reduced the extent of both cerebral contusion (MDL28170 vs. vehicle group, 16.90 ± 1.01 mm3 and 17.20 ± 1.17 mm3 vs. 9.30 ± 1.05 mm3 and 9.90 ± 1.17 mm3, both P < 0.001) and edema (MDL28170 vs. vehicle group, 80.76 ± 1.25% and 82.00 ± 1.84% vs. 82.55 ± 1.32% and 83.64 ± 1.25%, both P < 0.05), improved neurological scores (MDL28170 vs. vehicle group, 7.50 ± 0.45 and 6.33 ± 0.38 vs. 12.33 ± 0.48 and 11.67 ± 0.48, both P < 0.001), and attenuated NVU damage resulting (including tight junction (TJ), basement membrane, BBB, and neuron) from CCI at 6 h and 24 h. Moreover, MDL28170 markedly downregulated nuclear factor-κB-related inflammation (tumor necrosis factor-α [TNF-α]: MDL28170 vs. vehicle group, 1.15 ± 0.07 and 1.62 ± 0.08 vs. 1.59 ± 0.10 and 2.18 ± 0.10, both P < 0.001; inducible nitric oxide synthase: MDL28170 vs. vehicle group, 4.51 ± 0.23 vs. 6.23 ± 0.12, P < 0.001 at 24 h; intracellular adhesion molecule-1: MDL28170 vs. vehicle group, 1.45 ± 0.13 vs. 1.70 ± 0.12, P < 0.01 at 24 h) and lessened both myeloperoxidase activity (MDL28170 vs. vehicle group, 0.016 ± 0.001 and 0.016 ± 0.001 vs. 0.024 ± 0.001 and 0.023 ± 0.001, P < 0.001 and 0.01, respectively) and matrix metalloproteinase-9 (MMP-9) levels (MDL28170 vs. vehicle group, 0.87 ± 0.13 and 1.10 ± 0.10 vs. 1.17 ± 0.13 and 1.25 ± 0.12, P < 0.001 and 0.05, respectively) at 6 h and 24 h after CCI.
Conclusions:
These findings demonstrate that MDL28170 can protect the structure of the NVU by inhibiting the inflammatory cascade, reducing the expression of MMP-9, and supporting the integrity of TJ during acute TBI.
Keywords: Calpain, Inflammation, Neurovascular Unit, Nuclear Factor-κB, Traumatic Brain Injury
Introduction
The neurovascular unit (NVU) is considered a structural and functional unit of the nervous system. The structural basis of the NVU includes neurons, astrocytes, brain endothelial cells that secrete basal lamina matrix, pericytes, vascular smooth muscle cells, microglia, and oligodendrocytes.[1,2,3] The NVU regulates the nutrient supply to the central nervous system (CNS) and prevents the passage of harmful substances from the intravascular space into the brain.[4] The blood-brain barrier (BBB) is composed of endothelium, the basal lamina matrix, and astrocyte end-feet and regulates the entry of certain molecules from the blood into the CNS.[5,6] Disruption of the NVU occurs in a number of pathophysiological processes including cerebral ischemia, traumatic brain injury (TBI), CNS infections, and multiple sclerosis and results in the development of brain edema, which is a frequent cause of mortality in patients with such conditions.[5,7,8] Thus, understanding the pathophysiological mechanism leading to NVU disruption and increased BBB permeability is crucial for the development of therapeutic strategies for these conditions.
Nuclear factor-κB (NF-κB), a pivotal transcription factor, is a cytoplasmic complex composed of hetero- and homo-dimmers from the Rel/NF-κB family of proteins. It is essential for immune and stress responses within the brain. These dimmers are complexed to a member of the inhibitory-κB (IκB), which inhibit the nuclear localization signal of NF-κB.[9] In pathology situation, active NF-κB translocates into the nucleus and stimulates the transcriptional activation of pro-inflammatory genes, such as tumor necrosis factor-α (TNF-α), intracellular adhesion molecule-1 (ICAM-1), inducible nitric oxide synthase (iNOS), and matrix metalloproteinase-9 (MMP-9).[9,10] Once activated, inflammatory cells can release a variety of cytotoxic agents including cytokines, MMPs, and nitric oxide, as well as other reactive oxygen species.[11] NF-κB activation has been shown to occur after experimental TBI in rodents; in addition, inhibiting the NF-κB signaling pathway through pharmacological and genetic approaches has been reported to be neuroprotective in experimental TBI models.[12,13,14] A growing body of literature has emerged indicating that NF-κB-related inflammation is involved in not only neurobehavioral deficits but also BBB disruption and apoptotic cell death.[7,14] NF-κB-driven transcription of pro-inflammatory cytokines and the secreted protease MMP-9 have both been implicated in NVU disruption and increased in BBB permeability in response to TBI and ischemic injury.[15,16] These data suggest that the NF-κB-inflammation signaling cascade is involved in the NVU degradation that occurs in TBI.
Calpains are calcium-dependent cysteine protease that contributes to necrotic and apoptotic cell death. They are activated by calcium and are regulated reversibly by calpastatin, which is an endogenous calpain inhibitor. Calpains are proposed to participate in the turnover of cytoskeletal proteins as well as regulation of kinases, membrane receptors, and many transcription factors, including NF-κB.[17] During TBI, calpains are activated and involved in necrotic and apoptotic neuron death, which has been demonstrated through calpain inhibition and calpain gene inactivation.[17,18,19] Calpastatin has been shown to be induced by TBI in rats.[20] Overexpression of calpastatin has been shown to protect the brain against TBI.[21] Moreover, it has been determined that calpain inhibitors reduce the proteasomal degradation of IκB and hence inhibit the translocation of NF-κB from the cytosol into the nucleus and the NF-κB-driven transcription of pro-inflammatory cytokines, chemotactic factors, and MMPs in a variety of disease models.[22,23,24,25]
Therefore, we suspect that calpain may influence traumatic NVU damage through upregulating NF-κB-related inflammation. In the present study, we investigated the mechanisms of calpain activation, leading to the increased NVU permeability observed after TBI. We examined both functional and structural changes related to the NVU disturbance after controlled cortical impact (CCI), a well-established experimental TBI model, as well as the effects of the administration of the calpain inhibitor, MDL28170, on the changes observed in mice.
Methods
Animals
One hundred and eight male BALB/c mice, weighing 25–30 g (Beijing Vital River Experimental Animals Technology, Ltd., Beijing, China), were used in this study. The animals were maintained in a controlled environment at a temperature of 23 ± 2°C on a 12-h light/dark cycle with access to food and water ad libitum. The study was performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee.
