

Abstract
We report here the draft genome sequences of eight bacterial strains of the genera Staphylococcus, Microbacterium, Mycobacterium, Plantibacter, and Pseudomonas. These isolates were obtained from aerosol sampling of bathrooms of five residences in the San Francisco Bay area. Taxonomic classifications as well as the genome sequence and gene annotation of the isolates are described. As part of the “Built Environment Reference Genome” project, these isolates and associated genome data provide valuable resources for studying the microbiology of the built environment.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-017-0223-9) contains supplementary material, which is available to authorized users.
Keywords: Built environment, Shower water, Airborne bacteria, Bacterial genomes
Introduction
Given that humans spend most of their lives in indoor environments [1], it is important to understand the microorganisms that can be found in these human-created structures. Previous work based on 16S rRNA gene surveys has described thousands of bacterial taxa from residences (e.g., [2]). Within these residences, periodically wet surfaces– such as shower walls, shower heads, sinks, drains – represent unique (compared to dryer areas within the home - [3–5]) and potentially medically important microbial communities [6]. Humans could readily interface with the microbial communities on these wet surfaces by direct contact or by inhalation from aerosolized particles. Focusing on these airborne microorganisms, Miletto & Lindow [7] collected aerosol particles from residences for genetic analysis and identified over 300 genera which they attributed to various sources including tap water, human occupants, indoor surfaces, and outdoor air.
An important tool in studying microbial communities involves culturing and genome sequencing. In order to expand our work on the microbiology of built environments [8] into a more experimental framework, we cultured bacteria from the air of residential bathrooms and report their genome sequences. Genome sequencing was utilized in order to provide insight into the basic biology of the bacteria collected in indoor environments and to aid with future metagenomic and transcriptomic efforts.
The eight isolates within five genera were isolated during a sampling campaign of residential bathrooms conducted in 2015. While simultaneously filtering aerosols for amplicon-based community composition analysis (which is in preparation and will be published elsewhere), petri dishes were exposed to the air to isolate viable bacteria. After an initial screening of multiple isolates by sequencing the full-length 16S rRNA gene and carrying out preliminary taxonomic classification, eight isolates were selected for further genomic sequencing based on an assessment of their putative importance in the built environment. Specifically, we favored strains that met the following criteria: they are commonly identified in indoor environments, they are likely inputs from a common source for indoor microbes (premise plumbing, outdoor origin [9]), and/or they (or their close relatives) can potentially impact human health. For instance, we include three species (four isolates) of staphylococci. CoNS are typically benign inhabitants of the human skin and mucous membranes, but they are associated with infections and can be pathogenic to humans with compromised immune systems [10]. Mycobacterium iranicum is a newly described species which has been isolated from clinical specimens originating in diverse countries including Iran, Greece, the Netherlands, Sweden and the USA [11], although genomic comparison indicated that this is likely an environmental bacterium [12]. Pseudomonas oryzihabitans (synonym Flavimonas oryzihabitans) has been isolated from water and damp environments such as rice paddies and sink drains [13]. The only two described species of the genus Plantibacter, P. auratus and P. flavus, have been detected as a tree endophyte [14] and a component from the phyllosphere of grass [15], respectively. Organisms within the genus Microbacterium belong to the class Actinobacteria in which some species are known for the production of a broad spectrum of secondary metabolites. The chemical ecology of microorganisms on indoor surfaces is a component of our ongoing research efforts in the built environment.
Here we report a summary classification and the features of these eight isolates collected as part of the Built Environment Reference Genomes initiative. Strains and their genomes have been deposited according to the following accessions: Staphylococcus capitis strain H36 (DSM-103511; GenBank ID LWCQ00000000), S. capitis strain H65 (DSM-103512; LWCP00000000), S. cohnii strain H62 (DSM-103510; LWAC00000000), S. hominis strain H69 (DSM-103553; LVVO00000000), Microbacterium sp. strain H83 (DSM-103506; LWCU00000000, Pseudomonas oryzihabitans strain H72 (DSM-103505; LWCR00000000), Mycobacterium iranicum strain H39 (DSM-103542; LWCS00000000), and finally Plantibacter sp. strain H53 (DSM-103507; LWCT00000000).
Organism information
Classification and features
Two growth media were used for the initial isolation of bacteria: lysogeny broth agar (LB, Difco Laboratories, Detroit, MI) and R2A agar (Difco Laboratories, Detroit, MI). Petri dishes were exposed to residential bathroom air for 1 h; 30 min during which shower water was running to create shower mist and 30 min after the shower was turned off. Petri dishes were mounted on vertical surfaces (door, wall, cabinets) at a height of approximately 1.50 m. Petri dishes were brought back to the laboratory, where LB plates were incubated at 28 °C for 48 h, and R2A plates were incubated at 28 °C for 5 days and at 35 °C for 3 days. Except for Staphylococcus hominis strain H69, which was isolated on LB agar medium at 35 °C, all other strains were isolated on R2A medium (Additional file 1). Research was approved by the University of California Committee for the Protection of Human Subjects Protocol ID 2015-02-7135, and the sampling was conducted in March, 2015.
Taxonomic classification of these isolates was undertaken after genome sequencing, either using the full-length 16S rRNA gene sequences or a concatenated marker gene approach. For Microbacterium, Mycobacterium, and Plantibacter there were insufficient publicly available genome sequences of close relatives for a concatenated marker approach. In these cases, the full length 16S rRNA gene sequence was uploaded to the Ribosomal Database Project [16] and added to alignments containing representatives of all close relatives (as estimated from BLAST [17]). These alignments were downloaded, cleaned with a custom script [18], and an approximately maximum likelihood tree was inferred using the default setting in FastTree [19]. Outgroups for all trees were type strains of another genus or genera within the same family. The sequence alignments supporting the phylogenetic trees of this article are available in the FigShare repository [20].
All strains were given a specific identifier (e.g., H83) based on our internal culture collection. The 16S rRNA gene trees for both Microbacterium and Plantibacter genera were poorly resolved (e.g., low bootstrap values), and these isolates were placed into polyphyletic clades with respect to the names of taxa in the genera (Additional files 2 and 3). In addition, while Microbacterium sp. H83 falls within a clade that contains mostly M. foliorum, this name also occurs outside the clade. Therefore we have not attempted to assign these isolates to a particular species. On the other hand, the rRNA gene for one isolate is found in a monophyletic clade with other M. iranicum isolates (Additional file 4) and thus we have assigned this the name M. iranicum H39, For the Pseudomonas and Staphylococcus isolates, the 16S rRNA gene trees were inadequate for taxonomic classification at the species level, but the genomes of numerous sequenced representatives of close relatives were available for further analysis. All available genome sequences of close relatives (to a max of 20 randomly selected genomes per species) were downloaded from NCBI. The file names and sequences were reformatted for easier visualization. The assemblies were then screened for 37 core maker genes [21] using PhyloSift [22] in search and align mode using “isolate” and “besthit” flags. PhyloSift concatenates and aligns the hits of interest so the sequences are subsequently extracted from the PhyloSift output files and added to a single file for tree-building. An approximately maximum-likelihood tree was then inferred using FastTree.
