

Abstract
Probiotic Lactobacillus species offer various health benefits, thus have been employed in treatment and prevention of various diseases. Due to the differences in the isolation source and the site of action, most of the lactobacilli tested in-vitro for probiotics properties fail to extend similar effects in-vivo. Consequently, the search of autochthonous, efficacious and probably population specific probiotics is a high priority in the probiotics research. In this regards, whole genome sequencing of as many Lactobacillus as possible will help to deepen our understanding of biology and their health effects. Here, we provide the genomic insights of two coherent oxalic acid tolerant Lactobacillus species (E2C2 and E2C5) isolated from two different healthy human gut flora. These two isolates were found to have higher tolerance towards oxalic acid (300 mM sodium oxalate). The draft genome of strain E2C2 consists of 3,603,563 bp with 3289 protein-coding genes, 94 RNA genes, and 43.99% GC content, while E2C5 contained 3,615,168 bp, 3293 coding genes (93.4% of the total genes), 95 RNA genes and 43.97% GC content. Based on 16S rRNA gene sequence analysis followed by in silico DNA-DNA hybridization studies, both the strains were identified as Lactobacillus plantarum belonging to family Lactobacillaceae within the phylum Firmicutes. Both the strains were genomically identical, sharing 99.99% CDS that showed 112 SNPs. Both the strains also exhibited deconjugation activity for the bile salts while genome analysis revealed that the L. plantarum strains E2C2 and E2C5 also have the ability to produce vitamins, biotin, alpha- and beta- glucosidase suggesting potential probiotic activities of the isolates. The description presented here is based on the draft genomes of strains E2C2 and E2C5 which are submitted to GenBank under the accession numbers LSST00000000.1 and LTCD00000000.1, respectively.
Keywords: Human Stool, Bacteria, Firmicutes, Lactobacillus plantarum
Introduction
The genome of lactobacilli is highly diversified which endorses them to occupy wide range of ecological habitats, including carbohydrate-rich environments [1], fermented meats [2], sourdoughs [3], plant-derived substrates [4] and different niches on and in the human body namely respiratory, gastrointestinal and urogenital tract [5, 6]. Owing to the beneficial effects offered by lactobacilli, they have been used as a gold standard in probiotic preparations. Consequently, many strains of lactobacilli such as Lactobacillus acidophilus, L. amylovorus, L. brevis, L. bulgaricus, L. casei, L. fermentum, L. lactis, L. pentosus , and L. rhamnosus have been well characterized for their ability to produce extracellular proteins, exopolysaccharides, and lipoteichoic acids, which influence the health and physiology of the host by interacting with the epithelial cells and enhancing the host immune system [7–12].
From the array of various Lactobacillus species, Lactobacillus plantarum, an organism found in a variety of ecological environments, is a well characterized probiotic species. Recent genome analysis of Lactobacillus plantarum WCFS1 indicates that this organism is endowed with sets of genes essential for survival in gastrointestinal tract, interactions with other organisms in the gut, interactions with the host epithelial barrier and immune system, making it an extremely versatile probiotic bacterium [13] and that the genome of this organism is highly plastic [14]. Despite the extraordinary features possessed by L. plantarum, it suffers from some drawbacks. First, a study involving the pharmacokinetics of L. plantarum has indicated that it is a transient passenger in the gut [15]. Secondly, significant genome editing is required in order to gain the improved probiotic properties [16]. Both of these could be attributed to the incompatibility of the isolation source e.g. human saliva [17] and its implied target (gut). Thus, the search of indigenous L. plantarum strains (e.g. from human gut) is a thrust area in probiotic research and its implications to human health.
Microbial communities in the human gut are complex and astonishingly diverse in nature [18]. Despite the fact that lactobacilli contribute minutely to these trillions of cells, due to their beneficial roles in gut ecology, they are gaining attention in biomedical research [19]. Consequently, we focused on the isolation of oxalate tolerant Lactobacilli from healthy stool samples. Out of the 16 Lactobacillus isolates grown on MRS media, two isolates E2C2 and E2C5 showed comparatively higher tolerance to oxalic acid and bile salt. Owing to the fact that hyperoxaluria leads to dysbiosis in the human gut [20], these strains of L. plantarum, GRAS category organism, may specifically be useful in ameliorating the hyperoxaluria and associated complications. We, therefore, sequenced the genomes of these isolates using Illumina Miseq platform and compared their metabolic potentials.
Organism information
Classification and features
The two oxalic acid tolerant isolates, E2C2 and E2C5, were isolated from human stool samples by double enrichment method (100 and 200 mM/L sodium oxalate) using MRS (10 g enzymatic digest of animal tissue, 10 g beef extract, 5 g yeast extract, 20 g dextrose, 5 g sodium acetate, 1 g polysorbate 80, 2 g potassium phosphate, 2 g ammonium citrate, 0.1 g magnesium sulfate, 0.05 g manganese sulfate) medium. These bacterial isolates were maintained on MRS agar at the incubation temperature of 30 °C and at pH 6.8.