Controlled cortical impact
The mice were anesthetized by isoflurane inhalation and were maintained at 37.0 ± 0.5°C by a thermal mat throughout the surgical procedure. CCI was produced in the mice using a PCI3000 PinPoint Precision Cortical Impactor (Hatteras Instruments, Cary, NC, USA). After the skull was exposed with a central skin incision and soft tissue was removed with a cotton tip, a circular craniotomy approximately 4 mm in diameter was made in the middle of the right parietal bone, approximately 0.5 mm from the sagittal, coronal, and lambdoid sutures, while leaving the dura intact. The CCI parameters were as follows: impact tip diameter, 3 mm; velocity, 2 m/s; compression time, 85 ms; and compression distance, 1 mm.[26] According to these impact parameters, we established a model of moderate injury. The impact tip was wiped clean with alcohol after each impact. Sham-operated mice underwent the same procedure without percussion. After surgery, the incision was closed with nylon sutures, and 2% lidocaine jelly was applied to the lesion site to minimize discomfort.[27]
Experimental protocols
Referring to the doses of MDL28170 used in experimental TBI, 20 mg/kg of MDL28170 (Millipore Co., Billerica, MA, USA; dissolved in 0.9% NaCl) was used intraperitoneally (i.p.) 5 min, 3 h, and 6 h after injury in this study.[28,29] One hundred and eight mice were used in this study. To evaluate the effects of MDL28170 on neuron death and NVU deficits, the mice were randomly assigned to the following groups and were treated with either MDL28170 or vehicle: (1) sham: vehicle (n = 6); (2) CCI 24 h: vehicle (n = 6); and (3) CCI 24 h + MDL28170: 20 mg/kg of MDL28170 (n = 6). The mice were decapitated 24 h after CCI; then, the ultrastructure and histopathology were examined. Neurological severity score (NSS), contusion volume, Evans blue leakage, brain water content, calpain and myeloperoxidase (MPO) activity, and the levels of calpastatin, NF-κB, IκB, TNF-α, ICAM-1, iNOS, MMP-9, occludin, and the extracellular matrix (ECM) proteins laminin were all assessed by randomly assigning the mice into the following groups to be treated with MDL28170 or vehicle: (1) sham: vehicle (n = 18); (2) CCI 6 h and CCI 24 h: vehicle (n = 36); and (3) CCI 6 h + MDL28170 and CCI 24 h + MDL28170: 20 mg/kg of MDL28170 (n = 36). The mice were decapitated, and the injured brain tissues of right parietal lobe were dissected at 6 h or 24 h after CCI (CCI 6 h or CCI 24 h) in the vehicle- or MDL28170-treated mice. The regions from the right hemisphere that corresponded to the injured brain tissues were dissected at 24 h after surgery in the sham-operated mice. The mice sacrificed at 6 h after CCI received two doses of MDL28170 or its vehicle (5 min and 3 h after injury), whereas the mice sacrificed at 24 h received three doses of MDL28170 or vehicle (5 min, 3 h, and 6 h after injury). The cytosolic, mitochondrial, and nuclear fractions were prepared and used to determine the activities of calpain and MPO and the levels of calpastatin, NF-κB, IκB, TNF-α, ICAM-1, iNOS, MMP-9, occludin, and laminin.
Sample collection and preparation
As has been previously described by Tao et al., the tissues of right parietal lobe were dissected according to the experimental protocols at 4°C. Briefly, animals were sacrificed with an overdose of chloral hydrate (600 mg/kg, i.p.) and brains were removed. An MPO activity assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) was used to determine MPO activity according to the manufacturer's instructions; the tissue samples were weighed, homogenized, and used to measure MPO activity. For determining calpain activity and for the Western blotting analysis, protein samples (n = 6 per group) were prepared by the method used in our previous studies.[14] The protein concentrations in the cytosolic, nuclear, and mitochondrial fractions were determined using a bicinchoninic acid assay.
Calpain activity assay
As has been previously described, the calpain activity assay was performed using fluorescent calpain I substrate.[30] In brief, cytosolic and mitochondrial proteins (30 μg) were incubated with calpain reaction buffer (20 mmol/L HEPES, 1 mmol/L EDTA, 50 mmol/L NaCl, and 0.1% (v/v) 2-mercaptoethanol, containing 10 μmol/L calpain I fluorescent substrate [Calbiochem Co., La Jolla, CA, USA], pH 7.6). The reaction was initiated by the addition of CaCl2 (final concentration of 5 μmol/L) and the mixture was incubated at 37°C for 30 min. The quantification of the substrate cleavage resulted in the release of free 7-amino-4-methylcoumarin (AMC; Millipore Co., Billerica, MA, USA), which was then detected using a microplate reader (Infinite® M200 Pro; Tecan Co., Männedorf, Switzerland) set at excitation and emission wavelengths of 335 nm and 500 nm, respectively. Fluorescence arbitrary units were converted into micromoles of AMC released per hour and milligrams of protein using a standard curve of free AMC (Millipore Co.).
Myeloperoxidase activity assay
The injured region was dissected according to the experimental protocols at 4°C. The brain was weighted and homogenated using an MPO activity assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), and the MPO activity was determined. One unit (U) of MPO activity was defined as the amount that degrades 1 μmol hydrogen peroxide at 37°C and was normalized to the wet tissue weight (U/g wet tissue).
Western blot analysis
Calpastatin, NF-κB, IκB, TNF-α, ICAM-1, iNOS, MMP-9, occludin, and laminin were determined from cytosolic and nuclear fraction separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as previously described.[31] Equal amounts of proteins (50 μg) were separated by SDS-PAGE, and the proteins on the gel were subsequently transferred onto a polyvinylidene fluoride membrane. Blots were probed with antibodies reactive with calpastatin (1:1000; Abcam Co., Cambridge, MA, USA), NF-κB (1:1000; Millipore Co.), IκB (1:500; Millipore Co.), TNF-α (1:400; R&D Systems, Inc., Minneapolis, MN, USA), ICAM-1 (1:400; R&D Systems, Inc.), iNOS (1:500; Millipore Co.), MMP-9 (1:1000; Millipore Co.), occludin (1:400; Invitrogen, Inc., Minneapolis, MN, USA), and laminin (1:400; Sigma-Aldrich, Inc.,) and were subsequently incubated with secondary antibody conjugated with horseradish. Finally, the antibody binding was visualized by enhanced chemiluminescence (ECL) (BioSpectrum 500 Imaging System; UVP Co., Upland, CA, USA). The membrane was then washed and probed with antibodies reactive with β-actin (1:1000; Millipore Co.) or histone H3 (1:1000; Millipore Co.), which were used as internal controls for the cytosolic and nuclear fractions, respectively. After the membrane was washed in TBST, the protein bands were visualized using an ECL detection kit. The relative band intensities were quantified by densitometric analysis. The densitometric plots of the results were normalized to the intensity of the actin or histone H3 band. The optical density of the intensity of the bands was performed using the Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Neurobehavioral evaluation
Animals were examined for neurological deficits at 6 h and 24 h after CCI by an investigator who was blinded to the treatment conditions. Neurological function was measured in terms of the NSS, a 21-point scale that assesses the motor, sensory (visual, tactile, and proprioceptive), beam balance, beam walking, and reflex tests. Table 1 shows a set of the modified NSS.[14,32,33]
Table 1.