The concatenated marker genes for one isolate placed it in a well-supported clade of P. oryzihabitans isolates (Additional file 5) and thus we have named this P. oryzihabitans H72. Based on this tree, we believe that one of the (unpublished) strains of P. psychrotolerans has been misclassified and should also be considered P. oryzihabitans. Four of the isolates were Staphylococcus species, for which we created a single concatenated marker tree containing the relevant close relatives of the isolates (Fig. 1). Two of our Staphylococcus isolates placed within a well-supported (i.e., high bootstrap support) monophyletic clade of S. capitis strains and thus we have named these S. capitis H36 and S. capitis H65. One Staphylococcus isolate placed within a well-supported clade of S. cohnii strains and thus we have named it S. cohnii H62. Our fourth Staphylococcus isolate was placed within a well-supported clade containing mostly S. hominis isolates but which also contains a few S. haemolyticus isolates. Because this tree shows a distinct clade containing many S. haemolyticus isolates, we have named this isolate S. hominis H69. It is unclear from this tree alone whether these few S. haemolyticus isolates are misnamed or whether further taxonomic revision of this group is needed.
Fig. 1.

Maximum Likelihood tree based on concatenated markers from Staphylococcus spp. genomes. The tree was inferred using FastTree from an Hmmalign alignment in Phylosift of 37 highly conserved marker genes. Numbers at the nodes represent local support values. The tree was rooted to Macrococcus caseolyticus as an outgroup (not shown) since this species is a close relative to Staphylococcus
General description of the isolates are summarized in Table 1, and micrographs appear in Fig. 2.
Table 1.
Classification and general features of the eight isolates in accordance with the MIGS recommendations [60]
| MIGS ID | Property | Microbacterium sp. H83 | Evidence codea | Mycobacterium iranicum H39 | Evidence code | Plantibacter sp. H53 | Evidence code | Pseudomonas oryzihabitans H72 | Evidence code | Staphylococcus capitis H36 | Evidence code | S. capitis H65 | Evidence code | S.cohnii H62 | Evidence code | S. hominis H69 | Evidence code |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Classification | |||||||||||||||||
| Domain | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | Bacteria | TAS [61] | |
| Phylum | Actinobacteria | TAS [62] | Actinobacteria | TAS [62] | Actinobacteria | TAS [62] | Proteobacteria | TAS [63] | Firmicutes | TAS [64] | Firmicutes | TAS [64] | Firmicutes | TAS [64] | Firmicutes | TAS [64] | |
| Class | Actinobacteria | TAS [65] | Actinobacteria | TAS [65] | Actinobacteria | TAS [65] | Gammaproteobacteria | TAS [66, 67] | Bacilli | TAS [68, 69] | Bacilli | TAS [68, 69] | Bacilli | TAS [68, 69] | Bacilli | TAS [68, 69] | |
| Order | Micrococcales | TAS [70, 71] | Corynebacteriales | TAS [72, 73] | Micrococcales | TAS [70, 71] | Pseudomonadales | TAS [71, 74] | Bacillales | TAS [70, 71] | Bacillales | TAS [70, 71] | Bacillales | TAS [70, 71] | Bacillales | TAS [70, 71] | |
| Family | Microbacteriaceae | TAS [75, 76] | Mycobacteriaceae | TAS [71, 77] | Microbacteriaceae | TAS [75, 76] | Pseudomonadaceae | TAS [71, 78] | Staphylococcaceae | TAS [79] | Staphylococcaceae | TAS [79] | Staphylococcaceae | TAS [79] | Staphylococcaceae | TAS [79] | |
| Genus | Microbacterium | TAS [71, 80] | Mycobacterium | TAS [71, 81] | Plantibacter | TAS [15] | Pseudomonas | TAS [71, 82] | Staphylococcus | TAS [71, 83] | Staphylococcus | TAS [71, 83] | Staphylococcus | TAS [71, 83] | Staphylococcus | TAS [71, 83] | |
| Species | Microbacterium sp. | NAS | M. iranicum | TAS [11] | Plantibacter sp. | NAS | P. oryzihabitans | TAS [84] | S. capitis | TAS [71, 85] | S. capitis | TAS [71, 85] | S. cohnii | TAS [71, 86] | S. hominis | TAS [71, 85] | |
| Strain | H83 | IDA | H39 | IDA | H53 | IDA | H72 | IDA | H36 | IDA | H65 | IDA | H62 | IDA | H69 | IDA | |
| Gram stain | Positive | TAS [23] | Positive | TAS [11] | Positive | TAS [87] | Negative | TAS [30] | Positive | TAS [88] | Positive | TAS [88] | Positive | TAS [88] | Positive | TAS [88] | |
| Cell shape | Rod | TAS [23] | Rod | TAS [11] | Rod | TAS [87] | Rod | TAS [30] | Coccus/grape-like clusters | TAS [88] | Coccus/grape-like clusters | TAS [88] | Coccus/grape-like clusters | TAS [88]] | Coccus/grape-like clusters | TAS [88] | |
| Motility | nd | Non-motile | TAS [11] | Non-motile | TAS [87] | Motile | TAS [30] | Non-motile | TAS [88] | Non-motile | TAS [88] | Non-motile | TAS [88] | Non-motile | TAS [88] | ||
| Sporulation | Non-spore forming | TAS [23] | Non-spore forming | TAS [11] | Non-spore forming | TAS [87] | Non-spore forming | NAS | Non-spore forming | TAS [88] | Non-spore forming | TAS [88] | Non-spore forming | TAS [88]] | Non-spore forming | TAS [88] | |
| Temperature range | Mesophile | TAS [23] | 25-40° | TAS [11] | Mesophile | IDA | Mesophile | TAS [30] | 18-45 °C | TAS [88] | 18-45 °C | TAS [88] | 15-45 °C | TAS [88] | 20-45 °C | TAS [88] | |
| Optimum temperature | nd | 37° | TAS [11] | 30 °C | TAS [87] | nd | nd | IDA | nd | nd | 30-40 °C | TAS [88] | |||||
| pH range; Optimum | 5-11; nd | TAS [23] | nd | nd | nd | nd | nd | nd | nd | ||||||||
| Carbon source | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Peptone, Dextrose, Starch, Casamino acids | IDA | Yeast extract, Tryptone | IDA | |
| GS-6 | Habitat | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS | Indoor air | NAS |
| 6.3 | Salinity | Normal | IDA | 5% NaCl (w/v) | TAS [11] | Normal | IDA | 6.5% NaCl (w/v) | TAS [84] | 10% NaCl (w/v) | TAS [88] | 10% NaCl (w/v) | TAS [88] | 10% NaCl (w/v) | TAS [88] | 10% NaCl (w/v) | TAS [88] |
| 22 | Oxygen requirement | Aerobic | TAS [23] | Aerobic | TAS [89] | Aerobic | TAS [87] | Aerobic | TAS [30] | Facultative anaerobes | TAS [88] | Facultative anaerobes | TAS [88] | Facultative anaerobes | TAS [88] | Facultative anaerobes | TAS [88] |
| 15 | Biotic relationship | Free living | NAS | Symbiont | TAS [11] | Free living | TAS [87] | Free living; symbiont | TAS [84] | Free living | NAS | Free living | NAS | Free living | NAS | Free living | NAS |
| 14 | Pathogenicity | nd | nd | nd | nd | nd | nd | nd | nd | ||||||||
| 4 | Geographic location | USA: California: Piedmont | NAS | USA: California: Oakland | NAS | USA: California: Walnut Creek | NAS | USA: California: Oakland | NAS | USA: California: Oakland | NAS | USA: California: Milpitas | NAS | USA: California: Milpitas | NAS | USA: California: Milpitas | NAS |
| 5 | Sample collection | 2015-03-16 | NAS | 2015-03-18 | NAS | 2015-03-17 | NAS | 2015-03-18 | NAS | 2015-03-18 | NAS | 2015-03-31 | NAS | 2015-03-31 | NAS | 2015-03-31 | NAS |
| 4.1 | Latitude | 37°49'25.6" | NAS | 122°16'21.9" | NAS | 122°03'50.1" | NAS | 122°16'21.9" | NAS | 122°16'21.9" | NAS | 121°53'59.0" | NAS | 121°53'59.0" | NAS | 121°53'59.0" | NAS |
| 4.2 | Longitude | 122°13'53.9" | NAS | 37°48'41.1" | NAS | 37°54'49.4" | NAS | 37°48'41.1" | NAS | 37°48'41.1" | NAS | 37°25'57.7" | NAS | 37°25'57.7" | NAS | 37°25'57.7" | NAS |
| 4.4 | Altitude | 100 m | NAS | 13 m | NAS | 40 m | NAS | 13 m | NAS | 13 m | NAS | 6 m | NAS | 6 m | NAS | 6 m | NAS |
aEvidence codes – IDA inferred from direct assay, TAS traceable author statement, NAS non-traceable author statement, nd not determined. These evidence codes are from the Gene Ontology project [90]
Fig. 2.