The strains were tested for phenotypic and biochemical characterization (Table 1). L. plantarum E2C2 and E2C5 isolates are Gram-positive, non-motile, non-spore forming and rod-shape in morphology (Fig. 1 and Table 1). While, in the case of bile salts, both the strains could grow up to 0.40% w/v of Oxgall (Sigma-Aldrich) tested for 24 h incubation at 30 °C. It was observed that these isolates have the ability to deconjugate the glycodeoxycolate (bile salt) and this activity was confirmed by plate assay and TLC assay methods [21]. Ninhydrin assay [22] was performed to quantitate the bile salt hydrolase production ability which was found to be maximum at the 72 h, 5.22 U and 5.27 U for glycodeoxycholic acid as a substrate for E2C2 and E2C5 isolates, respectively (Fig. 2). They were able to utilize a large number of carbon compounds, namely dextrose, fructose, galactose, inulin, L-arabinose, maltose, mannose, mannitol, melibiose, Na-gluconate, raffinose, salicin, sorbitol, sucrose, trehalose, xylose, etc. during their growth (Table 1).
Table 1.
Classification and general features of L. plantarum E2C2 and L. plantarum E2C5
| MIGS ID | Property | L. plantarum E2C2 | L. plantarum E2C5 | Evidence codea |
|---|---|---|---|---|
| Domain | Bacteria | Bacteria | TAS [41] | |
| Phylum | Firmicutes | Firmicutes | TAS [42, 43] | |
| Class | Bacilli | Bacilli | TAS [44] | |
| Order | Lactobacillales | Lactobacillales | TAS [45] | |
| Family | Lactobacillaceae | Lactobacillaceae | TAS [46] | |
| Genus | Lactobacillus | Lactobacillus | TAS [43, 47–50] | |
| Species | Lactobacillus plantarum | Lactobacillus plantarum | TAS [43, 47, 51] | |
| Strain | E2C2 | E2C25 | ||
| Gram stain | Positive | Positive | TAS [43] | |
| Cell shape | Rod | Rod | IDA | |
| Motility | non-motile | non-motile | TAS [43] | |
| Sporulation | spore forming | spore forming | IDA | |
| Temperature range | 25 °C −39 °C | 25 °C −39 °C | NAS | |
| Optimum temperature | 30 °C | 30 °C | TAS [43] | |
| pH range; Optimum | 3.5–6.5; 5 | 3.5–6.5; 5 | TAS [43] | |
| Carbon source | Xylose, Maltose, Fructose, Dextrose, Galactose, Raffinose, Melibiose, Trehalose, Sucrose, L-Arabinose, Mannose, Inulin, Na-gluconate, Salicin, Sorbitol, Mannitol, Cellobiose, Melezitose, ONPG, Esculin, Citrate, Malonate | Xylose, Maltose, Fructose, Dextrose, Galactose, Raffinose, Melibiose, Trehalose, Sucrose, L-Arabinose, Mannose, Inulin, Na-gluconate, Salicin, Sorbitol, Mannitol, Cellobiose, Melezitose, ONPG, Esculin, Citrate, Malonate | IDA | |
| MIGS-6 | Habitat | Human stool | Human stool | IDA |
| MIGS-6.3 | Salinity tolerance | 5- 8% | 5- 8% | TAS [45] |
| MIGS-22 | Oxygen requirement | Facultatively anaerobic | Facultatively anaerobic | TAS [43] |
| MIGS-15 | Biotic relationship | Free-living | Free-living | TAS [45] |
| MIGS-14 | Pathogenicity | non-pathogen | non-pathogen | NAS |
| MIGS-4 | Geographic location | India/Asia | India/Asia | IDA |
| MIGS-5 | Sample collection | November 2015 | November 2015 | IDA |
| MIGS-4.1 | Latitude | 18.5204° N | 18.5204° N | IDA |
| MIGS-4.2 | Longitude | 73.8567° E | 73.8567° E | IDA |
| MIGS-4.4 | Altitude | 562 m a.s.l. | 562 m a.s.l. | IDA |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [52]
Fig. 1.