Neurological severity score of mice after neurotrauma
| Motor tests | Score |
|---|---|
| Raising rat by the tail | |
| Flexion of forelimb | 1 |
| Flexion of hind limb | 1 |
| Head moved 10° to vertical axis within 30 s | 1 |
| Placing rat on the floor (normal = 0; maximum = 3) | |
| Normal walk | 0 |
| Inability to walk straight | 1 |
| Circling toward the paretic side | 2 |
| Fall to the paretic side | 3 |
| Sensory tests | |
| Placing test (visual and tactile test) | 1 |
| Proprioceptive test (deep sensation, pushing the paw against the table edge to stimulate limb muscles) | 1 |
| Beam balance tests (normal = 0; maximum = 6) | |
| Balances with steady posture | 0 |
| Grasps side of beam | 1 |
| Hugs the beam and one limb falls from the beam | 2 |
| Hugs the beam and two limbs fall from the beam, or spins on beam(>60 s) | 3 |
| Attempts to balance on the beam but falls off (>40 s) | 4 |
| Attempts to balance on the beam but falls off (>20 s) | 5 |
| Falls off: No attempt to balance or hang on to the beam (<20 s) | 6 |
| Beam walking tests | |
| Inability to walk on a 1 cm wide beam | 1 |
| Inability to walk on a 2 cm wide beam | 2 |
| Inability to walk on a 3 cm wide beam | 3 |
| Reflexes absent and abnormal movements | |
| Pinna reflex (head shake when touching the auditory meatus) | 1 |
| Corneal reflex (eye blink when lightly touching the cornea with cotton) | 1 |
| Startle reflex (motor response to a brief noise from snapping a clipboard paper) | 1 |
| Seizures, myoclonus, myodystony | 1 |
| Maximum points | 21 |
One point is awarded for the inability to perform the tasks or for the lack of a tested reflex; 13–18, severe injury; 7–12, moderate injury; 1–6, mild injury.
Measurement of cerebral contusion volume
The mice were anesthetized by with chloral hydrate (400 mg/kg, i.p.) and perfusion-fixed with 4% paraformaldehyde at 6 h and 24 h after CCI. The brains were then removed and immersed in the same paraformaldehyde for 1 day. Coronal sections, 5 μm thick, were cut with a cryostat. The sections were stained with hematoxylin and eosin and examined using a light microscopy. Images were then captured using a digital camera. Since brain edema might affect the accuracy of contusion estimation, the corrected infarct volume was calculated. The contusion volume in every experimental mouse was calculated as sum of the contusion areas × 0.3 mm3, as previously described.[7] All contusion area analyses were performed by an independent investigator who was blinded to the treatment status of the animals.
Evaluation of blood–brain barrier integrity
Evans blue dye (EBD) leakage was measured 6 h and 24 h after CCI to assess the BBB permeability (n = 8). According to a previous study, EBD (2% in saline) was injected intravenously (4 mg/kg) 4 h and 22 h after CCI and allowed to circulate for 2 h.[34] The animals were then perfused with saline until clear perfusion fluid was obtained from the right atrium. The tissue levels of EBD were assessed using a multifunctional microplate reader at excitation and emission wavelengths of 620 nm and 680 nm, respectively. The levels of EBD in the hemispheres were expressed as μg/mg dry weight.
Measurement of water content
The water content of the brain was measured using the wet–dry weight method as the procedures described in our previous paper.[14] In brief, at 6 h and 24 h after CCI, mice (n = 6) were sacrificed with an overdose of chloral hydrate (600 mg/kg, i.p.). The right hemisphere was dissected. The sample was first weighed to determine the wet weight and then dried in an incubator at 120°C for 24 h and reweighed (dry weight). Tissue water content (%) was calculated using the formula: brain water content = ([wet weight − dry weight]/wet weight) × 100%.
Electron microscopy analysis
Sample processing and electron microscopy (EM) were conducted as previously described.[35] In brief, anesthetized animals were perfused, and brain tissue samples were taken to produce sections. The sections were postfixed with 2.5% glutaraldehyde for 2 h, washed with 0.1 M phosphate-buffered saline, and then exposed to 1% osmium tetroxide for 2 h. Subsequently, the sections were washed several times with water, dehydrated using an alcohol gradient, and embedded in Epon resin. Randomly selected, ultrathin sections were stained with uranyl acetate and lead citrate and then examined using a transmission electron microscope (H-7650, HITACHI, Tokyo, Japan).
Histopathological analysis
The cresyl violet staining was used to assess the normal neurons and damaged neurons. Animals were anesthetized with chloral hydrate and perfused with saline followed by 4% paraformaldehyde 24 h after CCI. Brains were removed, fixed, embedded in paraffin, and the 5 mm thick coronal sections were collected. Cresyl violet was performed following the procedures described in our previous studies.[14] The sections were observed using a light microscope and pictures were taken with a digital camera. The evaluations of the necrotic neurons in cresyl violet-stained sections were determined.
Statistical analysis
The data are presented as the mean ± standard deviation (SD). The comparisons between the groups were evaluated using one-way analysis of variance (ANOVA) with Fisher's post hoc test (the activities of calpain and MPO and the levels of calpastatin, NF-κB, IκB, TNF-a, ICAM-1, iNOS, MMP-9, occludin, and laminin) or Kruskal–Wallis ANOVA (NSS, contusion volume, EBD, water content, and necrotic and apoptotic cell death). The level of statistical significance was set at P < 0.05.
Results
Effects of MDL28170 on calpain activity in cytosolic and mitochondrial fractions
The calpain activity assay was performed using fluorescent calpain I substrate in the cytosolic and mitochondrial fractions at 6 h and 24 h after CCI. As shown in Figure 1a, the MDL28170 treatment markedly reduced calpain activity in the cytosolic fractions at 6 h and 24 h after CCI (P < 0.05 and 0.001 compared with the vehicle group, respectively). Figure 1b illustrates that the MDL28170 treatment markedly reduced calpain activity in the mitochondrial fractions at 6 h and 24 h after CCI (P < 0.001 compared with the vehicle group).
Figure 1.

MDL28170 treatment suppresses the calpain activity in the cytosolic and mitochondrial fractions and upregulates the expression of calpastatin in the cytosolic fractions. (a and b) The bar graphs reflect the calpain activity in the cytosolic fractions and mitochondrial fractions at 6 h and 24 h. (c) Representative Western blots of calpastatin and β-actin from each group; (d) the results were quantified and are shown as the mean ± SD. *P < 0.05 compared with the sham group. †P < 0.05 compared with the vehicle group. SD: Standard deviation.
Effects of MDL28170 on calpastatin levels
The calpastatin protein levels in the cytosolic fractions were determined by Western blot analysis. As illustrated in Figure 1c and 1d, the MDL28170 treatment significantly enhanced the calpastatin protein levels at 6 and 24 h after CCI (P < 0.01 and 0.05 compared with the vehicle group, respectively).