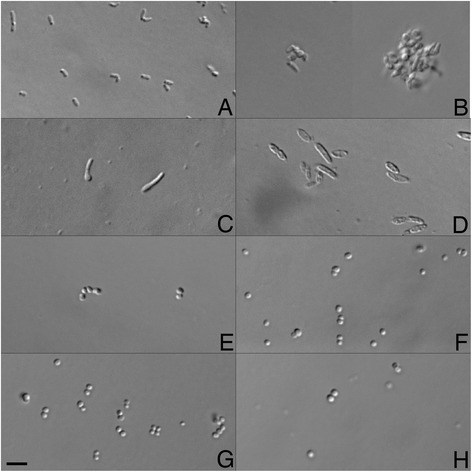
Transmitted light microscope images of the eight isolates. Bar is 5 μm. a Rod-shaped cells of Microbacterium sp. H83 b Mycobacterium iranicum H39; note, this organism was sparse in the images and tended to be highly clumped, so two snapshots were used for the sake of visualization c pleomorphic, rod-shaped cells of Plantibacter sp. H53 d Pseudomonas oryzihabitans H72, rods with rounded ends typically occurring as solitary cells but rarely also in pairs, e Staphylococcus capitis H36, occurring in pairs or strings of cells f Staphylococcus capitis H65, as single cells and pairs g Staphylococcus cohnii H62, as single cells, pairs, and occasionally threes or tetrads, h Staphylococcus hominis H69, as single cells and pairs. Images were collected using a Zeiss M1 AxioImager equipped with DIC and a Hamamatsu Orca 03 camera run by BioVision’s iVision software. Images were cropped and organized into a plate using Adobe Photoshop CS6
Staphylococcus are non-spore-forming, non-motile round-shaped cells (Fig. 2 e-h). They demonstrate habitat preference in the human body with S. capitis mainly being found on the adult head and S. cohnii on the feet [10]. S. hominis is the main colonizer of head, axillae, arms, and legs, and is frequently encountered in nosocomial infections.
Organisms within the genus Microbacterium spp. are yellow-pigmented, aerobic, rod-shaped, Gram-positive bacteria [23] (Fig. 2a). They have been isolated from numerous and variable environments, including soil and water [24], the phyllosphere [25], human patients [26], and a residential toilet [27], and they have been associated with endophthalmitis [28] and catheter infections [29].
Pseudomonas oryzihabitans (Fig. 2d) is a Gram-negative, non-fermenting, yellow-pigmented bacterium [30]. Despite its environmental origin, P. oryzihabitans has been recognized as a potential pathogen in recent years, especially in immunocompromised hosts, both in nosocomial or community-level settings. It can form biofilms in aquifers in association with suspended particulate matter, which can be subsequently entrained into the drinking water distribution systems, posing a potential risk for human health given their resistance to chlorine compared to their planktonic counterparts [13]. This species has been associated with catheter [31] and bloodstream infections, endophthalmitis [32], necrotic enteritis [33], and peritonitis ([34] and references therein). There are two instances in which the source of human infection has been well documented, and the source has been found to be a synthetic sponge, one used by an immunocompromised individual [31] and another in the milk kitchen of a neonatal intensive care unit [33].
Mycobacterium iranicum (Fig. 2b) is a newly described, rapidly growing, orange-pigmented scotochromogenic, non-tuberculous mycobacterial species. Its clinical significance is still under study but it has been associated with patients with pulmonary infections, such as pneumonia, chronic obstructive airway disease, and bronchiectasis [11, 35].
Lastly, Plantibacter (Fig. 2c) are pleomorphic, rod-shaped, yellow-pigmented, aerobic, Gram-positive bacteria that belong to the class of Actinobacteria.
Genome sequencing information
Genome project history
These genomes were generated as part of a project to sequence reference genomes from the built Environment, funded by the Alfred P. Sloan Foundation through their “Microbiology of the Built Environment” Program. Sequencing and assembly of all isolates were performed at the University of California, Davis. The genome sequences were deposited in GenBank and given a Genome On-Line Database identifier [36]. Project information and association with MIGS version 2.0 are presented in Table 2.
Table 2.
Project information
| MIGS ID | Property | Microbacterium sp. H83 | Mycobacterium iranicum H39 | Plantibacter sp. H53 | Pseudomonas oryzihabitans H72 | Staphylococcus capitis H36 | S. capitis H65 | S. cohnii H62 | S. hominis H69 |
|---|---|---|---|---|---|---|---|---|---|
| MIGS 31 | Finishing quality | Permanent Draft | Permanent Draft | Permanent Draft | Permanent Draft | Permanent Draft | Permanent Draft | Permanent Draft | Permanent Draft |
| MIGS-28 | Libraries used | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) | Illumina PE library (300 bp insert size) |
| MIGS 29 | Sequencing platforms | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq | Illumina MiSeq |
| MIGS 31.2 | Fold coverage | 239x | 95x | 115x | 112x | 258x | 170x | 157x | 64x |
| MIGS 30 | Assemblers | A5-miseq | A5-miseq | A5-miseq | A5-miseq | A5-miseq | A5-miseq | A5-miseq | A5-miseq |
| MIGS 32 | Gene calling method | IMG | IMG | IMG | IMG | IMG | IMG | IMG | IMG |
| Locus Tag | A4X16 | A4X20 | A4X17 | A4X15 | A4X14 | A4X13 | A4A82 | A3836 | |
| Genbank ID | LWCU01000000.1 | LWCS01000000.1 | LWCT01000000.1 | LWCR01000000.1 | LWCQ01000000.1 | LWCP01000000.1 | LWAC01000000.1 | LVVO01000000.1 | |
| Genbank Date of Release | May 24, 2016 | May 24, 2016 | May 24, 2016 | May 24, 2016 | May 24, 2016 | May 24, 2016 | May 24, 2016 | May 24, 2016 | |
| GOLD ID | Gp0147178 | Gp0147183 | Gp0147185 | Gp0147186 | Gp0147187 | Gp0147188 | Gp0147192 | Gp0147190 | |
| BIOPROJECT | PRJNA317658 | PRJNA317657 | PRJNA317656 | PRJNA317602 | PRJNA317600 | PRJNA317599 | PRJNA316869 | PRJNA316465 | |
| MIGS 13 | Source Material Identifier | DSM-103506 | DSM-103542 | DSM-103507 | DSM-103505 | DSM-103511 | DSM-103512 | DSM-103510 | DSM-103553 |
| Project relevance | Built Environment Reference Genomes | ||||||||
Growth conditions and genomic DNA preparation
Strains were initially collected through environmental sampling (see Classification and features section) and were subsequently deposited into the DMSZ. Glycerol stocks of all isolates were initially grown at 28 °C on LB plates. A single colony was then inoculated in LB and incubated at 28 °C for 18 h (except for M. iranicum strain H39, grown at 37 °C for 5 days). DNA was subsequently extracted from the cultures using the DNeasy Blood and Tissue kit (Qiagen), and the quality was assessed using a NanoDropTM spectrophotometer.