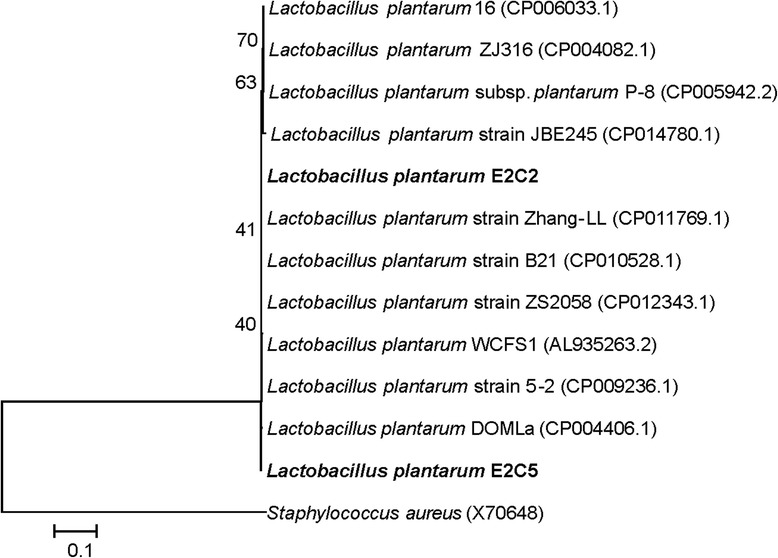
Neighbour-joining phylogenetic tree is constructed based on 16S rRNA gene sequence. The tree is constructed using Jukes–Cantor distances. Then 1000 bootstraps analyses are conducted. Sequences represented in bold font are derived from isolated strains of this study
Fig. 2.
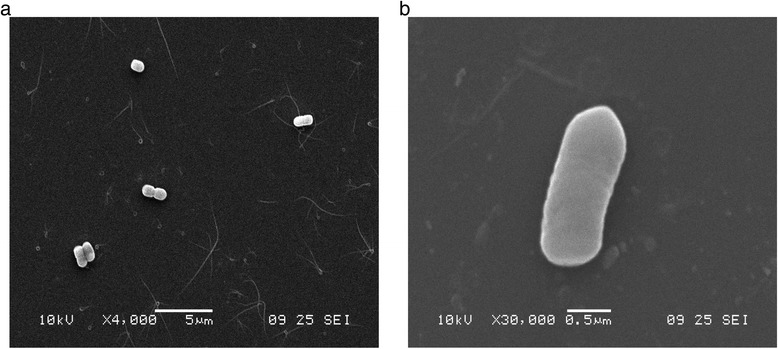
Scanning electron microscopic (SEM) analysis of bacterial isolates (a) Lactobacillus plantarum E2C2 and (b) Lactobacillus plantarum E2C5
16S rRNA gene sequencing and isDDH were used for the identification for isolates. 16S rRNA gene sequences were used for phylogenetic analysis using neighbour-joining method, which reveals that the two isolates E2C2 and E2C5 isolates are the members of Lactobacillaceae family, including Lactobacillus plantarum WCFS1, a previously reported probiotic bacterium isolated from human saliva [23] and Lactobacillus plantarum strain 5–2 [24], earlier isolated and identified from fermented foods (Fig. 3). The isDDH analysis was performed against type strain L. plantarum ATCC 14197 T for ANI and GGDC [25, 26]. Both the isolates congruently showed 98.91% ANI and 93.60% GGDC score to the type strain, which are more than recommended thresholds (95% for ANI and 70% for GGDC) for the identification of the species, confirming both isolates as L. plantarum, belonging to the phylum Firmicutes and class Bacilli. Both the strains are deposited in National Collection of Industrial Microorganisms, Pune with accession no. NCIM 5603 (L. plantarum E2C2) and NCIM 5602 (L. plantarum E2C5). The isolates were also deposited in Microbial Culture Collection, Pune with accession no. MCC 3016 (L. plantarum E2C2) and MCC 3190 (L. plantarum E2C5).
Fig. 3.
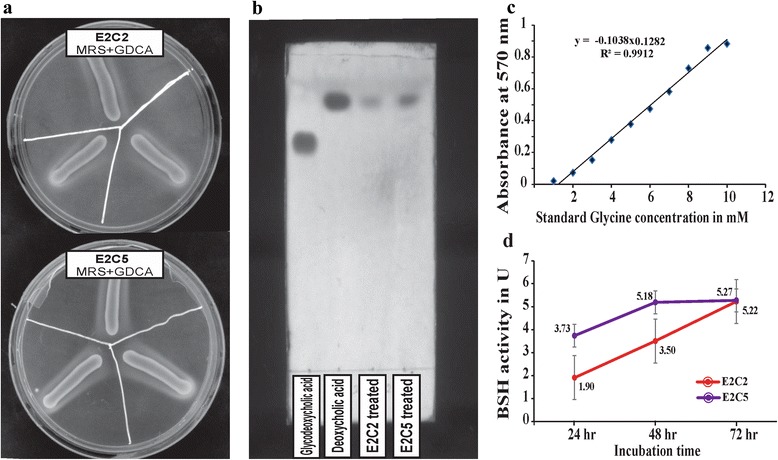
Bile salt hydrolase activity of Lactobacillus plantarum E2C2 and E2C5 isolates (a) Plate assay showing precipitation zones around the line of inoculation in triplicates (b) TLC plate assay showing deconjugation ability and (c & d) Ninhydrin assay indicating quantification of glycine removal by deconjugation ability
Genome sequencing information
Genome project history
The isolates were selected for sequencing as part of an ongoing project investigating the association of gut microbiota with hyperoxaluric condition. Based on metabolic versatility and oxalate tolerance, strains E2C2 and E2C5, were selected and sequenced by Illumina MiSeq platform at Institute of Medical Microbiology, Germany. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession LSST00000000.1 and LTCD00000000.1 (Table 2). The version described in this paper is version LSST00000000.1 and LTCD00000000.1.