Effects of MDL28170 on nuclear factor-κB levels in the cytosolic and nuclear fractions
We assayed the levels of NF-κB in the cytosolic and nuclear fractions to determine the extent of NF-κB activation as shown in Figure 2. The MDL28170 treatment markedly reduced the levels of NF-κB in the cytosolic fractions at 24 h after CCI and in the nuclear fractions at both 6 h and 24 h after CCI (P < 0.05, 0.001, and 0.001 compared with the vehicle group, respectively). In addition, although there were differences between the MDL28170- and vehicle-treated groups in the NF-κB levels of the cytosolic fractions at 6 h after CCI, this did not reach statistical significant. These data confirmed that MDL28170 suppressed the induction and activation of NF-κB in traumatic brain tissue after CCI.
Figure 2.

MDL28170 specifically inhibits the expression of NF-κB in the cytosolic and nuclear fractions and enhances the protein levels of IκB in the cytosolic fractions. (a, b, and c) the results were quantified and are shown as the mean ± SD, normalized to the ratio of the NF-κB and IκB band to β-actin or histone band of the sham group. *P < 0.05 compared with the sham group. †P < 0.05 compared with the vehicle group. NF-κB: Nuclear factor-κB; IκB: Inhibitory-κB; SD: Standard deviation.
Effects of MDL28170 on inhibitory-κB levels in the cytosolic fractions
IκB, an endogenous inhibitor protein of NF-κB, exists in the cytoplasm as a complex with NF-κB. Thus, the levels of IκB in the cytosolic fractions were determined by Western blot analysis, and the results of which are illustrated in Figure 2. MDL28170 markedly enhanced the levels of IκB at 6 h and 24 h after CCI (both P < 0.001 compared with the vehicle group).
Effects of MDL28170 on inflammatory mediator levels
Western blot analysis was performed to determine the levels of TNF-α, iNOS, and ICAM-1 in traumatic brain tissues at 6 h and 24 h after CCI. The protein levels of TNF-α, iNOS, and ICAM-1 increased significantly at 6 h and 24 h after CCI compared with those in the sham-operated mice [Figure 3a, 3b, and 3c; all P < 0.001]. Compared with the vehicle-treated mice, the MDL28170-treated mice markedly reduced TNF-α protein levels at 6 h and 24 h after CCI (MDL28170 vs. vehicle group, 1.15 ± 0.07 and 1.62 ± 0.08 vs. 1.59 ± 0.10 and 2.18 ± 0.10) (both P < 0.001), as well as iNOS and ICAM-1 levels at 24 h after CCI iNOS [4.51 ± 0.23 vs. 6.23 ± 0.12; ICAM-1, 1.45 ± 0.13 vs. 1.7 ± 0.12; (P < 0.001 and 0.01, respectively)]. In addition, although the MDL28170 treatment reduced the levels of iNOS and ICAM-1 at 6 h after CCI compared with the vehicle treatment, this did not reach statistical significant.
Figure 3.

MDL28170 effectively inhibits the expression of TNF-α, iNOS and ICAM-1 and MPO activity. (a, b, and c) the results were quantified and are shown as the mean ± SD, normalized to the ratio of the TNF-α, iNOS, and ICAM-1 band to the β-actin band of the sham group as unity. (d) The bar graphs reflect MPO activity in the injured hemisphere at 6 h and 24 h after CCI. *P < 0.05 compared with the sham group. †P < 0.05 compared with the vehicle group. TNF-α: Tumor necrosis factor-α; iNOS: Inducible nitric oxide synthase; ICAM-1: Intracellular adhesion molecule-1; MPO: Myeloperoxidase; CCI: Controlled cortical impact; SD: Standard deviation.
Effects of MDL28170 on myeloperoxidase activity
We measured the activity of MPO to determine the extent of neutrophil infiltration into the injured hemisphere. As shown in Figure 3d, the MDL28170 treatment markedly reduced MPO activity at 6 h and 24 h after CCI (MDL28170 vs. vehicle group, 0.016 ± 0.001 and 0.016 ± 0.001 vs. 0.024 ± 0.0.001 and 0.023 ± 0.001) (P < 0.001 and 0.01 compared with the vehicle group, respectively).
Effects of MDL28170 on matrix metalloproteinase-9, occludin, and laminin levels
Western blot analysis was used to assay the levels of MMP-9, occludin, and laminin in traumatic brain tissues at 6 h and 24 h after CCI. The representative protein bands are displayed in Figure 4a, 4c, and 4e. The MMP-9 protein levels increased significantly at 6 h and 24 h after CCI compared with those in the sham-operated mice [Figure 4b; MDL28170 vs. vehicle group, 0.87 ± 0.13 and 1.1 ± 0.13 vs. 1.17 ± 0.13 and 1.25 ± 0.12 P < 0.01]. However, the protein levels of occludin and laminin decreased significantly at 6 h and 24 h after CCI compared with those in the sham-operated mice [Figure 4d and 4f; P < 0.001]. Compared with the vehicle treatment, the MDL28170 treatment markedly reduced the levels of MMP-9 and enhanced the levels of occludin and laminin at 6 h and 24 h after CCI (MMP-9: P < 0.001 and 0.05, respectively; occludin: both P < 0.05; laminin: both P < 0.01).
Figure 4.

The effects of MDL28170 on the levels of MMP-9, occludin, laminin, and TJ. (a, c, and e) Representative Western blots of MMP-9, occludin, laminin, and β-actin; (b, d, and f) the results of the MMP-9, occludin, and laminin band were quantified and are shown as the mean ± SD. *P < 0.05 compared with the sham group. †P < 0.05 compared with the vehicle group. (g) Representative electron micrographs showing the TJ and basement membrane (original magnification: ×80,000 or ×40,000). The TJ is indicated by black arrowheads. The gray arrowhead indicates the damaged structure of the basement membrane. MMP-9: Matrix metalloproteinase-9; TJ: Tight junction.
Effects of MDL28170 on neurological defects, cerebral contusion volume, and blood–brain barrier integrity
In the present study, we used an NSS to evaluate the neurological deficits of the mice. The vehicle-treated mice existed obvious neurologic deficits at 6 h and 24 h (MDL28170 vs. vehicle group, 7.5 ± 0.45 and 6.33 ± 0.38 vs. 12.33 ± 0.48 and 11.67 ± 0.48). However, the MDL28170 treatment markedly reduced the NSS at 6 h and 24 h after CCI ([Figure 5a]; P < 0.001 compared with the sham group; P < 0.001 compared with the vehicle group). The CCI caused a focal lesion in the cortex at, around, and beneath the impact site. The contusion volume was 16.90 ± 1.01 mm2 and 17.20 ± 1.17 mm2 at 6 h and 24 h after CCI in the vehicle-treated mice [Figure 5b] and was significantly reduced by the MDL28170 treatment (9.30 ± 1.05 mm2 and 9.90 ± 1.17 mm2; both P < 0.001). Disruption of BBB integrity was reflected by EBD leakage and cerebral edema. Figure 5c and 5d depicts the EBD content (μg/mg dry weight) and water content (%) at 6 h and 24 h after CCI in the sham-, vehicle-, and MDL28170-treated mice. The MDL28170 treatment significantly reduced the EBD and water contents at 6 h and 24 h after CCI in the injured hemisphere (EBD, 428.13 ± 61.99 and 655.62 ± 76.68 vs. 966.96 ± 106.64 and 1202.213 ± 108.90; edema, 80.76% ± 1.25% and 82% ± 1.84% vs. 82.55% ± 1.32% and 83.64% ± 1.25%; P < 0.001 and 0.05 compared with the vehicle group, respectively).