Genome sequencing and assembly
Barcoded Illumina paired-end libraries were generated from all samples using the Nextera XT kit (Illumina). After pooling, the libraries were size-selected for a range of 600–900 bp on a Pippin Prep (Sage Science) and then sequenced on an Illumina MiSeq (Paired End 300 bp). After demultiplexing with a custom script, the reads from each sample were assembled using the A5-miseq pipeline, which automates the process of adapter removal, quality trimming, error-correction, and contig generation [37, 38]. The completeness and contamination of the assemblies was estimated using PhyloSift [22] and CheckM [39]. Across all strains, genome completeness was determined to be a minimum of 98.9%, and the maximum contamination was 0.99% (Additional file 1).
Genome annotation
Isolates were predominantly annotated using the IMG system [40] with no additional manual curation. Table 3 summarizes genome statistics and Table 4 the COG functional categories for the eight isolates according to IMG. Additional annotations were performed with PGAP [41] and RAST [42]. The full-length 16S rRNA gene sequences for each isolate, used for tree building (see above), were extracted from RAST.
Table 3.
Genome statistics
| Microbacterium sp. H83 | Mycobacterium iranicum H39 | Plantibacter sp. H53 | Pseudomonas oryzihabitans H72 | Staphylococcus capitis H36 | S. capitis H65 | S. cohnii H62 | S. hominis H69 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Attribute | Value | %a | Value | % | Value | % | Value | % | Value | % | Value | % | Value | % | Value | % |
| Genome size (bp) | 3,531,197 | 100 | 6,470,840 | 100 | 4,012,045 | 100 | 5,316,471 | 100 | 2,412,840 | 100 | 2,482,551 | 100 | 2,656,939 | 100 | 2,335,200 | 100 |
| DNA coding (bp) | 3,256,592 | 92 | 5,997,369 | 93 | 3,678,027 | 92 | 4,723,080 | 89 | 2,064,637 | 86 | 2,146,765 | 86 | 2,213,412 | 83 | 2,024,342 | 87 |
| DNA G + C (bp) | 2,459,099 | 70 | 4,277,463 | 66 | 2,783,137 | 69 | 3,459,720 | 65 | 789,696 | 33 | 812,206 | 33 | 859,087 | 32 | 733,146 | 31 |
| DNA scaffolds/contigs | 52 | 100 | 91 | 100 | 50 | 100 | 78 | 100 | 24 | 100 | 31 | 100 | 262 | 100 | 143 | 100 |
| Total genes | 3522 | 100 | 6227 | 100 | 3826 | 100 | 5005 | 100 | 2454 | 100 | 2476 | 100 | 2761 | 100 | 2450 | 100 |
| Protein coding genes | 3462 | 98 | 6162 | 99 | 3763 | 98 | 4897 | 98 | 2355 | 96 | 2378 | 96 | 2666 | 97 | 2350 | 96 |
| RNA genes | 60 | 2 | 65 | 1 | 63 | 2 | 108 | 2 | 99 | 4 | 98 | 4 | 95 | 3 | 100 | 4 |
| Pseudo genes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Genes in internal clusters | 874 | 25 | 2189 | 35 | 1075 | 28 | 1487 | 30 | 451 | 18 | 456 | 18 | 593 | 21 | 464 | 19 |
| Genes with function prediction | 2637 | 75 | 4691 | 75 | 2943 | 77 | 3969 | 79 | 1970 | 80 | 1970 | 80 | 2151 | 78 | 1907 | 78 |
| Genes assigned to COGs | 2271 | 64 | 3929 | 63 | 2567 | 67 | 3605 | 72 | 1759 | 72 | 1802 | 73 | 1877 | 68 | 1708 | 70 |
| Genes with Pfam domains | 2783 | 79 | 4989 | 80 | 3093 | 81 | 4209 | 84 | 2044 | 83 | 2057 | 83 | 2231 | 81 | 1979 | 81 |
| Genes with signal peptides | 148 | 4 | 366 | 6 | 164 | 4 | 496 | 10 | 67 | 3 | 70 | 3 | 55 | 2 | 95 | 3 |
| Genes with transmembrane helices | 953 | 27 | 1365 | 22 | 1117 | 29 | 1138 | 23 | 637 | 26 | 632 | 26 | 671 | 24 | 575 | 23 |
| CRISPR repeats | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
aThe percentage of total is based on either the size of the genome in base pairs or the total number of genes in the annotated genome
Table 4.
Numbers of genes associated with general COG functional categories
| Microbacterium sp. H83 | Mycobacterium iranicum H39 | Plantibacter sp. H53 | Pseudomonas oryzihabitans H72 | Staphylococcus capitis H36 | S. capitis H65 | S. cohnii H62 | S. hominis H69 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Code | Description | Value | %a | Value | % | Value | % | Value | % | Value | % | Value | % | Value | % | Value | % |
| J | Translation, ribosomal structure and biogenesis | 167 | 4.8 | 191 | 3.1 | 172 | 4.6 | 242 | 4.9 | 188 | 8.0 | 191 | 8.0 | 192 | 7.2 | 187 | 8.0 |
| A | RNA processing and modification | 1 | 0.0 | 1 | 0.0 | 1 | 0.0 | 1 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| K | Transcription | 265 | 7.7 | 403 | 6.5 | 306 | 8.1 | 328 | 6.7 | 125 | 5.3 | 132 | 5.6 | 141 | 5.3 | 128 | 5.4 |
| L | Replication, recombination and repair | 109 | 3.1 | 120 | 1.9 | 105 | 2.8 | 134 | 2.7 | 86 | 3.7 | 95 | 4.0 | 89 | 3.3 | 101 | 4.3 |
| B | Chromatin structure and dynamics | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 0.0 | 1 | 0.0 | 1 | 0.0 | 1 | 0.0 | 0 | 0.0 |
| D | Cell cycle control, Cell division, chromosome partitioning | 24 | 0.7 | 33 | 0.5 | 24 | 0.6 | 38 | 0.8 | 24 | 1.0 | 26 | 1.1 | 25 | 0.9 | 27 | 1.1 |
| V | Defense mechanisms | 50 | 1.4 | 118 | 1.9 | 71 | 1.9 | 80 | 1.6 | 50 | 2.1 | 41 | 1.7 | 49 | 1.8 | 38 | 1.6 |
| T | Signal transduction mechanisms | 87 | 2.5 | 189 | 3.1 | 121 | 3.2 | 286 | 5.8 | 67 | 2.8 | 68 | 2.9 | 65 | 2.4 | 66 | 2.8 |
| M | Cell wall/membrane biogenesis | 115 | 3.3 | 222 | 3.6 | 140 | 3.7 | 245 | 5.0 | 96 | 4.1 | 101 | 4.2 | 104 | 3.9 | 108 | 4.6 |
| N | Cell motility | 31 | 0.9 | 11 | 0.2 | 9 | 0.2 | 153 | 3.1 | 6 | 0.3 | 8 | 0.3 | 5 | 0.2 | 4 | 0.2 |
| U | Intracellular trafficking and secretion | 28 | 0.8 | 22 | 0.4 | 17 | 0.5 | 77 | 1.6 | 19 | 0.8 | 20 | 0.8 | 14 | 0.5 | 18 | 0.8 |
| O | Posttranslational modification, protein turnover, chaperones | 97 | 2.8 | 138 | 2.2 | 92 | 2.4 | 161 | 3.3 | 75 | 3.2 | 78 | 3.3 | 78 | 2.9 | 74 | 3.1 |
| C | Energy production and conversion | 141 | 4.1 | 331 | 5.4 | 128 | 3.4 | 243 | 5.0 | 111 | 4.7 | 111 | 4.7 | 109 | 4.1 | 99 | 4.2 |
| G | Carbohydrate transport and metabolism | 270 | 7.8 | 244 | 4.0 | 392 | 10.4 | 275 | 5.6 | 131 | 5.6 | 133 | 5.6 | 150 | 5.6 | 118 | 5.0 |
| E | Amino acid transport and metabolism | 274 | 7.9 | 337 | 5.5 | 304 | 8.1 | 403 | 8.2 | 174 | 7.4 | 188 | 7.9 | 202 | 7.6 | 170 | 7.2 |
| F | Nucleotide transport and metabolism | 77 | 2.2 | 98 | 1.6 | 82 | 2.2 | 91 | 1.9 | 79 | 3.4 | 78 | 3.3 | 82 | 3.1 | 80 | 3.4 |
| H | Coenzyme transport and metabolism | 145 | 4.2 | 295 | 4.8 | 156 | 4.1 | 207 | 4.2 | 136 | 5.8 | 136 | 5.7 | 125 | 4.7 | 126 | 5.4 |
| I | Lipid transport and metabolism | 113 | 3.3 | 478 | 7.8 | 134 | 3.6 | 168 | 3.4 | 91 | 3.9 | 88 | 3.7 | 93 | 3.5 | 78 | 3.3 |
| P | Inorganic ion transport and metabolism | 166 | 4.8 | 244 | 4.0 | 171 | 4.5 | 261 | 5.3 | 136 | 5.8 | 143 | 6.0 | 141 | 5.3 | 136 | 5.8 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 58 | 1.7 | 322 | 5.2 | 65 | 1.7 | 94 | 1.9 | 40 | 1.7 | 43 | 1.8 | 46 | 1.7 | 36 | 1.5 |