Table 2.
Project information
| MIGS ID | Property | L. plantarum E2C2 | L. plantarum E2C25 |
|---|---|---|---|
| MIGS 31 | Finishing quality | High-quality draft | High-quality draft |
| MIGS-28 | Libraries used | 300 bp | 300 bp |
| MIGS 29 | Sequencing platforms | Illumina MiSeq | Illumina MiSeq |
| MIGS 31.2 | Fold coverage | 100 × | 100 × |
| MIGS 30 | Assemblers | DNASTAR assembler v. 11.2.1.25 | DNASTAR assembler v. 11.2.1.25 |
| MIGS 32 | Gene calling method | RAST | RAST |
| Locus Tag | AYO51 | AZJ01 | |
| Genbank ID | LSST00000000.1 | LTCD00000000.1 | |
| GenBank Date of Release | 03/23/2016 | 03/25/2016 | |
| GOLD ID | Gs0118511 | Gs0120378 | |
| BIOPROJECT | PRJNA311909 | PRJNA313343 | |
| MIGS 13 | Source Material Identifier | NCIM 5603, MCC 3016 | NCIM 5602, MCC 3190 |
| Project relevance | Human stool bacteria | Human stool bacteria |
Growth conditions and genomic DNA preparation
The E2C2 and E2C5 bacterial strains of L. plantarum were cultured in MRS agar (MA; Difco) at 30 °C under the aerobic condition for 3 days of incubation. Genomic DNA of the bacterial strains were isolated using a Qiagen DNA extraction kit (Hilden, Germany) following manufacturer’s instructions. Extracted DNA quality was assessed by 1.0% agarose gel electrophoresis, concentration and purity (A260/A280) were measured using NanoDrop ND-1000 (NanoDrop technologies, Willingminton, USA). Extracted DNA samples of the strains were preserved at −20 °C until further processing.
Genome sequencing and assembly
The bacterial genomes of L. plantarum E2C2 and L. plantarum E2C5 were sequenced by Illumina MiSeq platform using 2x300 paired-end libraries. Sequence quality of both the genomes was analyzed for quality control using FastQC software [27]. After analysis, raw sequences were trimmed and assembled using de novo assemblers SPAdes 3.5.0 [28] and DNA star assembler v. 11.2.1.25. More than 6 million good quality paired-end reads were obtained from both the strains, which accounted for an approximate 100x sequencing coverage. After assembly, it was found that the draft genomes of L. plantarum E2C2 and L. plantarum E2C5 contained 94 and 99 scaffolds respectively.
Genome annotation
Assembled genomes of both the strains were annotated using RAST version 2.0 [29] and the NCBI Prokaryotic Genome Annotation Pipeline [30]. Protein-encoding genes, tRNA and rRNA genes of the genomes were predicted using Glimmer version 3.02 [31], tRNA_scan-SE [32], and RNAmmer [33], respectively. Protein coding genes were analyzed by COG database [34] on WebMGA [35] and Pfam domains were predicted using NCBI Batch CD-Search Tool [36]. Transmembrane helix and signal peptide prediction of the genome was identified by using Phobius [37]. The presence of CRISPR repeats was predicted using the CRISPRFinder tools [38] (Table 4).
Table 4.