Figure 5.

MDL28170 decreases the NSS and contusion volume after CCI in mice and lessens the EBD and water contents of the injured tissue. Vehicle or MDL28170 was injected i.p. at 5 min, 3 h, and 6 h after CCI. The data are expressed as the mean ± SD; n = 8 per group. (a) Effects of MDL28170 on NSS. (b) The bar graph presents the contusion volumes in each group determined by H and E staining. (c) EBD content. (d) Water content. *P <0.05 compared with the sham group. †P < 0.05 compared with the vehicle group. NSS: Neurological severity score; EBD: Evans blue dye; CCI: Controlled cortical impact; SD: Standard deviation.
Effects of MDL28170 on neurovascular unit ultrastructural changes and blood–brain barrier permeability
The EM images clearly showed components of the BBB, including endothelial cells, basal membrane, and astrocytic foot processes. In the vehicle-treated mice, the electron-dense tight junction (TJ) between the capillary endothelial cells and the basement membrane was deformed and the gap junctions were also observed in the intercellular clefts. The MDL28170 treatment markedly protected the normal structures of the TJ and the basement membrane after CCI [Figure 4g]. Figure 6 shows representative images of the BBB, neuron death, and brain edema after CCI in the sham-, vehicle-, and MDL28170-treated mice. The astrocyte end-feet in the ischemic brain regions in the vehicle-treated mice was observed varying degrees of swelling at 24 h after CCI [Figure 6a]. In many of these cases, the intracellular organelles were absent or scarce as indicated in Figure 6a-6c.
Figure 6.

MDL28170 effectively decreases BBB destruction, neuron death and brain edema. Electron micrographs showing the BBB, neurons, and brain edema in an injured cortex from each group. (a) Representative images of the BBB in animals from all groups. Vascular deformation and swelling of astrocytic foot processes are indicated by black arrowheads. (b) Representative images of neuron death. The gray arrowhead indicates structural neuron damage. (c) Representative images of brain edema. Scale bar = 5.0 μm. The red arrowhead indicates an area of edema. BBB: Blood–brain barrier.
Effects of MDL28170 on necrotic cell death
The results of cresyl violet staining are illustrated in Figure 7. The neurons in the sham-operated mice exhibited intact morphology. In contrast, most neurons in the cortical lesions of the vehicle-treated mice exhibited some or all of the following features: Nissl bodies’ reduction, chromatolysis, nuclear pyknosis, eosinophilic cytoplasm, or a lack of cellular structure. The necrotic cell death scores were significantly lower in the MDL28170-treated mice than in the vehicle-treated mice [Figure 7; P < 0.01].
Figure 7.
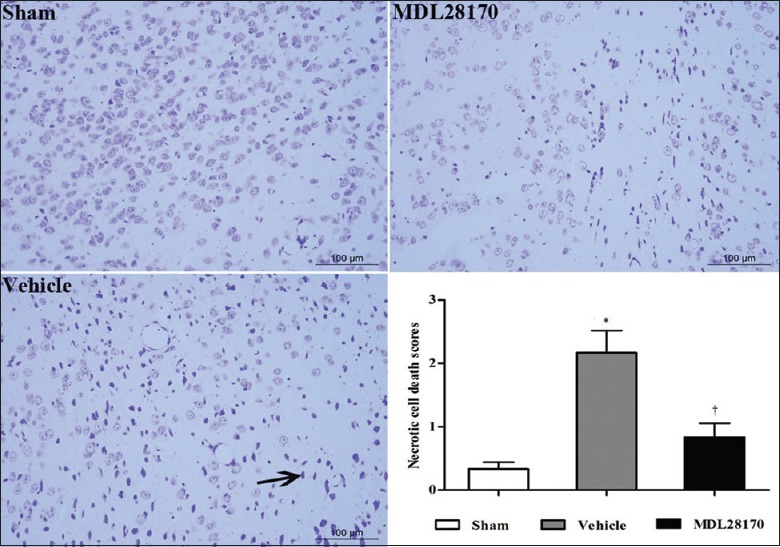
MDL28170 decreases the necrotic cell death. Representative images of necrotic cell death as shown by cresyl violet staining. Black arrowheads indicate reduced Nissl bodies or cellular structure as well as increased leukocyte infiltration. The bar graph presents the necrotic cell death score; n = 8. *P < 0.05 compared with the sham group. †P <0.05 compared with the vehicle group.
Discussion
In this study, we found that MDL28170, an efficient calpain inhibitor, reduced BBB permeability and brain edema, improved neurological functions, and attenuated cell death in the mouse model of CCI. Moreover, MDL28170 suppressed the over-activation of calpain, enhanced the levels of calpastatin and IκB, lessened the nuclear translocation of NF-κB, reduced the production of TNF-α, iNOS, ICAM-1, and MMP-9, lessened the degradation of endothelial TJ proteins and ECM proteins, and decreased the leukocyte infiltration into the brain. These data indicate that MDL28170 may be an important therapeutic drug for protecting of the NVU against TBI insults through downregulating NF-κB-related inflammation [Figure 8].
Figure 8.
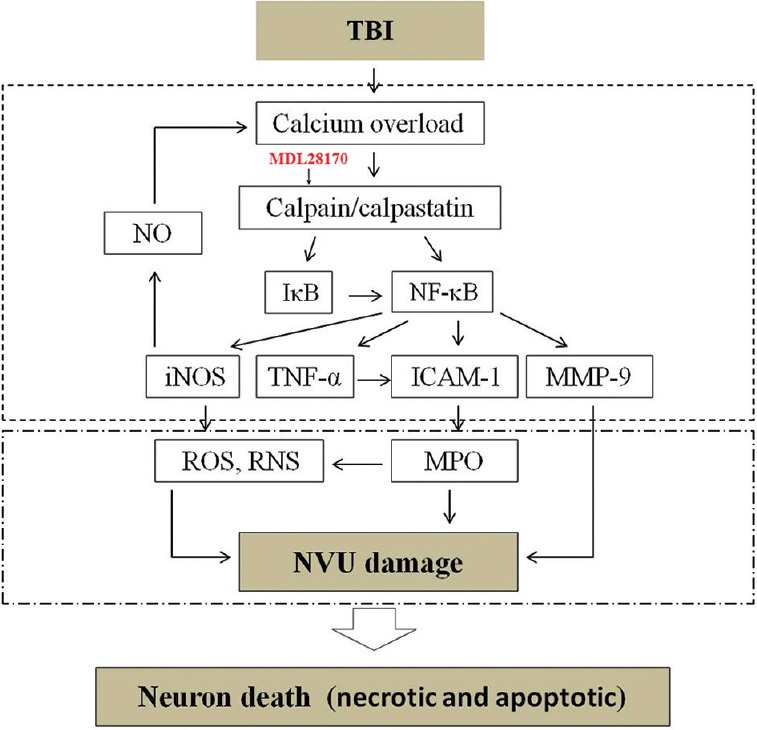
Schematic diagram showing the relationship of calpain/NF-κB/inflammation/NVU damage after CCI in mice. Traumatic brain injury induces calcium overload, which, in turn, upregulates calpain. Calpain may downregulate IκB and activate NF-κB. NF-κB induces activation of TNF-α, iNOS, ICAM-1, and MMP-9. These inflammatory substances induce degradation of basal lamina and tight junction proteins, resulting in NVU disruption, leading to brain edema. MDL28170 could reverse those changes. NF-κB: Nuclear factor-κB; NVU: Neurovascular unit; CCI: Controlled cortical impact; IκB: Inhibitory-κB; TNF-α: Tumor necrosis factor-α; iNOS: Inducible nitric oxide synthase; ICAM-1: Intracellular adhesion molecule-1; MMP-9: Matrix metalloproteinase-9.