| R | General function prediction only | 233 | 6.7 | 571 | 9.3 | 285 | 7.6 | 348 | 7.1 | 183 | 7.8 | 184 | 7.7 | 197 | 7.4 | 164 | 7.0 |
| S | Function unknown | 107 | 3.1 | 224 | 3.6 | 138 | 3.7 | 217 | 4.4 | 139 | 5.9 | 143 | 6.0 | 156 | 5.9 | 128 | 5.4 |
| - | Not in COGs | 1251 | 36.1 | 2298 | 37.3 | 1259 | 33.5 | 1400 | 28.6 | 695 | 29.5 | 674 | 28.3 | 884 | 33.2 | 742 | 31.6 |
a Percent of annotated genes. The total is based on the total number of protein coding genes in the genome
Genome properties
Genome sizes were smallest for the Staphylococcus isolates at approximately 2.5 Mbps and largest for M. iranicum H39 at nearly 6.5 Mbps (Table 3). Similarly, the DNA G + C content was lowest for the Staphylococcus isolates (approximately 31%) and much higher for the other four isolates (at least 65% content). Predicted coding regions accounted for 83–93% of the genomes for all eight isolates, and the total number of predicted genes ranged from 2450 in S. hominis H69 to 6227 in M. iranicum H39. The percentage of genes with a functional prediction was fairly consistent across the genomes, ranging from 75 to 80%. The percentage of RNA genes for the Staphylococcus isolates ranged from 3 to 4% and were higher than the others isolates (1–2%). Conversely, the percentage of genes in internal clusters (an indicator of non-redundant sequences) ranged from 18 to 21% in the Staphylococcus isolates but ranged from 25 to 35% in the other isolates. The genome of P. oryzihabitans H72 encoded a much higher percentage of signal peptides than the other genomes (Tables 3 and 4). Neither pseudogenes nor CRISPR repeats were identified in any of the genomes.
For all strains, 27–37% of the proteins were not predicted to be part of a COG category (Table 4). P. oryzihabitans was the only recognized motile organism (Table 1), and P. oryzihabitans H72 showed a much greater percentage of genes related to motility (Table 4). M. iranicum H39 harbored a much higher percentage of genes for the COG categories of lipid transport/metabolism and secondary metabolites biosynthesis/transport/catabolism than the other isolates. There was no observed relationship between genome coverage (Table 2) and the percentage of unassigned proteins (Table 4).
Insights from the genome sequences
Phylogenetic comparisons
The genomes of the sequenced isolates were compared to publicly available closely related genomes to determine the ANI values [43]. For those six isolates in which a species epithet was given based on gene trees, ANI values were greater than 90% (Additional file 6), and were greater than 96% for the Staphylococcus isolates. The genomes of those isolates that were assigned to genera based on gene trees were compared to closely related publicly available genomes. For Microbacterium sp. H83, the ANI value with M. hydrocarbonoxydans was 84.1% and for Plantibacter sp. H53 was 87.8% with another Plantibacter sp. (Additional file 6).
Virulence and biofilm production
CoNS are opportunistic pathogens and they do not encode for virulence factors (e.g., exotoxins) commonly found in pathogenic species such as S. aureus. However, they do encode genes related to biofilm formation, persistence and immune invasion [44]. The attachment to a surface is the first step to successful colonization and a precursor for the establishment of infection. In the IMG annotation, we found genes with predicted functions to be associated with cell wall-associated FBP, such as fbe, and several other surface-associated proteins such as a bifunctional autolysin and putative adhesins. However, the gene fbe was not found in S. capitis H36, and another gene known to be important for surface adhesion in Staphylococcus, ebh [44], was not observed in any isolate. Both Ebh and FBP act as adhesins but FBP also acts as an invasin, facilitating binding and internalization in host cells [45]. Additionally, we found genes with predicted functions to be associated with Microbial Surface Components Recognizing Adhesive Matrix Molecules, such as the sdrG gene. Further biofilm accumulation is mediated by exopolysaccharides such as PNAG and PGA. Genes related only to PGA (cap operon), which have been shown to provide resistance to phagocytosis and to a host’s antimicrobial peptides in S. epidermidis [46], were identified. Genes encoding predicted pro-inflammatory molecules with cytolytic and antimicrobial properties such as β-type phenol soluble modulins (PSM) [44] were found in all four staphylococci strains, along with genes encoding their accessory regulator B (Agr) [47]. Other systems important for the regulation of virulence in staphylococci that were found in our strains included the staphylococcal accessory regulator Sar, one of the two components of each of the regulatory systems, SaeRS and ArlRS, and an infection-related protease, ClpC [44].
Antibiotic resistance
We used the Resistance Gene Identifier of CARD [48] to explore possible genes related to antimicrobial resistance. Microbial genome sequencing has the potential to be used as a prediction tool of antibiotic resistance in clinical settings [49, 50], and in fact has been shown to be a promising approach in S. aureus [51, 52] as well as other bacteria [53]. However, at the moment, clinical testing of antibiotic resistance is restricted to PCR-based targeting of specific genes [54, 55], and many of the genes in antibiotic databases have not been verified in clinical settings and are subject to errors in annotation (e.g., [56]). Nevertheless, we surveyed genes predicted to confer antibiotic resistance in order to explore commonalities across the different isolates. Additional file 7 details the Gene ID and other information stemming from the IMG annotation of putative antibiotic resistance genes identified in CARD. Limiting the results to “perfect” and “strict” hits, many of these genes included efflux pumps predicted to confer resistance to more than one class of antimicrobials (e.g., fluoroquinolones, tetracyclines, polymyxins) as well as genes predicted to be associated with resistance to specific antimicrobials (e.g., beta-lactams, aminocoumarins, chloramphenicol, aminoglycosides, and fosfomycin). Some antimicrobial genes were common to many strains; others were limited to specific taxonomic groups. For example, all eight strains were found to contain genes predicted to confer resistance to mupirocin and fosfomycin, while genes for fusidic acid resistance were only observed in S. capitis H65 (Additional file 7).
