

Abstract
Somatic cells harbor random heteroplasmic mitochondrial DNA mutations, which are considered to contribute to aging. In this issue of The EMBO Journal, Perales‐Clemente et al (2016) show that mtDNA mutations, present at low levels in the starting fibroblasts, become enriched in iPS cells and lead to functional defects in iPS‐derived cells. In another recent study, Kang et al (2016) demonstrated that accumulation of mtDNA mutations of somatic origin in iPSCs is age related.
Subject Categories: Stem Cells
Genetic integrity of induced pluripotent stem (iPS) cells is important for both therapeutic and research applications. Several studies have shown that different nuclear DNA aberrations, point mutations, copy number variation, and chromosomal rearrangements are frequent in iPS cells and that the majority of these arise during the reprogramming process (Oliveira et al, 2014). Mitochondrial DNA mutations have also been shown to be present in iPS cells (Prigione et al, 2011), but their extent and origin, as well as their effects on reprogramming to pluripotency, remain unknown. The mitochondrial genome is small but very gene‐rich. It encodes genes essential for oxidative phosphorylation and cellular energy production. Mutations in the mitochondrial DNA (mtDNA) cause a variety of human inherited diseases and are implicated in many age‐related conditions (Park & Larsson, 2011). The mitochondrial genome is a multicopy genome with tens or hundreds of copies present in a cell, depending on the cell type. Pathogenic mutations in mtDNA are often heteroplasmic, meaning that both the healthy and the mutated allele exist in the same cell, and different tissues have different threshold levels for the mutations that they tolerate before manifesting defects (Larsson & Clayton, 1995). Aging is associated with an increase in random mtDNA mutations, either due to replication errors or accumulated damage, and the rate of mtDNA mutations is significantly higher than that of nuclear DNA mutations (Wallace, 2010). Two new studies shed light on the prevalence of mtDNA mutations in iPS cells and their consequences on differentiated cells derived from the iPS cells.
Perales‐Clemente and colleagues studied iPS cells and fibroblasts from mitochondrial disease patients and from healthy donors and report that the majority of iPS clones, independent of the donors' disease status, harbor mtDNA mutations, some at high frequencies, and that all these mutations are present at low frequencies in the parental fibroblasts. Kang et al (2016) reported similar results and showed that while some mtDNA mutations are present at low levels in blood and fibroblasts of healthy adult donors, iPS cells harbor elevated mutation loads. Further, they showed that cloned fibroblast lines show comparable mtDNA mutation levels to the iPS cells, suggesting that although undetectable in whole tissues, the mutations are already present in the individual starting cells and not generated during the reprogramming process.
Kang et al (2016) reported a clear age‐related increase in the mtDNA mutation frequency, whereas no correlation to the donor age was seen in the Perales‐Clemente study. However, the study cohort in the Perales‐Clemente study (0–40 years) falls within the young group (27–50 years) of the Kang study, whereas the increase was seen in the older individuals (60–72 years), suggesting that this age‐related increase in mtDNA mutations is seen only in cells derived from elderly donors. Kang et al (2016) also studied the origin of the mtDNA mutations by looking for shared mutations between blood and fibroblasts of single individuals as well as between a mother–daughter pair. No common variants were shared between the mother and the daughter, and only a small portion (~10%) of the changes were common to blood and skin within individuals, suggesting that the majority of the mutations are of somatic origin and that most do not arise during early development but later in life. Further, they collected oocytes from the same females whose blood, fibroblasts and iPSCs were studied and generated ES lines by in vitro fertilization. Whereas in the iPSCs most of the mtDNA mutations are in protein‐coding regions, in the ES cells the mutations are mostly non‐coding, further suggesting somatic origin of the adult mtDNA mutations. This difference in the mutation types between ES and iPS cells is in accordance with previous animal studies, where selection against detrimental protein‐coding mutations is known to take place in germ cells (Stewart et al, 2008).
Figure 1. iPS cells harbor mitochondrial DNA mutations.