Number of genes associated with general COG functional categories
| Code | Lactobacillus plantarum | Description | ||||
|---|---|---|---|---|---|---|
| E2C2 | E2C5 | WCFS1 | ||||
| Value | % age | Value | % age | Value | ||
| J | 213 | 6.47 | 213 | 6.47 | 197 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0 | 0 | 0 | 0 | RNA processing and modification |
| K | 313 | 9.51 | 310 | 9.41 | 259 | Transcription |
| L | 166 | 5.04 | 168 | 5.10 | 103 | Replication, recombination and repair |
| B | 0 | 0 | 0 | 0 | 0 | Chromatin structure and dynamics |
| D | 49 | 1.48 | 48 | 1.45 | 33 | Cell cycle control, Cell division, chromosome partitioning |
| V | 92 | 2.79 | 92 | 2.79 | 76 | Defense mechanisms |
| T | 106 | 3.22 | 104 | 3.15 | 86 | Signal transduction mechanisms |
| M | 186 | 5.65 | 184 | 5.58 | 158 | Cell wall/membrane biogenesis |
| N | 22 | 0.66 | 22 | 0.66 | 10 | Cell motility |
| U | 26 | 0.79 | 26 | 0.78 | 17 | Intracellular trafficking and secretion |
| O | 99 | 3.01 | 100 | 3.03 | 83 | Posttranslational modification, protein turnover, chaperones |
| C | 118 | 3.58 | 118 | 3.58 | 101 | Energy production and conversion |
| G | 286 | 8.69 | 286 | 8.68 | 265 | Carbohydrate transport and metabolism |
| E | 218 | 6.62 | 219 | 6.65 | 183 | Amino acid transport and metabolism |
| F | 103 | 3.13 | 104 | 3.15 | 89 | Nucleotide transport and metabolism |
| H | 116 | 3.52 | 116 | 3.52 | 86 | Coenzyme transport and metabolism |
| I | 97 | 2.94 | 96 | 2.91 | 80 | Lipid transport and metabolism |
| P | 121 | 3.67 | 121 | 3.67 | 105 | Inorganic ion transport and metabolism |
| Q | 19 | 0.57 | 20 | 0.60 | 23 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 189 | 5.74 | 189 | 5.74 | 174 | General function prediction only |
| S | 235 | 7.14 | 236 | 7.16 | 201 | Function unknown |
| X | 94 | 2.85 | 97 | 2.94 | 55 | Mobilome: prophages, transposons |
| - | 421 | 12.80 | 424 | 12.87 | - | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Genome properties
The draft genome sequence of L. plantarum strains E2C2 and E2C5 contained 3603,563 bp and 3615,168 bp, with GC content 43.99% and 43.97%, respectively. The reads of L. plantarum strains E2C2 and E2C5 were assembled into 94 and 99 contigs (N 50, 235,913 bp, and 256,152 bp, respectively). The genome sequence of L. plantarum strain E2C2 included a total of 3504 genes and 3289 candidate CDS, giving a coding intensity of 94%. The genome was shown to encode at least 94 predicted RNAs, including 15 rRNAs and 75 tRNAs, and also 121 pseudogenes. Whereas, L. plantarum E2C5 genome which contained total 3523 genes and 3293 candidate CDS. L. plantarum E2C5 genome contained 95 predicted RNAs including 16 rRNAs and 75 tRNAs, and also 135 pseudogenes (Table 3). The draft genome size of the strains E2C2 and E2C5 was more than average of L. plantarum genome size that has been reported in public databases. It was found that most of the predicted genes (87.19% and 87.15% of strains E2C2 and E2C5, respectively) code for proteins which involved in major metabolic pathways were assigned to one of the 25 functional COG categories while the remaining genes were assigned as unknown functional proteins (Table 4).
Table 3.
Genome statistics
| Species Attribute | L. plantarum E2C2 | L. plantarum E2C5 | ||
|---|---|---|---|---|
| Value | % of Total | Value | % of Total | |
| Genome size (bp) | 3,603,563 | 100.00 | 3,615,168 | 100.00 |
| DNA coding (bp) | 2,684,877 | 74.5 | 2690, 385 | 74.4 |
| DNA G + C (bp) | 1,585,330 | 43.9 | 1589, 803 | 43.9 |
| DNA scaffolds | 94 | 99 | ||
| Total genes | 3504 | 100 | 3523 | 100 |
| Protein coding genes | 3289 | 93.8 | 3293 | 93.4 |
| RNA genes | 94 | 2.6 | 95 | 2.6 |
| Pseudo genes | 121 | 3.4 | 135 | 3.8 |
| Genes in internal clusters | NA | NA | ||
| Genes with function prediction | 2416 | 68.9 | 2426 | 68.9 |
| Genes assigned to COGs | 2868 | 81.8 | 2869 | 81.4 |
| Genes with Pfam domains | 2952 | 89.7 | 2969 | 90.1 |
| Genes with signal peptides | 278 | 7.9 | 275 | 7.8 |
| Genes with transmembrane helices | 755 | 21.5 | 755 | 21.4 |
| CRISPR repeats | 1 | 0.028 | 1 | 0.028 |
Insights from the genome sequences
Genome sequence analysis of L. plantarum strains E2C2 and E2C5 showed a presence of common subsystem structure, i.e., carbohydrate and protein metabolisms, iron acquisition and metabolism, chemotaxis, stress response, secondary metabolism, nitrogen metabolism, dormancy and sporulation. Genome analysis of both the strains showed that more than 800 genes are present for carbohydrate metabolism indicating a diverse carbohydrate utilization pattern or abilities that include C1- metabolism, organic acids, mono-, di- and polysaccharides metabolisms. Lactobacillus is well known for its capability to grow in protein-rich environments and contains protein degradation enzymes/machinery, therefore it is well adapted to these conditions. It was observed that both the strains have more than 50 protein degrading enzymes/transport systems that include metallo-carboxypeptidases, dipeptidase, proteasome and many ATP-dependent uptake systems. A large number of stress response systems that include oxidative stress, heat shock and cold shock are present in both the strains. Stress response genes, namely sodA, sodB, HPI, HPII and CCP for reactive oxygen species; PRP, Rex, OxyR, Fnr, ZUR and FUR for oxidative stress; HrcA, GrpE and fam for heat shock response were identified. In L. plantarum strains E2C2 and E2C5, genes for alpha-glucosidase, choloylglycine hydrolase, alpha-L-rhamnosidase essential for antidiabetic, hydrolysis of bile salt in the small intestine, adaptation to changing nutritional resources are noted. Therefore, the analysis suggests that both the L. plantarum strains (E2C2 and E2C5) can be used in multi-therapeutic aspects. The presence of biotin and other cofactors, vitamins, prosthetic groups and pigment synthesis genes are observed in the genome of both the strains, suggesting their ability to produce bioactive compounds. Considerable variation was not observed in the remaining subsystems that indicates biochemical homogeneity and similar capabilities of the strains in substrate utilization and processing. In addition, both L. plantarum E2C2 and L. plantarum E2C5 contain sulfur cycling, cobalt, zinc, and cadmium resistance genes.