In addition to general targeting of antibiotic resistance genes, we also looked specifically for genes related to triclosan resistance. TCS is a synthetic antimicrobial agent that is commonly used in home and personal care products such as hand soaps, toothpastes, deodorants, body washes, hand creams, body lotions, and cosmetics. It has been directly associated with the development of multidrug antibiotic resistance in a variety of primarily pathogenic bacteria via in vitro assays [57]. TCS induces resistance through mutations in the gene (fabI) that encodes TCS’s target enzyme (enoyl-acyl carrier protein reductase FabI) through overexpression, or through efflux pumps, with the latter only to be associated with multi-antibiotic resistance [57]. The fabI gene was identified only in one out of four staphylococci isolates, S. capitis H65, as well as in the M. iranicum H39 and P. oryzihabitans H72 genomes. We found several genes related to non-specific multidrug efflux pumps, such as mex genes (mexJKL) in their genomes. The MexJK efflux pump can efflux triclosan, but also requires the outer membrane protein channel composed of the OprM in order to efflux other antibiotics in Pseudomonas aeruginosa [58]. MexJK-OprM was found through CARD in all our genomes, except for Plantibacter sp. H53 that did not carry OprM. The triclosan efflux transporter TriABC–OpmH [59] was only partially present in P. oryzihabitans H72 (TriB was absent). Additionally, P. oryzihabitans H72 was the only isolate to contain an efflux pump predicted to offer triclosan resistance (Additional file 7). Susceptibility to TCS or other antibiotics has not been experimentally tested for the strains described here.
Conclusions
The genomes of these eight isolates of bacteria collected from a residential environment will be valuable tools for exploring the basic microbiology of indoor microbes (e.g., overexpression of genes targeted by drugs/antimicrobial agents, such as triclosan, can provide insight into the mode of action of antibiotics and the associated development of resistance) as well as interpreting future metagenomic and transcriptomic datasets. These isolates represent seven species across five genera and likely originate from the dominant sources of indoor bacteria: the outdoor environment, human commensals, and premise plumbing.
Acknowledgements
We would like to thank the UC Davis DNA Technologies Core for help with sequencing.
Funding
This work was supported by the Alfred P. Sloan Foundation to the Microbiology of the Built Environment Network (microBE.net) at UC Davis and to the Berkeley Indoor Microbial Ecology Research Consortium (BIMERC) at UC Berkeley.
Authors’ contributions
Conceived and designed the experiments: DSL SEL. Performed the experiments, maintained the isolates, and extracted DNA: DSL. Prepared the sequencing libraries: DAC. Contributed reagents/materials/analysis tools: DSL DAC SEL GJ JAE. Performed imaging: DS. Analyzed the data: DSL DAC GJ RIA. Drafted the manuscript: DSL DAC RIA. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- ANI
Average nucleotide identity
- BLAST
Basic local alignment search tool
- CARD
Comprehensive antibiotic resistance database
- COG
Clusters of orthologous groups
- CoNS
Coagulase-negative staphylococci
- FBP
Fibronectin/fibrinogen binding protein
- IMG
Integrated microbial genomes
- Pfam
Protein families
- PGA
Poly-γ-glutamate
- PNAG
Poly-N-acetylglucosamine
- RAST
Rapid annotation using subsystem technology
- TCS
Triclosan
Additional files
Additional strain information, including growth conditions and genome completeness/contamination. (XLSX 36 kb)
Phylogenetic tree of Microbacterium sp. H83. (PDF 153 kb)
Phylogenetic tree of Plantibacter sp. H53. (PDF 153 kb)
Phylogenetic tree of Mycobacterium iranicum H39. (PDF 146 kb)
Phylogenetic tree of Pseudomonas oryzihabitans H72. (PDF 111 kb)
ANI values between these eight strains and selected genomes in the IMG database. (XLSX 8 kb)
Antibiotic genes detected in the eight genomes using CARD. (XLSX 47 kb)
References
- 1.Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, et al. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J Expo Sci Environ Epidemiol. 2001;11(3):231–52. doi: 10.1038/sj.jea.7500165. [DOI] [PubMed] [Google Scholar]
- 2.Barberán A, Dunn RR, Reich BJ, Pacifici K, Laber EB, Menninger HL, et al. The ecology of microscopic life in household dust. Proc R Soc Lond [Biol] 2015;282(1814):20151139. doi: 10.1098/rspb.2015.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams RI, Miletto M, Taylor JW, Bruns TD. The diversity and distribution of fungi on residential surfaces. PLoS One. 2013;8(11):e78866. doi: 10.1371/journal.pone.0078866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn RR, Fierer F, Henley JB, Leff JW, Menninger HL. Home life: factors structuring the bacterial diversity found within and between homes. PLoS One. 2013;8(5):e64133. doi: 10.1371/journal.pone.0064133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores GE, Bates S, Caporaso JG, Lauber CL, Leff JW, Knight R, et al. Diversity, distribution and sources of bacteria in residential kitchens. Environ Microbiol. 2013;15(2):588–96. doi: 10.1111/1462-2920.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR. Opportunistic pathogens enriched in showerhead biofilms. Proc Natl Acad Sci U S A. 2009;106(38):16393–98. doi: 10.1073/pnas.0908446106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miletto M, Lindow SE. Relative and contextual contribution of different sources to the composition and abundance of indoor air bacteria in residences. Microbiome. 2015;3:61. doi: 10.1186/s40168-015-0128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.American Academy of Microbiology. FAQ: Microbiology of Built Envrionments. In: Colloquim Reports. 2016. http://academy.asm.org/images/Colloquia-report/FAQ_Microbiology_of_Built_Environments.pdf. Accessed 2 Nov 2016.