The inflammatory responses have been reported to be a crucial mechanism in the second injury after TBI. The early responses of the inflammatory reactive cells result in a conspicuous accumulation of other inflammatory mediators such as cytokines and adhesion molecules.[7,36,37,38] TNF-α is one of the most important inflammatory factors of the body, secreted by various cell types. TNF-α has multiple pro-inflammatory functions and thus plays an important role in experimental TBI.[39] ICAM-1 is an adhesive molecule that can be produced by vascular endothelial cells and neutrophils after TBI. Moreover, iNOS can be secreted by inflammatory cells and vascular endothelial cells, which may produce large amount of harmful nitric oxide after TBI.[38] The significant neuroprotection that has been observed after neutrophils, cytokines, or iNOS inhibition demonstrates that these inflammatory elements exacerbate TBI.[40,41]
Pathophysiologic responses in the brain after TBI are extremely complex and affect multiple regions and cell types. All cellular and structural components of the NVU need to be rescued after TBI to reduce the resulting brain damage.[42] Various matrix proteases play key roles in the BBB perturbations that predominate during the early phase of neurovascular injury. In addition, recent studies have suggested that the manifestations of brain function and dysfunction begin at the level of cell–cell signaling between neuronal, glial, and vascular elements.[43] Thus, protecting neurons alone may not be an effective therapeutic approach for TBI and other neurological disorders. Inflammatory responses have long been known to disrupt BBB permeability.[44,45] In parallel with this knowledge, our results revealed that downregulating NF-κB-related inflammation markedly ameliorated ultrastructural NVU damage (including neuronal, glial, and BBB damage), reduced cerebral contusion volume, and improved neurological function in mice after TBI.
The BBB is mainly composed of capillary endothelial cells; TJ presents between endothelial cells and astrocytic foot processes. Of these, the TJ and basal membrane are the most important structures for maintaining the integrity of the BBB. The amount of EBD present in brain tissue is commonly used to evaluate BBB permeability. The hyperpermeability of the BBB and its preservation with MDL28170 treatment at 6 h and 24 h after CCI are directly demonstrated by the observed changes in EBD content and TJ proteins. MMPs belong to a zinc-dependent protease family and are capable of degrading basement membrane proteins as well as disrupting TJ assemblies, thus enhancing BBB permeability.[44] MMP-9 is one of the most important members of all MMP family in the brain tissue.[46] In accordance with our results, several investigators have reported that MMP-9 could degrade the TJ proteins and ECM proteins, such as occludin and laminin, disrupt the permeability of the BBB, and lead to vasogenic brain edema.[44,47]
Calpains are recognized as multifunctional enzymes that are involved in cell mitosis, migration, and differentiation. A lot of studies have suggested an important role of calpain in death of neurons after acute TBI. Bains et al. demonstrated in 2013 that administration of calpain inhibitor SNJ-1945 15min after injury reduced degradation of cytoskeleton.[48] Bralic and Stemberga reported in 2012 that sustained activation of calpain led to NVU injury after TBI.[49] Karklin Fontana et al. reported in 2016 that calpain inhibitor MS153 protected against TBI, possibly through inhibition of Ca2+ channel.[50] In addition, a total of three studies have applied calpain inhibitor MDL28170 in the treatment of TBI. Buki et al. found that injecting MDL28170 before TBI alleviated traumatic axonal injury, which proved potential efficacy of MDL28170 for TBI.[51] Ai et al. reported in 2007 that MDL28170 had certain therapeutic effect for TBI-caused injury of corpus callosum.[52] Thompson et al. proved the efficacy of MDL28170 for TBI using a mouse model of CCI. The results of these studies suggest that MDL28170 can reduce death of neurons but had no effect in reducing injured volume.[28] A growing number of studies suggest that calpains could participate in acute and chronic inflammatory processes under pathological conditions by acting as inflammatory regulators; in particular, calpains can indirectly promote NF-κB-mediated gene transcription through mediating proteolysis of the NF-κB-binding proteins, IκB.[24,25] Therefore, we focused on the action of calpain on NF-κB signaling during TBI, especially on the expression of NF-kB-driven MMP-9 genes. NF-κB-driven transcription of MMP-9 is induced during TBI, which has been demonstrated in our previous studies. These data suggest the involvement of calpain/NF-κB signaling in MMP-9 activation during TBI. Our data show that MMP-9 expression was markedly increased at 6 h and 24 h after CCI; in addition, MDL28170 reduced this MMP-9 increase as well as NF-κB activity.
The increased BBB permeability and NVU dysfunction at 6 h to 24 h were related to other events that damaged the vascular endothelium cell. We found that ICAM-1 expression and MPO activity were increased at these time points in our model. Both increased ICAM-1 expression and augmented MPO activity have been shown to be reliable estimates of the extent of neutrophil infiltration into the brain.[53] Administering antibodies against ICAM-1 or using antibodies to deplete neutrophils in the blood has been shown to protect the brain and BBB in different experimental models.[54,55] Recent studies have also shown the importance of neutrophil-derived MMP-9 in an experimental stroke model.[56] In our experiments, MDL28170 decreased ICAM-1 expression and MPO levels, reduced MMP-9 expression, and ultimately protected BBB permeability. These results are consistent with those of other studies, which showed that calpain inhibition protected against leukocyte infiltration in a variety of organs.[57,58]
Based on our results, we speculate that the NVU protection observed after TBI is an effect exerted directly on the cerebral endothelium. This view is supported by the fact that the attenuation of BBB permeability caused by MDL28170 was associated with decreased ICAM-1 and MPO expression, as well as preserved occludin and ECM levels, all of which are endothelial cell markers. One limitation of this study is the lack of data excluding the contribution of effects on damaged glia and immune cells.