Extended insights
Comparison of the strains E2C2 and E2C5 genome showed 99.99% shared CDS and only 112 SNPs among the core genome, thus overall demonstrating the high similarity of the two genomes (Tables 3 and 4). The high similarity of the two isolates, despite the different source of isolation, is an indication of their selective adaptation to the gut environment. But based on COG data analysis it was found that these two strains E2C2 and E2C5 were differed from each other with respect to number of protein coding genes namely signal transduction mechanisms, cell wall/membrane biogenesis, Mobilome: prophages, transposons, etc. Oxalate tolerance ability of the two isolates is an important feature to note. In the hyperoxaluric condition, human gut often acts as a primary excretory organ of oxalate [39] and higher oxalate concentration in the gut has been linked with dysbiosis [20]. In the light of oxalate tolerance ability of the E2C2 and E2C5 isolates, their use as probiotics for hyperoxaluric patients is anticipated. In addition, genomes of strains E2C2 and E2C5 were compared with the reference strain, Lactobacillus plantarum WCFS1 [17]. The comparison revealed that the three genomes comprised 2639 genes in common at 80% coverage and 90% sequence identity [40]. E2C2 and E2C5 both contained an additional 345 genes while WCFS1 strain contained additional 265 genes. Further, about 344 genes were exclusively found in strains E2C2 and E2C5 as compared to strain WCFS1. When COG categories compared, a significant difference was observed for the functional annotation of the genes. COGs functional categories could be assigned to 2868 and 2869 genes for E2C2 and E2C5 respectively, while in case of WCFS1 only 2384 genes could be categorised by COGs (Table 4).
Conclusions
Considering the high genetic versatility of Lactobacillus plantarum [14], it is important to sequence as many strains as possible to account for the genetic variability and their association with specific probiotic features such as oxalate tolerance. In this study, we provide the in-depth genome analysis of two oxalic acid and bile acid tolerant isolates- L. plantarum E2C2 and L. plantarum E2C5 obtained from healthy human stool samples. Genomic as well as phenotypic analysis reveals that both the isolates are coherent belonging to a single genetic lineage. The two strains described here can be an intriguing target to be explored further for their probiotics potentials in managing the specific metabolic disorders such as hyperoxaluria.
Acknowledgements
MS acknowledges Council of Scientific and Industrial Research, New Delhi, for fellowship. Authors are thankful to the Department of Biotechnology, Government of India; Microbial Culture Collection Project (BT/PR10054/NDB/52/94/2007) for financial aids. Authors are thankful to Dr. Shashi Ghodake, DDS, Middletown, CT 06457, USA for proofreading of the manuscript.