- 9.Prussin AJ, Marr LC. Sources of airborne microorganisms in the built environment. Microbiome. 2015;3(1):1–10. doi: 10.1186/s40168-015-0144-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piette A, Verschraegen G. Role of coagulase-negative staphylococci in human disease. Vet Microbiol. 2009;134(1–2):45–54. doi: 10.1016/j.vetmic.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Shojaei H, Daley C, Gitti Z, Hashemi A, Heidarieh P, Moore ER, et al. Mycobacterium iranicum sp. nov., a rapidly growing scotochromogenic species isolated from clinical specimens on three different continents. Int J Syst Evol Microbiol. 2013;63(4):1383–89. doi: 10.1099/ijs.0.043562-0. [DOI] [PubMed] [Google Scholar]
- 12.Tan JL, Ngeow YF, Wee WY, Wong GJ, Ng HF, Choo SW. Comparative genomic analysis of Mycobacterium iranicum UM_TJL against representative mycobacterial species suggests its environmental origin. Sci Rep. 2014;4:7169. doi: 10.1038/srep07169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dussart L, Dupont JP, Zimmerlin I, Lacroix M, Saiter JM, Junter GA, et al. Occurrence of sessile Pseudomonas oryzihabitans from a karstified chalk aquifer. Water Res. 2003;37(7):1593–600. doi: 10.1016/S0043-1354(02)00555-9. [DOI] [PubMed] [Google Scholar]
- 14.Izumi H, Anderson IC, Killham K, Moore ERB. Diversity of predominant endophytic bacteria in European deciduous and coniferous trees. Can J Microbiol. 2008;54(3):173–79. doi: 10.1139/W07-134. [DOI] [PubMed] [Google Scholar]
- 15.Behrendt U, Ulrich A, Schumann P, Naumann D, Suzuki K-i. Diversity of grass-associated Microbacteriaceae isolated from the phyllosphere and litter layer after mulching the sward; polyphasic characterization of Subtercola pratensis sp. nov., Curtobacterium herbarum sp. nov. and Plantibacter flavus gen. nov., sp. nov. Int J Syst Evol Microbiol. 2002;52(5):1441–54. doi: 10.1099/00207713-52-5-1441. [DOI] [PubMed] [Google Scholar]
- 16.Cole JR, Wang Q, Fish JA, Chai BL, McGarrell DM, Sun YN, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(D1):D633–D42. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 18.Dunitz MI, Lang JM, Jospin G, Darling AE, Eisen JA, Coil DA. Swabs to genomes: a comprehensive workflow. PeerJ. 2015;3. doi:10.7717/peerj.960. [DOI] [PMC free article] [PubMed]
- 19.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–50. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coil D, Jospin G. https://figshare.com/articles/Alignments_zip/3491945.
- 21.Wu DY, Jospin G, Eisen JA. Systematic identification of gene families for use as “markers” for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PLoS One. 2013;8(10):e77033. doi: 10.1371/journal.pone.0077033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darling AE, Jospin G, Lowe E, Matsen FIV, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2:e243. doi: 10.7717/peerj.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki K-I, Hamada M. Genus I. Microbacterium. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 5: The Actinobacteria. 2. New York: Springer; 2012. pp. 814–52. [Google Scholar]
- 24.Collins MD, Bradbury JF. The genera Agromyces, Aureobacterium, Clavibacter, Curtobacterium, and Microbacterium. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer KH, editors. The Prokaryotes. 2. New York: Springer; 1992. pp. 1355–68. [Google Scholar]
- 25.Behrendt U, Ulrich A, Schumann P. Description of Microbacterium foliorum sp. nov. and Microbacterium phyllosphaerae sp. nov., isolated from the phyllosphere of grasses and the surface litter after mulching the sward, and reclassification of Aureobacterium resistens (Funke et al. 1998) as Microbacterium resistens comb. nov. Int J Syst Evol Microbiol. 2001;51(4):1267–76. doi: 10.1099/00207713-51-4-1267. [DOI] [PubMed] [Google Scholar]
- 26.Funke G, Falsen E, Barreau C. Primary identification of Microbacterium spp. encountered in clinical specimens as CDC coryneform group A-4 and A-5 bacteria. J Clin Microbiol. 1995;33(1):188–92. doi: 10.1128/jcm.33.1.188-192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bendiks ZA, Lang JM, Darling AE, Eisen JA, Coil DA. Draft genome sequence of Microbacterium sp. strain UCD-TDU (phylum Actinobacteria) Genome Announc. 2013;1(2):e00120–13. doi: 10.1128/genomeA.00120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funke G, Haase G, Schnitzler N, Schrage N, Reinert RR. Endophthalmitis due to Microbacterium species: case report and review of Microbacterium infections. Clin Infect Dis. 1997;24(4):713–16. doi: 10.1093/clind/24.4.713. [DOI] [PubMed] [Google Scholar]
- 29.Lau SKP, Woo PCY, Woo GKS, Yuen K-Y. Catheter-related Microbacterium bacteremia identified by 16S rRNA gene sequencing. J Clin Microbiol. 2002;40(7):2681–85. doi: 10.1128/JCM.40.7.2681-2685.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palleroni NJ. Genus I. Pseudomonas. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology Vol 2: The Proteobacteria. New York: Springer; 2005. pp. 323–79. [Google Scholar]
- 31.Marín M, de Viedma DG, Martín-Rabadán P, Rodríguez-Créixems M, Bouza E. Infection of Hickman catheter by Pseudomonas (formerly Flavimonas) oryzihabitans traced to a synthetic bath sponge. J Clin Microbiol. 2000;38(12):4577–79. doi: 10.1128/jcm.38.12.4577-4579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu E, Foster C. Chronic postoperative endophthalmitis due to Pseudomonas oryzihabitans. Am J Ophthalmol. 2002;134(4):613–14. doi: 10.1016/S0002-9394(02)01586-6. [DOI] [PubMed] [Google Scholar]
- 33.Prifti H, Oikonomidou D, Pappa O, Tryfinopoulou K, Vatzeli K, Karaiskos K, et al. Outbreak of Pseudomonas (Flavimonas) oryzihabitans bacteraemia in a neonatal intensive care unit. In conference proceedings: 21st European Society of Clinical Microbiology and Infectious Diseases; Milan, Italy. 2011
- 34.Dussart-Baptista L, Bodilis J, Barray S, Frébourg N, Fournier M, Dupont JP, et al. Recurrent recovery of Pseudomonas oryzihabitans strains in a karstified chalk aquifer. Water Res. 2007;41(1):111–17. doi: 10.1016/j.watres.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Balakrishnan N, Tortoli E, Engel SL, Breitschwerdt EB. Isolation of a novel strain of Mycobacterium iranicum from a woman in the United States. J Clin Microbiol. 2013;51(2):705–07. doi: 10.1128/JCM.02560-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy T, Thomas AD, Stamatis D, Bertsch J, Isbandi M, Jansson J, et al. The Genomes OnLine Database (GOLD) v. 5: a metadata management system based on a four level (meta) genome project classification. Nucleic Acids Res. 2014;43:D1099–106. [DOI] [PMC free article] [PubMed]
- 37.Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31(4):587–89. doi: 10.1093/bioinformatics/btu661. [DOI] [PubMed] [Google Scholar]
- 38.Tritt A, Eisen JA, Facciotti MT, Darling AE. An integrated pipeline for de novo assembly of microbial genomes. PLoS One. 2012;7(9):e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(D1):D115–D22. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (Meta) genomic annotation. Omics. 2008;12(2):137–41. doi: 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102(7):2567–72. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cameron DR, Jiang J-H, Hassan KA, Elbourne LDH, Tuck KL, Paulsen IT, et al. Insights on virulence from the complete genome of Staphylococcus capitis. Front Microbiol. 2015;6:980. doi: 10.3389/fmicb.2015.00980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinha B, Francois P, Que Y-A, Hussain M, Heilmann C, Moreillon P, et al. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect Immun. 2000;68(12):6871–78. doi: 10.1128/IAI.68.12.6871-6878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kocianova S, Vuong C, Yao Y, Voyich JM, Fischer ER, DeLeo FR, et al. Key role of poly-γ-DL-glutamic acid in immune evasion and virulence of Staphylococcus epidermidis. J Clin Invest. 2005;115(3):688–94. doi: 10.1172/JCI200523523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang R, Braughton KR, Kretschmer D, Bach T-HL, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nature Med. 2007;13(12):1510–14. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 48.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57(7):3348–57. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nature Rev Genet. 2012;13(9):601–12. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Köser CU, Ellington MJ, Cartwright EJP, Gillespie SH, Brown NM, Farrington M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8(8):e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McAdam PR, Holmes A, Templeton KE, Fitzgerald JR. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One. 2011;6(9):e24301. doi: 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon N, Price J, Cole K, Everitt R, Morgan M, Finney J, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol. 2014;52(4):1182–91. doi: 10.1128/JCM.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bradley P, Gordon NC, Walker TM, Dunn L, Heys S, Huang B, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun. 2015;6:10063. [DOI] [PMC free article] [PubMed]