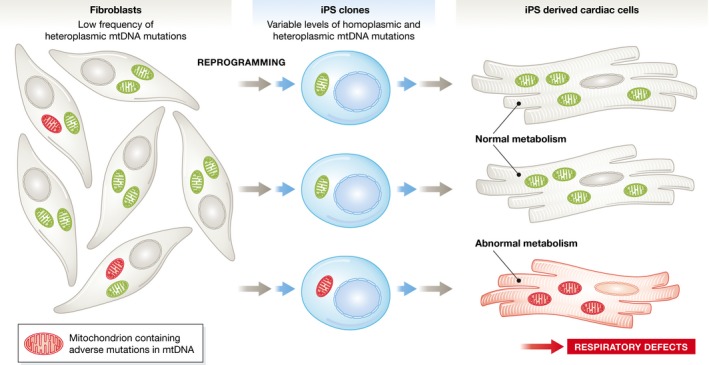
Low levels of heteroplasmic mtDNA mutations are present in somatic cells. Perales‐Clemente and colleagues show that these may become enriched in clonal iPS lines and lead to respiratory defects in differentiated cells derived from the iPS cells.
Both studies showed that most iPSC clones harbor hetero‐ or even homoplasmic mtDNA mutations, but do these mutations have functional consequences on the iPS cells and cells derived from them? To answer this, Perales‐Clemente et al (2016) differentiated the cells to cardiomyocytes and studied respiratory chain function in both the iPS and cardiac cells. The iPS cells are mostly glycolytic and do not need to rely on oxidative phosphorylation to produce energy; thus, no changes in the respiration levels were detected even with high mutation frequencies. On the other hand, cardiac cells have high mitochondrial content and rely heavily on respiration for their energy production. Accordingly, a decrease in both basal and maximal respiration was seen in cells with high (~70%) mutation load of certain mutations, whereas lower frequency (~40%) of the same mutation did not affect respiration. All protein‐coding mutations did not lead to respiratory defects even at high levels (~80%). In the Kang et al (2016) study, similar results with reduced respiration as a consequence of specific mutations were observed in iPSC‐derived fibroblasts.
These two new studies clearly demonstrate that iPSCs may harbor mtDNA mutations at high enough frequencies to cause functional effects. The authors of both papers suggest including mtDNA screens as selection criteria when choosing iPSC clones to work with, and based on their results this may well be justified. For cells to be used for therapeutic or other clinical purposes, mtDNA integrity should clearly be included alongside monitoring for nuclear DNA integrity, especially since the frequency of mtDNA mutations in these new studies is significantly higher than that previously published for the nuclear genome (Johannesson et al, 2014). A basic screen of the small 16‐kb mtDNA genome could also easily be included for clones used for research purposes. Further, for research purposes deep sequencing might not be needed, but Sanger sequencing of the mtDNA should reveal mutations present at high enough levels to cause respiratory problems.
See also: E Perales‐Clemente et al (September 2016) and E Kang et al (May 2016)
References
- Johannesson B, Sagi I, Gore A, Paull D, Yamada M, Golan‐Lev T, Li Z, LeDuc C, Shen Y, Stern S, Xu N, Ma H, Kang E, Mitalipov S, Sauer MV, Zhang K, Benvenisty N, Egli D (2014) Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell 15: 634–642 [DOI] [PubMed] [Google Scholar]
- Kang E, Wang X, Tippner‐Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, Hayama T, Luo S, Harding CO, Amato P, Jensen J, Battaglia D, Lee D, Wu D, Terzic A et al (2016) Age‐related accumulation of somatic mitochondrial DNA mutations in adult‐derived human iPSCs. Cell Stem Cell 18: 625–636 [DOI] [PubMed] [Google Scholar]
- Larsson NG, Clayton DA (1995) Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29: 151–178 [DOI] [PubMed] [Google Scholar]
- Oliveira PH, da Silva CL, Cabral JM (2014) Concise review: genomic instability in human stem cells: current status and future challenges. Stem Cells 32: 2824–2832 [DOI] [PubMed] [Google Scholar]
- Park CB, Larsson NG (2011) Mitochondrial DNA mutations in disease and aging. J Cell Biol 193: 809–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales‐Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA, Terzic A, Nelson TJ (2016) Natural underlying mtDNA heteroplasmy as a potential source of intra‐person hiPSC variability. EMBO J 35: 1979–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, Ralser M, Timmermann B, Adjaye J (2011) Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell‐like metabolic reprogramming. Stem Cells 29: 1338–1348 [DOI] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG (2008) Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 6: e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2010) Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen 51: 440–450 [DOI] [PubMed] [Google Scholar]