In summary, this study shows that calpain inhibition by MDL28170 reduces NVU disruption and brain edema in the mouse model of CCI. Moreover, MDL28170 suppresses the nuclear translocation of NF-κB and reduces the production of inflammatory factors, as well as the degradation of TJ and the ECM proteins. Our data suggest the involvement of calpain-mediated inflammation in the enhancement of BBB permeability during TBI as such calpain inhibition may be a potential therapeutic strategy for preserving the integrity of the NVU in acute TBI.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Min Chen
References
- 1.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–62. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo EH, Rosenberg GA. The neurovascular unit in health and disease:Introduction. Stroke. 2009;40(3 Suppl):S2–3. doi: 10.1161/STROKEAHA.108.534404. doi:10.1161/STROKEAHA.108.534404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacoste B, Gu C. Control of cerebrovascular patterning by neural activity during postnatal development. Mech Dev. 2015;138(Pt 1):43–9. doi: 10.1016/j.mod.2015.06.003. doi:10.1016/j.mod.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderón-Garcidueñas L, Reynoso-Robles R, Vargas-Martínez J, Gómez-Maqueo-Chew A, Pérez-Guillé B, Mukherjee PS, et al. Prefrontal white matter pathology in air pollution exposed Mexico City young urbanites and their potential impact on neurovascular unit dysfunction and the development of Alzheimer's disease. Environ Res. 2016;146:404–17. doi: 10.1016/j.envres.2015.12.031. doi:10.1016/j.envres.2015.12.031. [DOI] [PubMed] [Google Scholar]
- 5.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: An overview:Structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. doi:10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Wu C, Chen J, Chen C, Wang W, Wen L, Gao K, et al. Wnt/ß-catenin coupled with HIF-1a/VEGF signaling pathways involved in galangin neurovascular unit protection from focal cerebral ischemia. Sci Rep. 2015;5:16151. doi: 10.1038/srep16151. doi:10.1038/srep16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, et al. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. doi:10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barakat R, Redzic Z. The role of activated microglia and resident macrophages in the neurovascular unit during cerebral ischemia: Is the jury still out?Med Princ Pract. 2016;25(Suppl 1):3–14. doi: 10.1159/000435858. doi:10.1159/000435858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: Its role in health and disease. J Mol Med (Berl) 2004;82:434–48. doi: 10.1007/s00109-004-0555-y. doi:10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 10.Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. doi:10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Gao M, Zhu SY, Tan CB, Xu B, Zhang WC, Du GH. Pinocembrin protects the neurovascular unit by reducing inflammation and extracellular proteolysis in MCAO rats. J Asian Nat Prod Res. 2010;12:407–18. doi: 10.1080/10286020.2010.485129. doi:10.1080/10286020.2010.485129. [DOI] [PubMed] [Google Scholar]
- 12.Chu W, Li M, Li F, Hu R, Chen Z, Lin J, et al. Immediate splenectomy down-regulates the MAPK-NF-kB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg. 2013;74:1446–53. doi: 10.1097/TA.0b013e31829246ad. doi:10.1097/TA.0b013e31829246ad. [DOI] [PubMed] [Google Scholar]
- 13.Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, et al. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. doi:10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tao X, Chen X, Hao S, Hou Z, Lu T, Sun M, et al. Protective actions of PJ34, a poly(ADP-ribose)polymerase inhibitor, on the blood-brain barrier after traumatic brain injury in mice. Neuroscience. 2015;291:26–36. doi: 10.1016/j.neuroscience.2015.01.070. doi:10.1016/j.neuroscience.2015.01.070. [DOI] [PubMed] [Google Scholar]
- 15.Yuan F, Xu ZM, Lu LY, Nie H, Ding J, Ying WH, et al. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-kB p65 acetylation and activation. J Neurochem. 2016;136:581–93. doi: 10.1111/jnc.13423. doi:10.1111/jnc.13423. [DOI] [PubMed] [Google Scholar]
- 16.Won S, Sayeed I, Peterson BL, Wali B, Kahn JS, Stein DG. Vitamin D prevents hypoxia/reoxygenation-induced blood-brain barrier disruption via Vitamin D receptor-mediated NF-kB signaling pathways. PLoS One. 2015;10:e0122821. doi: 10.1371/journal.pone.0122821. doi:10.1371/journal.pone.0122821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: Therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–89. doi: 10.2174/1568007033482887. doi:10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- 18.Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. doi:10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada KH, Kozlowski DA, Seidl SE, Lance S, Wieschhaus AJ, Sundivakkam P, et al. Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J Biol Chem. 2012;287:13182–93. doi: 10.1074/jbc.M111.302612. doi:10.1074/jbc.M111.302612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ringger NC, Tolentino PJ, McKinsey DM, Pike BR, Wang KK, Hayes RL. Effects of injury severity on regional and temporal mRNA expression levels of calpains and caspases after traumatic brain injury in rats. J Neurotrauma. 2004;21:829–41. doi: 10.1089/0897715041526177. doi:10.1089/0897715041526177. [DOI] [PubMed] [Google Scholar]
- 21.Ma M, Shofer FS, Neumar RW. Calpastatin overexpression protects axonal transport in an in vivo model of traumatic axonal injury. J Neurotrauma. 2012;29:2555–63. doi: 10.1089/neu.2012.2473. doi:10.1089/neu.2012.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ponnappan S, Cullen SJ, Ponnappan U. Constitutive degradation of IkappaBalpha in human T lymphocytes is mediated by calpain. Immun Ageing. 2005;2:15. doi: 10.1186/1742-4933-2-15. doi:10.1186/1742-4933-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuzzocrea S, McDonald MC, Mazzon E, Siriwardena D, Serraino I, Dugo L, et al. Calpain inhibitor I reduces the development of acute and chronic inflammation. Am J Pathol. 2000;157:2065–79. doi: 10.1016/S0002-9440(10)64845-6. doi:10.1016/S0002-9440(10)64845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marzocco S, Di Paola R, Autore G, Mazzon E, Pinto A, Caputi AP, et al. Calpain inhibitor I reduces intestinal ischemia-reperfusion injury in the rat. Shock. 2004;21:38–44. doi: 10.1097/01.shk.0000095056.62263.b2. doi:10.1097/01.shk.0000095056.62263.b2. [DOI] [PubMed] [Google Scholar]
- 25.McDonald MC, Mota-Filipe H, Paul A, Cuzzocrea S, Abdelrahman M, Harwood S, et al. Calpain inhibitor I reduces the activation of nuclear factor-kappaB and organ injury/dysfunction in hemorrhagic shock. FASEB J. 2001;15:171–86. doi: 10.1096/fj.99-0645com. doi:10.1096/fj.99-0645com. [DOI] [PubMed] [Google Scholar]