- 54.Wolk D, Picton E, Johnson D, Davis T, Pancholi P, Ginocchio C, et al. Multicenter evaluation of the Cepheid Xpert methicillin-resistant Staphylococcus aureus (MRSA) test as a rapid screening method for detection of MRSA in nares. J Clin Microbiol. 2009;47(3):758–64. doi: 10.1128/JCM.01714-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hillemann D, Weizenegger M, Kubica T, Richter E, Niemann S. Use of the genotype MTBDR assay for rapid detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis complex isolates. J Clin Microbiol. 2005;43(8):3699–703. doi: 10.1128/JCM.43.8.3699-3703.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Apagyi KI, Ellington MJ. A survey of metallo-β-lactamase sequence accuracy before the data deluge. J Antimicrob Chemother. 2014 doi: 10.1093/jac/dku284. [DOI] [PubMed] [Google Scholar]
- 57.Carey DE, McNamara PJ. The impact of triclosan on the spread of antibiotic resistance in the environment. Front Microbiol. 2015;5:780. doi: 10.3389/fmicb.2014.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chuanchuen R, Narasaki CT, Schweizer HP. The MexJK efflux pump of Pseudomonas aeruginosa requires OprM for antibiotic efflux but not for efflux of triclosan. J Bacteriol. 2002;184(18):5036–44. doi: 10.1128/JB.184.18.5036-5044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mima T, Joshi S, Gomez-Escalada M, Schweizer HP. Identification and characterization of TriABC-OpmH, a Triclosan efflux pump of pseudomonas aeruginosa requiring two membrane fusion proteins. J Bacteriol. 2007;189(21):7600–09. doi: 10.1128/JB.00850-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–47. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–79. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodfellow M. Phylum XXVI. Actinobacteria phyl. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 5: The Actinobacteria. New York: Springer; 2012. pp. 33–2028. [Google Scholar]
- 63.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology Vol 2: The Proteobacteria. New York: Springer; 2005. p. 1. [Google Scholar]
- 64.Gibbons NE, Murray RGE. Proposals concerning the higher taxa of bacteria. Int J Syst Bacteriol. 1978;28(1):1–6. doi: 10.1099/00207713-28-1-1. [DOI] [Google Scholar]
- 65.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a New hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol. 1997;47(2):479–91. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 66.Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology Vol 2: The Proteobacteria. New York: Springer; 2005. p. 1. [Google Scholar]
- 67.List Editor Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol. 2005;55:2235–38. doi: 10.1099/ijs.0.64108-0. [DOI] [PubMed] [Google Scholar]
- 68.Ludwig W, Schleifer KH, Whitman WB. Class I. Bacilli class nov. In: Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 3: The Firmicutes. New York: Springer; 2009. pp. 19–1317. [Google Scholar]
- 69.List Editor List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol. 2010;60:469–72. doi: 10.1099/ijs.0.022855-0. [DOI] [PubMed] [Google Scholar]
- 70.Prévot AR. Dictionnaire des Bactéries Pathogènes. In: Hauduroy P, Ehringer G, Guillot G, Magrou J, Prevot AR, Rosset, Urbain A, editors. Dictionnaire des Bactéries Pathogènes. 2. Paris: Masson; 1953. p. 692. [Google Scholar]
- 71.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 72.Oren A, Garrity GM. List of new names and new combinations previously effectively, but not validly, published. List no. 164. Int J Syst Evol Microbiol. 2015;65(7):2017–25. doi: 10.1099/ijs.0.000317. [DOI] [PubMed] [Google Scholar]
- 73.Goodfellow M, Jones AL. Order V. Corynebacteriales ord. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 5: The Actinobacteria. New York: Springer; 2012. p. 235. [Google Scholar]
- 74.Orla-Jensen S. The main lines of the natural bacterial system. J Bacteriol. 1921;6(3):263–73. doi: 10.1128/jb.6.3.263-273.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park YH, Suzuki K-i, Yim DG, Lee KC, Kim E, Yoon J, et al. Suprageneric classification of peptidoglycan group B actinomycetes by nucleotide sequencing of 5S ribosomal RNA. Antonie Van Leeuwenhoek. 1993;64:307–13. doi: 10.1007/BF00873089. [DOI] [PubMed] [Google Scholar]
- 76.List Editor. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 53. Int J Syst Bacteriol. 1995;45(2):418–19. [DOI] [PubMed]
- 77.Chester FD. Report of mycologist: bacteriological work. Delaware Agricultural Experiment Station Bulletin. 1897;9:38. [Google Scholar]
- 78.Winslow CEA, Broadhurst J, Buchanan RE, Krumwiede C, Rogers LA, Smith GH. The families and genera of the bacteria: preliminary report of the Committee of the Society of American Bacteriologists on characterization and classification of bacterial types. J Bacteriol. 1917;2(5):505–66. doi: 10.1128/jb.2.5.505-566.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schleifer KH, Bell JA. Family VIII. Staphylococcaceae fam. nov. In: Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 3: The Firmicutes. 2. New York: Springer; 2009. [Google Scholar]
- 80.Orla-Jensen . The Lactic Acid Bacteria. Copenhagen: Host; 1919. [Google Scholar]
- 81.Lehmann KB, Neumann R. Atlas und Grundriss der Bakteriologie und Lehrbuch der speziellen bakteriologischen Diagnostik. München: J.F. Lehmann; 1896. [Google Scholar]
- 82.Migula W. Über ein neues System der Bakterien. Arbeiten aus dem Bakteriologischen Institut der Technischen Hochschule zu Karlsruhe. 1894;1:235–38.
- 83.Rosenbach FJ. Mikro-organismen bei den Wund-Infections-Krankheiten des Menschen. Wiesbaden: J.F. Bergmann; 1884. [Google Scholar]
- 84.Kodama K, Kimura N, Komagata K. Two new species of Pseudomonas: P. oryzihabitans isolated from rice paddy and clinical specimens and P. luteola isolated from clinical specimens. Int J Syst Evol Microbiol. 1985;35(4):467–74. [Google Scholar]
- 85.Kloos WE, Schleifer KH. Isolation and characterization of staphylococci from human skin II. Descriptions of four New species: Staphylococcus warneri, Staphylococcus capitis, Staphylococcus hominis, and Staphylococcus simulans. Int J Syst Evol Microbiol. 1975;25(1):62–79. [Google Scholar]
- 86.Schleifer KH, Kloos WE. Isolation and characterization of staphylococci from human skin I. Amended descriptions of Staphylococcus epidermidis and Staphylococcus saprophyticus and descriptions of three new species: Staphylococcus cohnii, Staphylococcus haemolyticus, and Staphylococcus xylosus. Int J Syst Evol Microbiol. 1975;25(1):50–61. [Google Scholar]
- 87.Behrendt U, Ulrich A, Schumann P. Genus XX. Plantibacter. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 5: The Actinobacteria. 2. New York: Springer; 2012. pp. 943–49. [Google Scholar]
- 88.Schleifer KH, Bell JA. Genus I. Staphylococcus. In: Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 3: The Firmicutes. New York: Springer; 2009. pp. 392–424. [Google Scholar]
- 89.Magee JG, Ward AC. Genus I. Mycobacterium. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology Vol 5: The Actinobacteria. New York: Springer; 2012. pp. 312–75. [Google Scholar]
- 90.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]