Authors’ contributions
Conceived and designed the experiments: MS, DP, SD, SB, RG, YS. Isolation, biochemical characterization, Genomic DNA, and microscopy: MS, DP, SB, TH. Genome sequencing: SD. Manuscript preparation: DP, MS, SB. Bioinformatics analysis: DP, MS, SD. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- ANI
Average nucleotide identity
- CDS
Coding DNA sequences or protein coding genes
- GGDC
Genome-to-genome distance calculator
- GRAS
Generally regarded as safe
- isDDH
In silico DNA-DNA hybridisation
- MRS medium
De Man, Rogosa and Sharpe medium
References
- 1.Pisano MB, Viale S, Conti S, Fadda ME, Deplano M, Melis MP, et al. Preliminary evaluation of probiotic properties of Lactobacillus strains isolated from Sardinian dairy products. Biomed Res Int. 2014;2014:286390. doi: 10.1155/2014/286390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Todorov SD, Vaz-Velho M, de Melo Franco BDG, Holzapfel WH. Partial characterization of bacteriocins produced by three strains of Lactobacillus sakei, isolated from salpicao, a fermented meat product from North-West of Portugal. Food Control. 2013;30:111–21. doi: 10.1016/j.foodcont.2012.07.022. [DOI] [Google Scholar]
- 3.Ilha EC, da Silva T, Lorenz JG, de Oliveira Rocha G, Sant’Anna ES. Lactobacillus paracasei isolated from grape sourdough: acid, bile, salt, and heat tolerance after spray drying with skim milk and cheese whey. Eur Food Res Technol. 2015;240:977–84. doi: 10.1007/s00217-014-2402-x. [DOI] [Google Scholar]
- 4.Gotteland M, Cires MJ, Carvallo C, Vega N, Ramirez MA, Morales P, et al. Probiotic screening and safety evaluation of Lactobacillus strains from plants, artisanal goat cheese, human stools, and breast milk. J Med Food. 2014;17:487–95. doi: 10.1089/jmf.2013.0030. [DOI] [PubMed] [Google Scholar]
- 5.Siezen RJ, Tzeneva VA, Castioni A, Wels M, Phan HTK, Rademaker JLW, et al. Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ Microbiol. 2010;12:758–73. doi: 10.1111/j.1462-2920.2009.02119.x. [DOI] [PubMed] [Google Scholar]
- 6.Liévin-Le Moal V, Servin AL. Anti-infective activities of lactobacillus strains in the human intestinal microbiota: from probiotics to gastrointestinal anti-infectious biotherapeutic agents. Clin Microbiol Rev. 2014;27:167–99. doi: 10.1128/CMR.00080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez B, Saad N, Schmitter J-M, Bressollier P, Urdaci MC. Adhesive properties, extracellular protein production, and metabolism in the Lactobacillus rhamnosus GG strain when grown in the presence of mucin. J Microbiol Biotechnol. 2010;20:978–84. doi: 10.4014/jmb.0911.11007. [DOI] [PubMed] [Google Scholar]
- 8.Segers ME, Lebeer S. Towards a better understanding of Lactobacillus rhamnosus GG-host interactions. Microb Cell Fact. 2014;13(Suppl 1):S7. doi: 10.1186/1475-2859-13-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiraishi T, Yokota S, Morita N, Fukiya S, Tomita S, Tanaka N, et al. Characterization of a Lactobacillus gasseri JCM 1131 T lipoteichoic acid with a novel glycolipid anchor structure. Appl Environ Microbiol. 2013;79:3315–8. doi: 10.1128/AEM.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki S, Kimoto-Nira H, Suganuma H, Suzuki C, Saito T, Yajima N. Cellular fatty acid composition and exopolysaccharide contribute to bile tolerance in Lactobacillus brevis strains isolated from fermented Japanese pickles. Can J Microbiol. 2014;60:183–91. doi: 10.1139/cjm-2014-0043. [DOI] [PubMed] [Google Scholar]
- 11.Farshad S, Norouzi F, Aminshahidi M, Heidari B, Alborzi A. Two cases of bacteremia due to an unusual pathogen, Comamonas testosteroni in Iran and a review literature. J Infect Dev Ctries. 2012;6:521–5. doi: 10.3855/jidc.2215. [DOI] [PubMed] [Google Scholar]
- 12.Abd El Ghany K, Hamouda R, Abd Elhafez E, Mahrous H, Salem-Bekhit M, Hamza HA. A potential role of Lactobacillus acidophilus LA1 and its exopolysaccharides on cancer cells in male albino mice. Biotechnol Biotechnol. 2015;29:977–83. doi: 10.1080/13102818.2015.1050455. [DOI] [Google Scholar]
- 13.van den Nieuwboer M, van Hemert S, Claassen E, de Vos WM. Lactobacillus plantarum WCFS1 and its host interaction: a dozen years after the genome. Microb Biotechnol. 2016;9:452–65. doi: 10.1111/1751-7915.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siezen RJ, van Hylckama Vlieg JET. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb Cell Fact. 2011;10(Suppl 1):S3. doi: 10.1186/1475-2859-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vesa T, Pochart P, Marteau P. Pharmacokinetics of Lactobacillus plantarum NCIMB 8826, Lactobacillus fermentum KLD, and Lactococcus lactis MG 1363 in the human gastrointestinal tract. Aliment Pharmacol Ther. 2000;14:823–8. doi: 10.1046/j.1365-2036.2000.00763.x. [DOI] [PubMed] [Google Scholar]
- 16.Bove P, Gallone A, Russo P, Capozzi V, Albenzio M, Spano G, et al. Probiotic features of Lactobacillus plantarum mutant strains. Appl Microbiol Biotechnol. 2012;96:431–41. doi: 10.1007/s00253-012-4031-2. [DOI] [PubMed] [Google Scholar]