- 26.Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, et al. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–73. doi: 10.1089/neu.2006.0011. doi:10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glushakov AV, Robbins SW, Bracy CL, Narumiya S, Doré S. Prostaglandin F2a FP receptor antagonist improves outcomes after experimental traumatic brain injury. J Neuroinflammation. 2013;10:132. doi: 10.1186/1742-2094-10-132. doi:10.1186/1742-2094-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson SN, Carrico KM, Mustafa AG, Bains M, Hall ED. A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J Neurotrauma. 2010;27:2233–43. doi: 10.1089/neu.2010.1474. doi:10.1089/neu.2010.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma M, Li L, Wang X, Bull DL, Shofer FS, Meaney DF, et al. Short-duration treatment with the calpain inhibitor MDL-28170 does not protect axonal transport in an in vivo model of traumatic axonal injury. J Neurotrauma. 2012;29:445–51. doi: 10.1089/neu.2011.2060. doi:10.1089/neu.2011.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vosler PS, Sun D, Wang S, Gao Y, Kintner DB, Signore AP, et al. Calcium dysregulation induces apoptosis-inducing factor release:Cross-talk between PARP-1- and calpain-signaling pathways. Exp Neurol. 2009;218:213–20. doi: 10.1016/j.expneurol.2009.04.032. doi:10.1016/j.expneurol.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun M, Xu C. Neuroprotective mechanism of taurine due to up-regulating calpastatin and down-regulating calpain and caspase-3 during focal cerebral ischemia. Cell Mol Neurobiol. 2008;28:593–611. doi: 10.1007/s10571-007-9183-8. doi:10.1007/s10571-007-9183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Li Y, Wang L, Lu M, Zhang X, Chopp M. Therapeutic benefit of intracerebral transplantation of bone marrow stromal cells after cerebral ischemia in rats. J Neurol Sci. 2001;189:49–57. doi: 10.1016/s0022-510x(01)00557-3. doi:10.1016/S0022-510X(01)00557-3. [DOI] [PubMed] [Google Scholar]
- 33.Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E. Dynamic changes in the recovery after traumatic brain injury in mice: Effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J Neurotrauma. 2008;25:324–33. doi: 10.1089/neu.2007.0452. doi:10.1089/neu.2007.0452. [DOI] [PubMed] [Google Scholar]
- 34.Chen G, Zhang S, Shi J, Ai J, Qi M, Hang C. Simvastatin reduces secondary brain injury caused by cortical contusion in rats: Possible involvement of TLR4/NF-kappaB pathway. Exp Neurol. 2009;216:398–406. doi: 10.1016/j.expneurol.2008.12.019. doi:10.1016/j.expneurol.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 35.Jasinska M, Siucinska E, Glazewski S, Pyza E, Kossut M. Characterization and plasticity of the double synapse spines in the barrel cortex of the mouse. Acta Neurobiol Exp (Wars) 2006;66:99–104. doi: 10.55782/ane-2006-1595. [DOI] [PubMed] [Google Scholar]
- 36.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. doi:10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation. 2012;9:236. doi: 10.1186/1742-2094-9-236. doi:10.1186/1742-2094-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51:966–79. doi: 10.1007/s12035-014-8752-3. doi:10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. doi:10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson CS, Gopinath SP, Valadka AB, Van M, Swank PR, Goodman JC. Variants of the endothelial nitric oxide gene and cerebral blood flow after severe traumatic brain injury. J Neurotrauma. 2011;28:727–37. doi: 10.1089/neu.2010.1476. doi:10.1089/neu.2010.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Wan Y, Fang Y, Yao E, Xu S, Ning Q, et al. Epoxyeicosanoid signaling provides multi-target protective effects on neurovascular unit in rats after focal ischemia. J Mol Neurosci. 2016;58:254–65. doi: 10.1007/s12031-015-0670-y. doi:10.1007/s12031-015-0670-y. [DOI] [PubMed] [Google Scholar]
- 42.Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2009;40(3 Suppl):S4–7. doi: 10.1161/STROKEAHA.108.534388. doi:10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan J, Lei X, Wang J, Huang S, Wang Y, Zhang Y, et al. Effects of Kaixinjieyu, a Chinese herbal medicine preparation, on neurovascular unit dysfunction in rats with vascular depression. BMC Complement Altern Med. 2015;15:291. doi: 10.1186/s12906-015-0808-z. doi:10.1186/s12906-015-0808-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12:223. doi: 10.1186/s12974-015-0434-1. doi:10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40:2519–25. doi: 10.1161/STROKEAHA.109.549592. doi:10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al. The role of hypoxia-inducible factor-1a, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. doi:10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- 47.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: A conserved 'calpain-cathepsin cascade' from nematodes to primates. Cell Calcium. 2004;36:285–93. doi: 10.1016/j.ceca.2004.03.001. doi:10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Bains M, Cebak JE, Gilmer LK, Barnes CC, Thompson SN, Geddes JW, et al. Pharmacological analysis of the cortical neuronal cytoskeletal protective efficacy of the calpain inhibitor SNJ-1945 in a mouse traumatic brain injury model. J Neurochem. 2013;125:125–32. doi: 10.1111/jnc.12118. doi:10.1111/jnc.12118. [DOI] [PubMed] [Google Scholar]
- 49.Bralic M, Stemberga V. Calpain expression in the brain cortex after traumatic brain injury. Coll Antropol. 2012;36:1319–23. [PubMed] [Google Scholar]
- 50.Karklin Fontana AC, Fox DP, Zoubroulis A, Valente Mortensen O, Raghupathi R. Neuroprotective effects of the glutamate transporter activator (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following traumatic brain injury in the adult rat. J Neurotrauma. 2016;33:1073–83. doi: 10.1089/neu.2015.4079. doi:10.1089/neu.2015.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buki A, Farkas O, Doczi T, Povlishock JT. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20:261–8. doi: 10.1089/089771503321532842. doi:10.1089/089771503321532842. [DOI] [PubMed] [Google Scholar]
- 52.Ai J, Liu E, Wang J, Chen Y, Yu J, Baker AJ. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J Neurotrauma. 2007;24:960–78. doi: 10.1089/neu.2006.0224. doi:10.1089/neu.2006.0224. [DOI] [PubMed] [Google Scholar]
- 53.Cuzzocrea S, McDonald MC, Mazzon E, Mota-Filipe H, Centorrino T, Terranova ML, et al. Calpain inhibitor I reduces colon injury caused by dinitrobenzene sulphonic acid in the rat. Gut. 2001;48:478–88. doi: 10.1136/gut.48.4.478. doi:10.1136/gut.48.4.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin M, Sun W, Gong W, Zhou Z, Ding Y, Hou Q. Methylophiopogonanone a protects against cerebral ischemia/reperfusion injury and attenuates blood-brain barrier disruption in vitro. PLoS One. 2015;10:e0124558. doi: 10.1371/journal.pone.0124558. doi:10.1371/journal.pone.0124558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, et al. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177. doi: 10.1186/s12974-014-0177-4. doi:10.1186/s12974-014-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, et al. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. 2014;45:857–64. doi: 10.1161/STROKEAHA.113.004100. doi:10.1161/STROKEAHA.113.004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trager N, Smith A, Wallace Iv G, Azuma M, Inoue J, Beeson C, et al. Effects of a novel orally administered calpain inhibitor SNJ-1945 on immunomodulation and neurodegeneration in a murine model of multiple sclerosis. J Neurochem. 2014;130:268–79. doi: 10.1111/jnc.12659. doi:10.1111/jnc.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blanc F, Furio L, Moisy D, Yen HL, Chignard M, Letavernier E, et al. Targeting host calpain proteases decreases influenza A virus infection. Am J Physiol Lung Cell Mol Physiol. 2016;310:L689–99. doi: 10.1152/ajplung.00314.2015. doi:10.1152/ajplung.00314.2015. [DOI] [PubMed] [Google Scholar]