- 17.Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci U S A. 2003;100:1990–5. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huttenhower C, Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl Environ Microbiol. 2008;74:4985–96. doi: 10.1128/AEM.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suryavanshi MV, Bhute SS, Jadhav SD, Bhatia MS, Gune RP, Shouche YS. Hyperoxaluria leads to dysbiosis and drives selective enrichment of oxalate metabolizing bacterial species in recurrent kidney stone endures. Sci Rep. 2016;6:34712. doi: 10.1038/srep34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar R, Grover S, Batish VK. Bile Salt Hydrolase (Bsh) Activity Screening of Lactobacilli: In Vitro Selection of Indigenous Lactobacillus Strains with Potential Bile Salt Hydrolysing and Cholesterol-Lowering Ability. Probiotics Antimicrob Proteins. 2012;4:162–72. doi: 10.1007/s12602-012-9101-3. [DOI] [PubMed] [Google Scholar]
- 22.Kumar RS, Brannigan JA, Prabhune AA, Pundle AV, Dodson GG, Dodson EJ, et al. Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum reveals an evolutionary relationship with penicillin V acylase. J Biol Chem. 2006;281:32516–25. doi: 10.1074/jbc.M604172200. [DOI] [PubMed] [Google Scholar]
- 23.Hayward AC, Davis GHG. The isolation and classification of Lactobacillus strains from italian saliva samples. Br Dent J. 1956;101:2733–41. [Google Scholar]
- 24.Liu C-J, Wang R, Gong F-M, Liu X-F, Zheng H-J, Luo Y-Y, et al. Complete genome sequences and comparative genome analysis of Lactobacillus plantarum strain 5–2 isolated from fermented soybean. Genomics. 2015;106:404–11. doi: 10.1016/j.ygeno.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 27.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010; Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 28.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–24. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27342282. [DOI] [PMC free article] [PubMed]
- 31.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–9. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 2011;12:444. doi: 10.1186/1471-2164-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 2007;35:W429–32. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369:649–58. doi: 10.1056/NEJMra1301564. [DOI] [PubMed] [Google Scholar]
- 40.Chaudhari NM, Gupta VK, Dutta C, Tettelin H, Rasko DA, Smokvina T, et al. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373. doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibbons NE, Murray RGE. Proposals concerning the higher taxa of bacteria. Int J Syst Evol Microbiol. 1978;28:1–6. [Google Scholar]
- 43.Bringel F, Castioni A, Olukoya DK, Felis GE, Torriani S, Dellaglio F. Lactobacillus plantarum subsp. argentoratensis subsp. nov., isolated from vegetable matrices. Int J Syst Evol Microbiol. 2005;55:1629–1634. doi: 10.1099/ijs.0.63333-0. [DOI] [PubMed] [Google Scholar]
- 44.Ludwig W, Schleifer K, Whitman W. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2009. Class I. Bacilli class nov; pp. 9–20. [Google Scholar]
- 45.Schleifer K, Whitman W. Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 3 (The Firmicutes) Second. New York: Springer; 2009. Order II. Lactobacillales ord. nov; p. 464. [Google Scholar]
- 46.Winslow CE, Broadhurst J, Buchanan RE, Krumwiede C, Rogers LA, Smith GH, et al. The Families and Genera of the Bacteria: Preliminary Report of the Committee of the Society of American Bacteriologists on Characterization and Classification of Bacterial Types. J Bacteriol. 1917;2:505–66. doi: 10.1128/jb.2.5.505-566.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skerman V, McGowan V, Sneath P. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 48.Haakensen M, Dobson C, Hill J, Ziola B. Reclassification of Pediococcus dextrinicus (Coster and White 1964) Back 1978 (Approved Lists 1980) as Lactobacillus dextrinicus comb. nov., and emended description of the genus Lactobacillus. Int J Syst Evol Micr. 2009;59:615–621. doi: 10.1099/ijs.0.65779-0. [DOI] [PubMed] [Google Scholar]
- 49.Cai Y, Pang H, Kitahara M, Ohkuma M. Lactobacillus nasuensis sp. nov., a lactic acid bacterium isolated from silage, and emended description of the genus Lactobacillus. Int J Syst Evol Micr. 2012;62:1140–1144. doi: 10.1099/ijs.0.031781-0. [DOI] [PubMed] [Google Scholar]
- 50.Bergey DH, Breed RS, Hammer BW, Huntoon FM, Murray EGD, Harrison FC. Genus Lactobacillus Beijerinck, 191. In: Bergey DH, Breed RS, Hammer BW, Huntoon FM, Murray EGD, Harrison FC, editors. Bergey’s Manual of Systematic Bacteriology. 4. Baltimore: The Williams and Wilkins Co; 1934. pp. 300–321. [Google Scholar]
- 51.Bergey DH, Harrison FC, Breed RS, Hammer BW, Huntoon FM. Bergey’s Manual of Determinative Bacteriology. 1923. p. 1. [Google Scholar]
- 52.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]